SEZIONE II
1 . S O G G E T T O D E L L O S T U D I O
Il soggetto MON, seguito in questo lavoro, faceva parte di un gruppo di individui che, in un precedente studio (Maggi et al., 2005), era stato arruolato per valutare le conseguenze dell’immunizzazione attiva sulla carica virale di TTV.
I soggetti arruolati erano stati in totale 22 (8 femmine, 14 maschi; età media 40.0 ± 12.7; range 28 - 70 anni), tutti negativi per il virus dell’epatite B (HBV) e per il virus dell’immunodeficienza umana (HIV), come dimostrato rispettivamente dall’assenza dell’antigene di superficie (HBsAg) e di specifici anticorpi.
Gli individui erano risultati tutti positivi per la presenza del DNA di TTV. I soggetti non presentavano segni clinici di malattie epatiche e non avevano assunto farmaci antivirali nel corso dell’ultimo anno; nei tre mesi precedenti la vaccinazione, nessuno dei soggetti aveva ricevuto un trattamento immunosoppressivo.
I vaccini furono effettuati contro il virus dell’epatite B e dell’influenza, a distanza di meno un mese l’uno dall’altro. Il soggetto MON fu incluso in entrambi i protocolli di vaccinazione.
In questo lavoro di tesi è stato studiato dettagliatamente il decorso dell’infezione da TTV in MON allo scopo di spiegare il rapido e intenso aumento della carica virale osservata al trentesimo giorno dopo la vaccinazione contro il virus dell’epatite B.
MON è una femmina di 33 anni, siero negativa per i virus dell’epatite B, C e dell’immunodeficienza umana.
I campioni dello studio sono stati ottenuti da MON con prelievi di sangue ad intervalli di tempo definiti: prima della vaccinazione e successivamente dopo 7, 15, 30, 45, 60 e 90 giorni dalla prima vaccinazione (HBV).
Al giorno 240 è stato somministrato il vaccino contro l’influenza e prelievi di sangue sono stati effettuati dopo gli stessi intervalli di tempo della prima vaccinazione.
2. IMMUNIZZAZIONE ATTIVA DEL
SOGGETTO MON
MON aveva ricevuto una formulazione standard dell’antigene di superficie del virus dell’epatite B, derivato da cellule geneticamente ingegnerizzate di lievito (Engerix-B preparation; Glaxo SmithKline Biologicals, Rixensart, Belgium). Il vaccino è stato inoculato per via intramuscolare in tre dosi, ciascuna di 20 µg di antigene ricombinante, lungo un arco di tempo di sei mesi. Quattro settimane dopo la prima iniezione è stata somministrata la seconda dose di vaccino e l’ultima al termine dello studio.
Lo stesso soggetto è stato anche immunizzato con 0,5 ml di vaccino trivalente inattivato del 2002/2003 contro l’influenza (Fluad preparation; Chiron, Siena, Italy) tramite una iniezione intramuscolare nel deltoide.
Nessuna reazione secondaria (febbre, rash cutanei o altro) è stata osservata in MON dopo le due vaccinazioni.
3. SEPARAZIONE DEI LINFOMONOCITI
PERIFERICI (PBMC)
I PBMC e il siero sono stati ottenuti dal sangue in seguito ad un’opportuna procedura di separazione.
La fase iniziale prevede la centrifugazione del sangue intero a 1800 rpm per 10 minuti a temperatura ambiente; grazie a tale processo è possibile ottenere infatti la separazione della componente cellulare del sangue dalla componente liquida. Il siero viene recuperato mentre le cellule, appositamente diluite con PBS, vengono poste a contatto con il Ficoll-Hypaque (Lympho separatium medium, ICN Biomedicals Inc., Aurora, Ohio) in un rapporto 1:1; questo reagente permette la formazione di un gradiente che consente l’ulteriore separazione delle cellule nelle loro sottopopolazioni. Infatti, a seguito di una centrifugata a 1800 rpm per 30 minuti, si assiste alla formazione di diversi strati: il primo, più leggero, costituito dal siero diluito, il secondo dato da un sottile anello di PBMC, il terzo costituito dal Ficoll stesso ed infine l’ultimo strato composto da globuli rossi e granulociti. Il siero diluito viene eliminato, mentre viene recuperato l’anello intermedio di PBMC, facendo attenzione a non prelevare anche il Ficoll.
A questo punto i PBMC vengono sottoposti ad un primo lavaggio con PBS 1X e centrifugati a 1800 rpm per 10 minuti. Il sovranatante viene eliminato mentre il pellet viene risospeso e trattato con 1 ml di tripsina allo 0,5%. Quest’ultimo trattamento è necessario per digerire il virus non internalizzato nella cellula e quindi rimasto all’esterno.
La tripsina viene fatta agire a 37°C per 20 minuti finché la sua azione non viene inibita dal contatto con 2 ml di terreno con siero a temperatura ambiente per alcuni minuti. Per eliminare ogni traccia del mezzo di separazione si effettuano altri due lavaggi con PBMC seguiti da centrifugazioni a 1800 rpm per 10 minuti. Al termine di tutto il procedimento il pellet dei PBMC viene risospeso in 200 µl di PBS e conservato a -80°C fino al momento dell’utilizzo.
4. ESTRAZIONE DEL DNA VIRALE
Utilizzare un efficiente metodo di estrazione e purificazione del DNA virale è fondamentale per ottenere risultati affidabili. Il kit commerciale utilizzato per eseguire le estrazioni del DNA dai campioni in esame è stato il QIAamp DNA mini kit (QIAgen, Chatsworth, CA). Esso fornisce infatti una semplice e rapida metodica che permette di purificare il DNA totale (genomico, mitocondriale e virale) da sangue intero, plasma, siero, fluidi corporei, linfociti, colture cellulari e tessuti, pronto per essere utilizzato nelle successive indagini di biologia molecolare.
L’estrazione è stata eseguita, secondo la procedura indicata dal kit per plasma, fluidi corporei e cellule, a partire da un volume di campione di 200 µl a cui è necessario aggiungere 20 µl di proteinasi K (20 mg/ml) e 200 µl di buffer AL per effettuare la lisi cellulare. Al fine di ottenere un’efficiente azione litica è essenziale agitare vigorosamente la miscela per produrre una soluzione omogenea e incubare poi per 10 minuti a 56°C. Successivamente si effettua una breve centrifugata per rimuovere dal tappo gli eventuali residui di
miscela evaporati e si aggiungono 200 µl di etanolo al 96-100 % per permettere la precipitazione degli acidi nucleici.
La miscela viene di nuovo mescolata con il vortex e centrifugata brevemente prima di essere trasferita in apposite colonnine da 2 ml fornite dal kit (QIAamp spin columns). Queste colonnine contengono al loro interno filtri di gel di silice con elevata affinità per gli acidi nucleici, mentre le condizioni saline e del pH nel lisato assicurano che proteine e altri contaminanti che potrebbero inibire la PCR o le altre reazioni enzimatiche non siano assorbite dal filtro.
Il legame degli acidi nucleici alla membrana contenuta nelle colonnine è garantito da una centrifugata di un minuto a 8000 rpm, che lascia fluire nella provetta di scarico i componenti della miscela non assorbiti dal filtro. Nei passaggi successivi, dopo aver scaricato il filtrato, si eseguono lavaggi con due diversi tamponi per incrementare la purezza del DNA estratto e per assicurare una completa rimozione di eventuali contaminanti, senza agire sul DNA legato. Con il primo lavaggio si aggiungono alla colonnina 500 µl di buffer AW1 (20 mM di NaCl, 2 mM di Tris-HCl, pH 7.5 e etanolo al 57%), si centrifuga a 8000 rpm per un minuto e si getta poi il filtrato ottenuto; il secondo lavaggio prevede l’aggiunta di buffer AW2, che differisce dal precedente solo nel contenuto di alcool
(etanolo al 70%), una centrifugata alla massima velocità per tre minuti e la rimozione del filtrato.
Per eliminare ogni possibile residuo di tampone si esegue un’altra centrifugata di un minuto alla massima velocità e si trasferisce poi ogni colonnina in una eppendorf pulita.
In caso di estrazione da cellule mononucleate del sangue periferico (PBMC) è preferibile eseguire ogni centrifugata alla massima velocità al fine di evitare l’intasamento della colonna. Il DNA trattenuto viene infine eluito con 55 µl di un apposito tampone AE (10 mM Tris-HCl, 0.5 mM EDTA, pH 9) che, oltre a consentire il rilascio del DNA dalla membrana, ne previene anche la degradazione. Dopo aver incubato per circa un minuto a temperatura ambiente, si centrifuga a 8000 rpm per un minuto affinché l’eluato possa filtrare attraverso la membrana delle colonnine. Gli estratti così ottenuti sono conservati a -80°C fino al momento dell’utilizzo.
5. SAGGI DI PCR
5.1 GENERALITA’
La reazione a catena della polimerasi (polymerase chain reaction), comunemente nota con l'acronimo PCR, è una tecnica di biologia molecolare che consente l’amplificazione di frammenti di acidi nucleici dei quali si conoscano le sequenze nucleotidiche iniziali e terminali. Tale metodica, ideata nel 1985 da Kary B. Mullis, consente di ottenere in vitro molto rapidamente la quantità di materiale genetico necessaria per le successive applicazioni, a partire da una sequenza bersaglio presente in concentrazioni minime in un campione di DNA eterogeneo. La PCR permette la sintesi di un segmento di DNA a doppia elica a partire da un filamento a singola elica; il filamento mancante viene ricostruito a partire da una serie di nucleotidi che vengono disposti nella corretta sequenza, complementare a quella del DNA interessato. È necessario che le estremità della sequenza da amplificare siano conosciute con sufficiente precisione per poter sintetizzare degli oligonucleotidi da utilizzare come innesco. Tali sequenze oligonucleotidiche, chiamate primers o amplimeri, sono
complementari a regioni dei filamenti opposti che fiancheggiano la sequenza del DNA bersaglio e sono di solito lunghi da 15 a 25 nucleotidi ciascuno. I primers devono potersi ibridare in maniera specifica ed efficiente alla sequenza d'interesse, tralasciando quelle aspecifiche, e devono avere le proprie estremità ossidriliche orientate l’una verso l’altra per poter agire da innesco nei confronti dell’attività catalitica della DNA polimerasi. Essa infatti è in grado di mediare l’aggiunta di deossiribonucleotidi all’estremità 3’ di una catena di DNA solo in presenza di un primosoma che renda disponibile un 3’-OH. Per avviare la reazione della polimerasi (fase di prolungamento del filamento a partire dal primer 5’) è prima necessario provvedere alla separazione dei filamenti del DNA (fase di denaturazione), quindi alla creazione del legame tra i primers e le regioni loro complementari sui filamenti di DNA denaturati (fase di annealing). Questo processo risulta però incompatibile con la DNA polimerasi umana, che viene distrutta alle temperature necessarie per la denaturazione (96-99 °C). Per ovviare a questo inconveniente si fa ricorso alle polimerasi appartenenti a organismi termofili che non sono inattivate dalle alte temperature, ad esempio la "Taq polimerasi" proveniente dal batterio termofilo Thermus aquaticus.
Ciò consente di realizzare più cicli di PCR in sequenza, in ciascuno dei quali viene duplicato anche il DNA sintetizzato nelle fasi precedenti, ottenendo una reazione a catena che consente una moltiplicazione estremamente rapida del materiale genetico di interesse. Al termine del processo si ottengono 2(n-1) copie della sequenza bersaglio specifica, dove n
rappresenta il numero di cicli.
I requisiti essenziali per ogni PCR sono quindi :
• due primers sintetici che non presentino al loro interno strutture secondarie né regioni di complementarietà reciproche;
• una DNA polimerasi termostabile;
• una quantità opportuna di nucleotidi liberi per costituire i nuovi filamenti;
• un tampone contenente un’adeguata concentrazione di ioni Mg2+,indispensabili al riconoscimento del substrato
da parte dell’enzima.
L’amplificazione viene effettuata con un processo ciclico in tre tempi:
1. Denaturazione: il campione viene portato a una temperatura di 95°C, alla quale la doppia elica del DNA
viene completamente scissa e i due filamenti di cui essa è composta sono liberi e pronti per l’appaiamento dei primers.
2. Appaiamento: la temperatura viene abbassata fino a 55 °C circa al fine di permettere il legame dei primers alle regioni loro complementari dei filamenti di DNA denaturati. La temperatura di annealing va scelta accuratamente, considerando che una temperatura più elevata aumenta la specificità della reazione ma ne può pregiudicare l'efficienza poiché favorisce la separazione dei primers dal bersaglio. Se la temperatura viene abbassata, le condizioni diventano meno stringenti ma viene favorita la formazione di ibridi e quindi di amplificati aspecifici.
3. Sintesi del DNA: la temperatura viene alzata fino a 70-75 °C al fine di massimizzare l'azione della DNA polimerasi che determina un allungamento dei primers legati, utilizzando come stampo il filamento singolo di DNA. Il tempo di questa fase varia a seconda della lunghezza del frammento da amplificare, considerando che, a questa temperatura, la Taq polimerasi catalizza l’aggiunta di 60 basi al secondo.
Il ciclo descritto viene ripetuto generalmente per circa 20-30 volte. In genere non si superano i 50 cicli in quanto ad un certo punto la quota di DNA ottenuto raggiunge un plateau. Ciò avviene, ad esempio, per carenza degli oligonucleotidi usati come inneschi o per diminuzione dei dNTP. Bisogna inoltre considerare che si potrebbe amplificare in maniera eccessiva anche eventuale materiale genomico contaminante. Normalmente un ciclo completo di PCR prevede anche un’ultima fase di estensione a 72°C per 5-15 minuti affinché la Taq polimerasi completi eventuali frammenti tronchi.
5.2 PCR QUANTITATIVA (REAL TIME PCR)
I campioni estratti sono stati analizzati per verificare la presenza e la carica totale di TTV. A tale scopo è stata utilizzata la tecnica di real time PCR, che prevede l’amplificazione dei campioni e la loro misura in tempo reale tramite analisi della fluorescenza.
La tecnica si basa sul principio che maggiore è il numero di molecole stampo presenti all’inizio della reazione e minore sarà il numero dei cicli necessari per raggiungere un determinato valore minimo (soglia) di quantità del prodotto. La concentrazione iniziale della sequenza in esame può quindi essere espressa come numero di cicli richiesti per raggiungere il valore soglia (Ct).
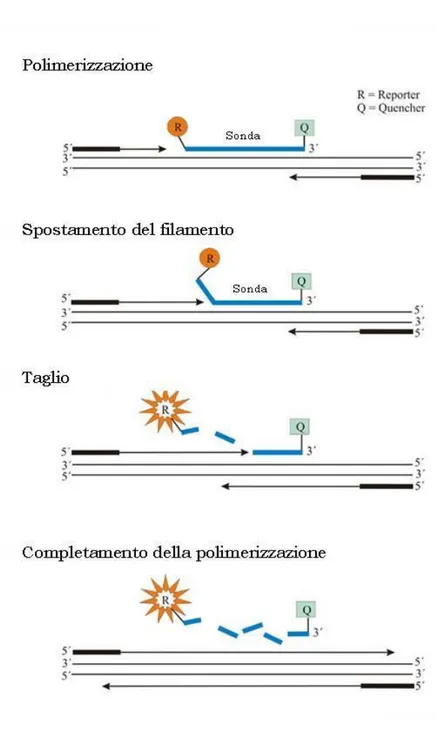
In particolare, per la determinazione della carica virale di TTV, è stata utilizzata la tecnologia TaqMan. Questa tecnica consente la rilevazione dell’amplificato contemporaneamente alla sua amplificazione, producendo un segnale fluorescente con intensità direttamente proporzionale al target di reazione. Essa si avvale dell’uso di una sonda complementare alla sequenza target posta tra i siti di legame dei primers e marcata alle due estremità con due differenti fluorocromi, al 5′
il reporter carbossi-fluoresceina, FAM) ed al 3′ il quencer (6-carbossi-tetrametil-rodamina, TAMRA).
La molecola quencer ha la capacità di inibire la fluorescenza dell’altra molecola fin quando entrambe si trovano sulla stessa sonda integra. Nel corso dell’amplificazione però la sonda viene distrutta mediante idrolisi dall’attività esonucleasica 5’-3’ associata alla polimerasi AmpliTaqGoldTM. L’enzima infatti,
quando incontra l’estremità 5’ della sonda, effettua il distacco del reporter; in questo modo il fluoroforo va in soluzione, non subisce più l’inibizione del quencher ed emette fluorescenza. In base a questo meccanismo l’intensità di fluorescenza emessa dipende dai ripetuti cicli di PCR ed è direttamente proporzionale alla quantità del target di reazione amplificato. L’incremento dell’intensità del segnale emesso è misurato ed interpretato dal sistema Applied Biosystems Prism 7700. Tale apparecchiatura include un termociclizzatore, un laser per indurre la fluorescenza, un rilevatore CCD (charge-coupled device) e un software per la rilevazione in tempo reale.
Il software rileva l’emissione della fluorescenza in base a una precedente normalizzazione, necessaria per correggere le inevitabili fluttuazioni della fluorescenza dovute alle variazioni di concentrazione o volume dei campioni in esame.
A questo scopo è presente, in concentrazione costante nel tampone di reazione, un controllo ottico passivo costituito da un terzo colorante non coniugato (rodamina, ROX).
Questo fluorocromo non è sottoposto all’azione nucleasica della polimerasi e rappresenta, quindi, un riferimento interno con cui può essere normalizzato il segnale emesso dal reporter. La normalizzazione viene effettuata dividendo l’intensità di emissione del reporter per quella del controllo passivo, ottenendo così un indice Rn (reporter normalizzato)
per ogni campione analizzato.
I parametri calcolati dal software sono:
- Rn+ : valore di Rn in una reazione contenente il DNA
bersaglio;
- Rn- : valore di Rn in una reazione senza il DNA bersaglio. Si
ricava nei primi 3-15 cicli di amplificazione, quando l’incremento di fluorescenza non è ancora rilevabile;
- ∆Rn : differenza tra Rn+ e Rn- . Indica la grandezza del segnale
generato in una reazione di PCR;
- CT o ciclo soglia: primo ciclo di amplificazione in cui si ha un
incremento statisticamente significativo del ∆Rn. E’ il valore utilizzato in genere per quantificare i campioni.
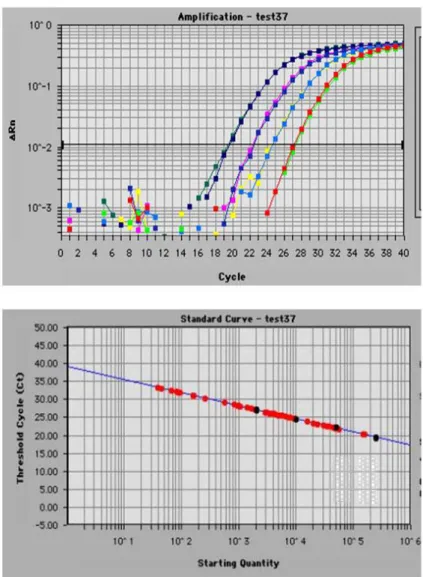
Quest’ultimo parametro viene determinato, all’interno di ciascuna reazione, anche per una serie di standard per i quali
è noto il numero di copie. Ne deriva una curva di taratura dalla quale viene dedotta la concentrazione dei diversi campioni in esame (figura II.2).
Ogni campione viene ripetuto in triplo e se si osservano coefficienti di variazione maggiori del 50% la valutazione non è ritenuta attendibile.
La quantificazione della carica virale di TTV nei campioni in esame è stata effettuata tramite real time PCR; i primers e la sonda TaqMan sono stati dedotti sulla base di una regione del genoma non tradotta (UTR, untraslated region), altamente conservata in tutte le sequenze di TTV fino ad oggi identificate. Prima del suo utilizzo, la metodica di real-time è stata sperimentata per verificarne le performances in termini di linearità, specificità e sensibilità. A tale scopo sono stati prodotti degli standard di riferimento opportunamente quantificati, utilizzati poi nei diversi esperimenti. Questi standard sono stati ottenuti con diluizioni seriali e valutati allo spettrofotometro per produrre da un minimo di 103 fino ad
un massimo di 106 copie. I prodotti di PCR sono stati poi
analizzati su gel di agarosio per confermarne la specificità. Il sistema si è dimostrato in grado di quantificare la carica virale dei campioni in esame con ottima precisione e con differenze minime fra il numero di copie analizzate e il numero
di copie calcolate (tra 880 e 1200 per gli standard di 103 copie
e tra 810.000 e 1.360.000 per gli standard di 106 copie) e con
variazioni intra e inter-saggio nei cicli soglia che non superavano rispettivamente il 2,4% e il 3,1%. Risultati simili, con variazioni dei titoli inferiori a 0,6 log, sono stati ottenuti anche quando l’analisi è stata effettuata su campioni biologici estratti e processati separatamente in diverse ed indipendenti reazioni. La specificità del saggio è stata confermata attraverso l’analisi di sequenza dei prodotti dell’amplificazione mentre, con le procedure standard utilizzate, il limite di sensibilità inferiore del sistema è stato stimato essere pari a 1.0x103
copie di DNA di TTV per ml di plasma o per µg di DNA estratto.
Come già detto in precedenza, è necessario avere a disposizione degli standard di riferimento nel processo di quantificazione. A questo scopo il DNA di TTV è stato estratto dal siero di un paziente ed amplificato mediante nested PCR con primers dedotti dalla UTR del genoma virale. I primers utilizzati sono:
CLONS primer senso I step: (nt 105-133) 5′-GTTTTCCACGCCCGTCCGC-3′;
CLONAS primer antisenso I step: (nt 236-253) 5′-AGAGCCTTGCCCATAGCC-3′;
AMTS primer senso II step: (nt 177-194) 5′-GTGCCGIAGGTGAGTTTA-3′;
AMTAS primer antisenso II step: (nt 226-239) 5′-AGCCGGCCAGTCC-3′.
L’amplificato ottenuto è stato inserito in un vettore plasmidico e transfettato in cellule competenti. I cloni ottenuti sono stati poi sequenziati e quantificati mediante lettura spettrofotometrica a 260 nm. Dalla misura della densità ottica, nota la lunghezza in nucleotidi del vettore ricombinante, è stato possibile determinare il numero esatto di copie ottenute con la purificazione e preparare, attraverso una serie di diluizioni successive, degli standard di riferimento a numero noto di copie di DNA.
La miscela di amplificazione deve avere un volume totale di 25 µl ed essere composta da:
Primers AMTS e AMTAS (Fornai et al., 2001), con concentrazione finale 0,9 µM;
Sonda AMTPU: 5’-FAM-TCAAGGGGCAATTCGGGCT-TAMRA-3’ (nt 205-223) alla concentrazione finale di 0,1 µM. FAM indica la molecola reporter e TAMRA il quencher. I primers e la sonda sono stati progettati utilizzando il programma “Primers Express Software” (versione 1.0 Perkin- Elmer Applied Biosystems).
Universal Master Mix alla concentrazione finale 1X, contenente la polimerasi AmpliTaq Gold, l’enzima AmpErase uracil-N-glycosilase (UNG) e i nucleotidi in cui dTTP è sostituito da dUTP, incorporato nella sintesi TaqMan al posto di dTTP. L’enzima UNG agisce sul DNA a doppio filamento tagliando la base uracilica e creando un sito apirimidinico che blocca la sintesi da parte della DNA polimerasi e rende la doppia elica molto sensibile all’idrolisi acido basica. In questo modo viene impedita l’ulteriore amplificazione dei prodotti delle precedenti reazioni di PCR, attraverso la degradazione di quei siti genomici che contengono dUTP (Longo et al., 1990). L’enzima UNG è attivo alla temperatura di 50°C e viene poi inibito alla temperatura di denaturazione, prima della caratteristica reazione di PCR.
5 µl del DNA estratto pari ad una concentrazione di 10-100 ng
X µl di acqua deionizzata per raggiungere il volume finale di 20 µl.
Nella reazione vengono anche utilizzati dei controlli negativi che non contengono DNA ma acqua, in modo tale da poter verificare che la miscela preparata non abbia subito
contaminazioni. L’amplificazione è condotta utilizzando il sistema ABI 7700 secondo il seguente profilo ciclico:
Fase di attivazione della UNG: 1 ciclo a 50°C per 2 minuti.
Fase di attivazione della polimerasi AmpliTaq GoldTM: 1
ciclo a 95°C per 10 minuti.
Fase di denaturazione: 40 cicli a 95°C per 15 secondi. Fase di ibridazione dei primers ed estensione: 60°C per 1 minuto.
Figura II.2: Esempio di curva di amplificazione e retta di taratura effettuate con la metodica TaqMan.
5.3 PCR QUALITATIVA GRUPPO SPECIFICA
Dopo la quantificazione della carica virale di TTV nei diversi campioni in esame, è stato possibile, tramite l’utilizzo di questa metodica di PCR, identificare quali genogruppi del virus fossero presenti nel plasma dei pazienti analizzati.
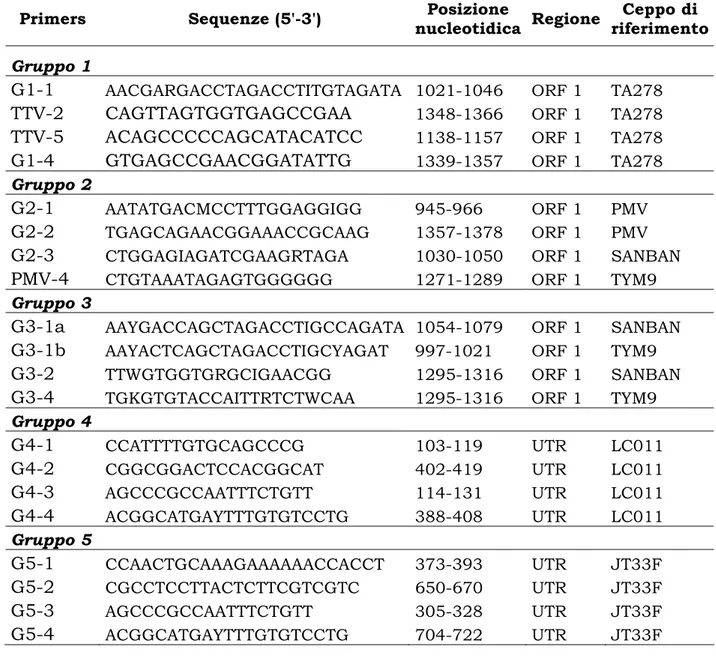
La determinazione dei gruppi in cui TTV è attualmente classificato è stata infatti realizzata con l’uso di 5 differenti reazioni di PCR progettate su regioni diverse del genoma virale. A questo scopo sono stati progettati primers specifici per ogni genogruppo cercando di evitare combinazioni di primers con strutture a forcina o formanti dimeri. Facendo riferimento a tutte le sequenze dei vari isolati virali depositate in banca dati, sono stati progettati oligonucleotidi sintetici nelle regioni ORF1 e UTR del genoma di TTV, capaci di discriminare i diversi genogruppi (tabella II.1). Per aumentare la specificità delle reazioni, le amplificazioni sono state eseguite con reazioni di nested o eminested PCR, condotte in due tempi successivi e con due diverse coppie di primers. Quelli utilizzati nel secondo step sono stati infatti studiati e costruiti in modo tale da appaiarsi internamente al frammento già amplificato durante il primo step.
Ogni coppia di primers necessita di una specifica temperatura di appaiamento con il doppio filamento di DNA e, generando frammenti di lunghezza ben definita, utilizza cicli di amplificazione con tempi diversi (tabella II.2 ).
Per tutte le amplificazioni viene utilizzato lo stesso protocollo, che differisce solo per i primers utilizzati e per la temperatura a cui essi si appaiano al doppio filamento.
Il primo step prevede la preparazione di una miscela di reazione del volume finale di 50 µl, composta da:
Tampone di reazione (10 mM Tris-HCL pH 8,2, 50 mM kCl, 1,5 mM MgCl2);
200 µM di ogni desossiribonucleotide trifosfato (dNTPs); 0,6 µM di ciascun primer (senso e antisenso);
5 U di Taq DNA polimerasi; 5 µl di DNA estratto;
X µl di acqua per raggiungere il volume di 50 µl.
Preparata la miscela vengono effettuati 35 cicli con il seguente profilo termico:
1. fase di denaturazione: 94°C per 30 secondi;
2. fase di appaiamento: temperatura di annealing per 30 secondi;
4. fase di estensione finale: al termini dei 35 cicli si termina l’estensione con 15 minuti a 72°C.
Il secondo step viene effettuato trasferendo 5 µl del prodotto del primo step in nuovi tubi di reazione contenenti una miscela analoga a quella del primo step ma con primers interni al frammento già amplificato. Il profilo di amplificazione del secondo step è stato quindi adattato ai nuovi primers utilizzati e comprende quindi le stesse fasi, ma con tempi e temperature diverse, per un totale di 25 cicli anziché 35. Le reazioni sono state eseguite in termociclizzatori automatici Perkin-Elmer 9600 (Cetus Corporation, Norwalk, CT, USA). La validità delle reazioni di PCR è stata determinata tramite la presenza di un controllo positivo mentre la presenza di eventuali contaminanti è stata esclusa inserendo in ogni reazione un numero di controlli negativi pari al 33% del totale dei campioni analizzati. Ogni campione è stato amplificato in doppio e in caso di discordanza il campione è stato nuovamente estratto e amplificato due volte.
I prodotti della reazione sono stati analizzati mediante corsa elettroforetica di 15 µl di amplificato uniti a 3 µl di colorante su gel d’agarosio al 3%, colorato con etidio bromuro. La dimensione dei prodotti di amplificazione è stata valutata per comparazione con un marker molecolare di riferimento.
Per ciascuna reazione di genotipizzazione è stata misurata la sensibilità utilizzando come standard diluizioni scalari di plasmidi ricombinanti per i frammenti d’interesse; il numero di copie di DNA per ogni diluizione è stato calcolato tramite lettura spettrofotometrica a 260 nm. I risultati ottenuti hanno dimostrato che il limite inferiore di sensibilità per tutte le reazioni di tipizzazione era al di sotto delle 100 copie di plasmide, equivalenti a circa 4000 copie di virus per ml di plasma.
Primers Sequenze (5'-3') Posizione nucleotidica Regione Ceppo di riferimento
Gruppo 1
G1-1 AACGARGACCTAGACCTITGTAGATA 1021-1046 ORF 1 TA278
TTV-2 CAGTTAGTGGTGAGCCGAA 1348-1366 ORF 1 TA278
TTV-5 ACAGCCCCCAGCATACATCC 1138-1157 ORF 1 TA278
G1-4 GTGAGCCGAACGGATATTG 1339-1357 ORF 1 TA278
Gruppo 2
G2-1 AATATGACMCCTTTGGAGGIGG 945-966 ORF 1 PMV
G2-2 TGAGCAGAACGGAAACCGCAAG 1357-1378 ORF 1 PMV
G2-3 CTGGAGIAGATCGAAGRTAGA 1030-1050 ORF 1 SANBAN
PMV-4 CTGTAAATAGAGTGGGGGG 1271-1289 ORF 1 TYM9
Gruppo 3
G3-1a AAYGACCAGCTAGACCTIGCCAGATA 1054-1079 ORF 1 SANBAN
G3-1b AAYACTCAGCTAGACCTIGCYAGAT 997-1021 ORF 1 TYM9
G3-2 TTWGTGGTGRGCIGAACGG 1295-1316 ORF 1 SANBAN
G3-4 TGKGTGTACCAITTRTCTWCAA 1295-1316 ORF 1 TYM9
Gruppo 4 G4-1 CCATTTTGTGCAGCCCG 103-119 UTR LC011 G4-2 CGGCGGACTCCACGGCAT 402-419 UTR LC011 G4-3 AGCCCGCCAATTTCTGTT 114-131 UTR LC011 G4-4 ACGGCATGAYTTTGTGTCCTG 388-408 UTR LC011 Gruppo 5 G5-1 CCAACTGCAAAGAAAAAACCACCT 373-393 UTR JT33F G5-2 CGCCTCCTTACTCTTCGTCGTC 650-670 UTR JT33F G5-3 AGCCCGCCAATTTCTGTT 305-328 UTR JT33F G5-4 ACGGCATGAYTTTGTGTCCTG 704-722 UTR JT33F
Tabella II.1: Caratteristiche dei primers utilizzati nella genotipizzazione di TTV.
Gruppo Primers Step Temperatura di annealing (°C) Dimensioni attese (bp) G1-1 TTV-2 I 50 346 TTV-5 1 G1-4 II 50 220 G2-1 G2-2 I 50 434 G2-3 2 PMV-4 II 50 260 G3-1a G3-1b G3-2 I 55 334 G3-1a G3-1b 3 G3-4 II 55 263 G4-1 G4-2 I 52 317 G4-3 4 G4-4 II 52 295 G5-1 G5-2 I 58 418 G5-3 5 G5-4 II 55 271-306
Tabella II.2: Temperature di appaiamento dei primers al doppio filamento e dimensioni attese del frammento amplificato.
Figura II.3: Rappresentazione schematica delle regioni bersaglio dell’amplificazione nel genoma di TTV.
6. ELETTROFORESI SU GEL DI AGAROSIO
L'elettroforesi è un metodo di separazione di molecole semplice e veloce che permette di identificare i frammenti di DNA amplificati con le reazioni di PCR. Questa metodica è basata sulla diversa velocità di migrazione di particelle elettricamente cariche attraverso una soluzione, sotto l'influenza di un campo elettrico. Essa viene condotta tramite un setaccio molecolare attraverso il quale vengono fatte passare le diverse molecole: la velocità di migrazione dipende da massa, dimensione, carica e forma delle varie particelle, ossia dalla loro mobilità elettroforetica. Può essere anche regolata dalla percentuale di agarosio nel gel e dal voltaggio applicato.
La maggior parte delle separazioni elettroforetiche di campioni di DNA viene eseguita utilizzando gel di agarosio. Questo è dovuto al fatto che l'agarosio, un polisaccaride ricavato dalle alghe marine, ha pori di dimensioni adeguate per la maggior parte delle molecole di DNA e dei frammenti di acido nucleico normalmente analizzati. I frammenti migrano, sotto l’influenza del campo elettrico che attraversa il gel, dal polo negativo a quello positivo, in funzione delle cariche elettriche negative conferite al DNA dai gruppi fosfato.
La separazione viene ottenuta in base alla resistenza al movimento dovuta alla matrice del gel; frammenti più piccoli migrano più velocemente rispetto a quelli più grandi, mentre le molecole più grosse incontreranno più difficoltà a passare attraverso le maglie del gel.
I gel di agarosio utilizzati in questo lavoro sono stati preparati sciogliendo al calore una quantità variabile di agarosio, (dallo 0,6 al 4% in base a dimensioni decrescenti dell’amplificato) nel buffer TAE ( Tris acetato 50X, 242g Tris base, 57,1 ml di acido acetico glaciale, 100 ml di EDTA 0,5M, pH 8.0), utilizzato anche come tampone di corsa. La miscela viene poi colata in un apposito lettino per elettroforesi orizzontale contenente due pettini con denti che dividono il gel in una o due file di pozzetti in cui verranno caricati i campioni da analizzare. A questo punto si lascia polimerizzare a temperatura ambiente.
I campioni di DNA derivati dalle reazioni di PCR vengono preparati aggiungendo un colorante (0,25% blu di bromofenolo, 0,25% xilene cianolo, 30% glicerolo) in un rapporto 1:10. Esso ha la duplice funzione di facilitare il controllo della corsa elettroforetica e di appesantire i campioni permettendone la deposizione sul fondo del pozzetto del gel, grazie al glicerolo in esso contenuto. Il lettino contenente il gel solidificato viene immerso nella cella elettroforetica contenente
il tampone di corsa e i campioni caricati nei pozzetti che si formano in seguito alla rimozione dei pettini. Viene poi applicata una differenza di potenziale costante, compresa tra 60 e 120 volt per un tempo variabile, in base al tipo di separazione richiesta.
La visualizzazione delle bande contenenti gli amplificati è resa possibile dall’aggiunta al gel, durante la fase di polimerizzazione, di bromuro d’etidio, un colorante fluorescente che viene stimolato utilizzando luce ultravioletta a una lunghezza d'onda compresa tra 254 e 306 nm.
Le dimensioni dei frammenti sono calcolate in base a un opportuno marcatore di peso molecolare caricato e corso nel gel insieme ai campioni. Il gel viene osservato a un transilluminatore (Fotodyne) e fotografato con un apparecchio Polaroid.
7. ESTRAZIONE DI DNA DA GEL
Dopo aver effettuato la corsa elettroforetica, per un tempo sufficiente a garantire una buona separazione dei frammenti, è possibile estrarre dal gel le singole bande, visibili al transilluminatore UV. Per il protocollo di estrazione è stato utilizzato il kit commerciale QIAquick Spin Hand (QIAGEN) con il quale i frammenti amplificati, di lunghezza compresa tra 70 e 10.000 basi, prima aderiscono a una membrana di gel di silice e poi sono rilasciati nell’eluato.
Per facilitare il taglio delle singole bande, i prodotti di PCR vengono sottoposti a una corsa elettroforetica su gel di agarosio al 3%, utilizzando pettini con pozzetti di caricamento più larghi. La banda viene tagliata con un bisturi sterile per evitare la contaminazione con altri amplificati e poi pesata. In base al peso in mg della parte recisa, viene calcolata la quantità di buffer QG che deve essere aggiunto per permettere la completa solubilizzazione del gel e il successivo legame del DNA alla membrana della colonna. Il kit suggerisce di utilizzare 3 volumi di buffer per ogni volume di gel, considerando che 100 mg corrispondano circa a 100 µl.
Il buffer QG contiene un indicatore di pH che rende possibile la determinazione del pH ottimale per il legame del DNA. L’assorbimento della maggior parte degli acidi nucleici richiede infatti un pH ≤ 7.5, valori a cui l’indicatore risulta essere giallo. Tale soluzione poi non interferisce nel processo di isolamento e viene rimossa nei successivi passaggi di lavaggio. A questo punto si lascia incubare la miscela a 50°C per 10 minuti. Per aumentare la resa dell’estrazione viene aggiunto un volume di isopropanolo in base al peso del gel e la miscela viene quindi trasferita in colonna (QIAquick column). Si centrifuga a 12.300 x g per 1 minuto, in modo tale che il DNA rimanga adeso al filtro, mentre i primers e i nucleotidi derivati dalla reazione di PCR ed eventuali impurità come sali, enzimi, agarosio, colorante fluiscano nella provetta di scarico.
Dopo aver rimosso l’eluato si esegue un lavaggio con 750 µl di buffer PE e si centrifuga a 12.300 x g per 1 minuto, affinché l’etanolo contenuto nel buffer PE elimini ogni residuo di sali. Per eliminare gli eventuali residui di buffer PE che potrebbero interferire con le successive reazioni enzimatiche, si effettua una nuova centrifugazione a 12.300 x g per 1 minuto.
Le colonne vengono quindi trasferite in eppendorf pulite e alla fine si eluisce il DNA con 40 µl di buffer EB (10 mM Tris HCl; pH 8,5); contrariamente a quanto avviene per l’assorbimento,
l’eluizione è più efficiente in condizioni basiche e a bassa concentrazione salina. Dopo aver incubato per qualche minuto a temperatura ambiente, affinché il filtro venga inumidito completamente e il processo sia reso più efficiente, si centrifuga a 6.000 x g per 1 minuto.
L’eluato ottenuto contiene i frammenti di DNA purificati e l’estrazione può essere verificata tramite una corsa su gel di controllo.
8. IL SEQUENZIAMENTO
8.1 LA METODICA
Il sequenziamento è una procedura che ha come scopo quello di determinare l'esatta sequenza di nucleotidi dell'acido nucleico in esame. L’utilizzo di questa metodica è stato necessario per confermare l’appartenenza dei nostri amplificati ai gruppi di TTV fino ad oggi conosciuti ed eventualmente valutare la presenza di mutazioni nelle regioni scelte come bersaglio di amplificazione. Il sequenziamento, infatti, definendo la sequenza nucleotidica di ogni campione analizzato, consente il confronto dei dati ottenuti con quelli presenti e continuamente aggiornati in banca dati.
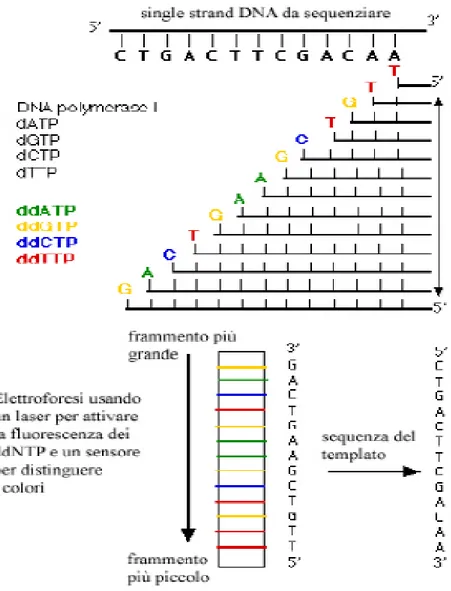
Sono state ideate diverse strategie per ottenere la sequenza nucleotidica del DNA. Il primo fu un metodo chimico, ideato da Maxam e Gilbert nel 1965, seguito nel 1977 dal metodo di sequenziamento enzimatico di Sanger, che è ormai il più utilizzato. Questa metodica viene anche definita a terminazione di catena o metodo dei dideossinucleotidi e richiede la presenza del DNA da sequenziare sotto forma di stampo a singolo filamento.
La DNA polimerasi, fornita dalla miscela di reazione, utilizza questo stampo per la sintesi di nuovi filamenti di DNA complementari; la reazione avviene utilizzando uno o più nucleotidi marcati ed un primer complementare a una regione del DNA adiacente al sito da analizzare. Oltre ai normali precursori nucleotidici (dNTPs), la sintesi del DNA avviene anche in presenza di dideossinucleotidi base-specifici (ddNTPs) che mancano di un gruppo ossidrile al carbonio in 2’ e 3’. I ddNTPs vengono incorporati facilmente nella catena nascente di DNA ma ne impediscono l’ulteriore allungamento. Essi infatti sono in grado di formare un legame fosfodiesterico tra l’atomo di carbonio in 5’ e quello in 3’ del nucleotide precedentemente incorporato ma, essendo privi del gruppo ossidrilico in 3’, non possono formare il legame fosfodiesterico con il nucleotide successivo e bloccano così la polimerizzazione della catena.
Questo metodo richiede la suddivisione del campione, opportunamente denaturato, in quattro aliquote e si portano quindi avanti quattro reazioni diverse base-specifiche. In ognuna delle quattro aliquote c’è ovviamente un ddNTP diverso e di conseguenza in ognuna di esse la sintesi si arresta dopo una specifica base. Ognuna delle quattro miscele di reazione sarà composta, oltre che dal campione da
sequenziare, anche dalla DNA polimerasi, primer, buffer e cloruro di magnesio, i quattro dNTPs e una piccola proporzione di uno dei quattro ddNTPs. Si producono così frammenti di DNA di lunghezza diversa poiché il nucleotide che blocca il processo, essendo anche in concentrazione minore rispetto al suo analogo dNTP, può essere internalizzato a livello di qualsiasi posizione del genoma.
Ciascuna delle quattro reazioni base-specifiche genera una collezione di frammenti di lunghezze diverse, con l’estremità 5’ comune, perché definita dal primer di sequenziamento, ed estremità 3’ variabili. I frammenti ottenuti, diversi in lunghezza anche per un singolo nucleotide, sono separabili su un gel denaturante di poliacrilammide ad alta risoluzione. I metodi tradizionali di sequenziamento prevedevano l’utilizzo di nucleotidi marcati con radioisotopi (35S o 33P) e la
visualizzazione delle bande tramite analisi autoradiografica. Oggi, invece, grazie al recente sviluppo di sequenziatori automatici che sfruttano la metodica di Sanger, il processo è effettuato molto più rapidamente. Il metodo automatico richiede l’uso di dideossinucleotidi marcati con fluorofori diversi a seconda della base azotata presente, che emettono fluorescenza quando sono sottoposti ad una certa lunghezza d’onda.
Il risultato di questa reazione produrrà dei frammenti di DNA nei quali le diverse basi saranno identificate da quattro colori diversi e ogni colorante reagisce alla luce emettendo una propria fluorescenza.
In sostanza, dopo l’avvenuta amplificazione, i campioni vengono purificati e inseriti nel sequenziatore; qui sono sottoposti ad una elettroforesi capillare, durante la quale il campione viene colpito da una sorgente luminosa (laser) e i vari coloranti emettono una fluorescenza che viene rilevata da un sensore; il segnale elaborato da un opportuno software mostra in forma grafica a quattro colori la sequenza del DNA (elettroferogramma).
8.2 REAZIONE DI SEQUENZA
Il protocollo di amplificazione del frammento da sequenziare prevede l’utilizzo di una miscela standard, contenente tutti gli elementi necessari alla reazione, alla quale vengono poi aggiunti il primer di innesco, il campione e acqua deionizzata per raggiungere il volume finale. Questa miscela, detta Mix Terminator Ready Reaction, contiene una particolare polimerasi, la AmpliTaq DNA polimerasi FS, simile all’enzima
prodotto da Thermus aquaticus, ma ottenuta da un gene con due mutazioni puntiformi sito-specifiche.
Una di queste mutazioni determina la sostituzione dell’aminoacido fenilalanina con una tirosina nel sito catalitico dell’enzima, portando ad un aumento di affinità nei confronti dei dideossinucleotidi, substrato non naturale per l’enzima. L’altra mutazione elimina l’attività esonucleasica 5’-3’, evitando così che i nucleotidi appena inseriti nel filamento di nuova sintesi vengano eliminati. La polimerasi viene associata ad una pirofosfatasi stabile al calore che determina il taglio del pirofosfato inorganico (PPi) generato dalla reazione di estensione del DNA, impedendone l’accumulo. Se così fosse, si favorirebbe infatti la reazione inversa alla polimerizzazione, cioè i nucleosidi monofosfato marcati potrebbero staccarsi dal filamento in crescita ed essere sostituiti da quelli non marcati, impedendo l’identificazione del nucleotide in quella posizione. La Mix Terminator contiene, oltre alla polimerasi, anche un tampone Tris HCl a pH 9, magnesio, deossinucleosidi trifosfato e i quattro dideossinucleotidi marcati. Tra i deossinucleotidi è incluso dITP al posto di dGTP e dUTP al posto di dTTP per facilitare l’incorporazione di ddTTP.
La miscela della reazione di sequenza ha un volume totale di 20 µl ed è costituita da:
4 µl di Mix Terminator Ready Reaction; 3,2 µl di primer diluito 1:20;
X µl di campione da valutare in seguito alla concentrazione di DNA presente;
• • •
Y µl di acqua deionizzata per raggiungere il volume finale stabilito.
La reazione di sequenza viene introdotta in un termociclizzatore automatico e sottoposta ad un programma di amplificazione che prevede 25 cicli così strutturati:
Denaturazione: 96° C per 10 secondi.
Ibridazione del primer: 50°C per 5 secondi. Estensione: 60°C per 4 minuti.
8.3 PURIFICAZIONE SU COLONNA
Una volta terminata l’amplificazione, il prodotto di PCR deve essere purificato prima di essere sottoposto ad elettroforesi. La purificazione è necessaria per eliminare eventuali residui di tampone, sali e nucleotidi marcati che potrebbero interferire con la rivelazione della fluorescenza durante l’elettroforesi.
Per tale processo sono state utilizzate le colonne Centri Sep (Princeton Separation), costituite da un gel liofilizzato a cui è necessario aggiungere 800 µl di acqua deionizzata per ottenere così un buon mezzo di purificazione.
Le colonne vengono lasciate ad idratare per circa due ore, prestando attenzione che nella colonna non si formino delle bolle; queste potrebbero infatti creare delle corsie preferenziali al momento del passaggio della soluzione nella colonna, impedendo un corretto processo di purificazione. Dopo l’idratazione viene prima eliminata l’acqua già sgocciolata per gravità nella colonna, poi viene effettuata una centrifugata a 650 x g per due minuti, per eliminare tutta l’acqua. A questo punto si trasferisce la miscela proveniente dalla reazione di amplificazione al centro delle colonne e si centrifuga di nuovo a 650 x g per due minuti.
Il campione è ora purificato dai prodotti di scarto e ne vengono trasferiti 5 µl in altri tubini, insieme a 15 µl di acqua. La miscela ottenuta viene sottoposta a denaturazione per circa due minuti a 94°C e posta poi in ghiaccio per mantenere i filamenti denaturati.
8.4 SEQUENZIAMENTO
Dopo la denaturazione, i frammenti di DNA, contenenti all’estremità 3’ uno dei quattro dideossinucleotidi marcati, sono pronti per essere analizzati dallo strumento ABI PRISM 310 DNA Sequencer. Tale strumento realizza l’elettroforesi e analizza, con l’aiuto di un software adeguato, l’emissione della fluorescenza.
L’apparecchio è costituito da due unità: una camera elettroforetica dotata di un raggio laser fisso e un computer esterno collegato alla camera. La camera è suddivisa in una zona contenente un carrello removibile su cui vengono posti i tubi con la miscela da sequenziare, i contenitori del tampone e dell’acqua, il catodo ed un’estremità del capillare in cui avviene l’elettroforesi. Un’altra zona contiene un recipiente con il tampone di corsa in cui è posto l’anodo ed un sistema costituito da una siringa che inietta un polimero nel capillare. L’ultima zona, infine, è quella in cui il capillare giunge in prossimità del raggio laser. Il capillare è un tubo di vetro di 50 µm di diametro e di lunghezza variabile, con un rivestimento opaco per tutta la sua lunghezza, eccetto che nella zona attraversata dal laser. Durante l’elettroforesi, i frammenti di DNA marcati a singolo filamento si muovono all’interno del
capillare contenente il polimero POP-6 (Performancer Optimizer Polymer 6%) in direzione dell’anodo e, colpiti dal laser, emettono fluorescenza. A contatto con il capillare e con gli elettrodi si trova il tampone di corsa (Genetic Analyzer Buffer), in modo tale che si possa generare la differenza di potenziale necessaria alla corsa elettroforetica. Una volta generata questa differenza di potenziale, gli elettrodi e il tampone vengono a contatto con il primo campione, il quale è iniettato all’interno del capillare per via elettrocinetica, cioè grazie a un flusso di corrente che si genera dal catodo verso l’anodo. Successivamente il catodo e il capillare sono di nuovo immersi nel tampone per generare una nuova differenza di potenziale che permetta la migrazione dei frammenti di DNA attraverso il polimero. Il sequenziatore è anche dotato di un contenitore con acqua deionizzata per lavare il capillare quando questo si sposta da un campione all’altro.
In prossimità della “zona finestra” del capillare, i frammenti di DNA vengono eccitati dal raggio laser ed emettono fluorescenza. Il raggio attraversa una serie di lenti e giunge ad uno spettrografo che separa le lunghezze d’onda e le indirizza verso una camera CCD (Charge-Coupled-Device) dotata di filtri virtuali diversi a seconda dei fluorocromi che devono essere analizzati.
Questa camera esamina lo spettro di emissione e converte il segnale luminoso in un segnale digitale che viene memorizzato sul computer per il successivo studio. In questo modo il nucleotide finale di ogni frammento viene identificato grazie al fluorocromo all’estremità 3’ e i frammenti di diversa grandezza permettono di discriminare gradatamente tutti i nucleotidi. Nel caso di regioni da sequenziare abbastanza corte, la corsa si protrae per circa 30 minuti a campione; alla fine i dati raccolti sono riportati su tipici grafici caratterizzati da picchi di quattro colori diversi, ognuno dei quali rappresenta l’intensità di fluorescenza emessa dai vari fluorocromi, riportata sull’asse y. L’asse x riporta invece la lunghezza della sequenza esaminata in paia basi.
8.5 ANALISI DELLE SEQUENZE
La sequenza consensus per ciascun amplificato, ottenuta dal confronto tra il filamento senso e quello antisenso, è stata confrontata in banca dati al fine di verificare la specificità dell’amplificazione (http://www.ncbi.nlm.nih.gov/BLAST/). Per l’analisi filogenetica, le sequenze ottenute sono state poi allineate con quelle omologhe presenti in banca dati, tramite il
programma Clustal W, disponibile sul server EBI (http://www.ebi.ac.uk/clustalw/). Le relazioni filogenetiche sono state stimate con il metodo FastMe fornito dal programma DAMBE (Data Analisys in Molecular Biology and Evolution, versione 4.2.13, Xia 2000).
Figura II.4: Rappresentazione schematica del metodo di sequenziamento di Sanger.
9. PRECIPITAZIONE IMMUNOCOMPLESSI
DI TTV
Per precipitare gli immunocomplessi di TTV è stata utilizzata una metodica descritta per altri virus (Kimura et al., 2000). Per prima cosa il siero è stato centrifugato a 6200 x g per 5 minuti in modo tale da pellettizzare ed eliminare proteine ed altri fattori del siero.
La quantità di siero utilizzata varia da 10 a 50 µl a seconda del carico virale del paziente. Dopo la centrifugata il sovranatante viene mescolato con 5 volumi di anti-immunoglobuline umane di coniglio diluite 1:10 (IgG; Dade Behiring, Marburg, Germany). Sono stati effettuati anche controlli negativi in cui non si usano le anti-IgG ma il siero di coniglio. Questa miscela viene incubata overnight a 4°C e poi centrifugata a 14.000 x g per 10 minuti. Il sovranatante viene quindi recuperato, mentre il pellet viene sottoposto a due lavaggi consecutivi e poi risospeso in 140 µl di soluzione fisiologica. Sia il sovranatante che il pellet sono stati poi sottoposti ad estrazione del DNA virale e quindi esaminati al Taqman per quantificare la carica di TTV.