Sezione 2
MATERIALI E METODI
2.1: Raccolta e conservazione dei campioni
Tutti i campioni utilizzati nel presente lavoro provengono da due ambienti di stagno costiero situati presso la foce del fiume Serchio (Stagno 1: 43°47’16’’N–10°16’02’’E; Stagno 2: 43°47’05’’N–10°16’06’’E). La salinità di tali ambienti registrata durante il periodo di studio è risultata essere compresa fra il 14‰ ed il 4‰ , nello Stagno 1, e fra il 3‰ e lo 0‰ nello Stagno 2; la temperatura dell’acqua fra 24,3°C e 26,9°C a livello della superficie del sedimento, e fra 27,2°C e 27,5°C a livello della colonna d’acqua a 5 cm dalla superficie (dati riferiti al solo Stagno 1).
In data 22-06-2006 sono state effettuate, nello Stagno 1, misurazioni della quantità di ossigeno disciolto nell’acqua interstiziale ed a vari livelli della colonna d’acqua: la media dei valori registrati è del 3,43 ± 1,55% a livello del sedimento, e 66,52 ± 22,79% nella colonna d’acqua a 5 cm dalla superficie1. Ciò è indicativo di una generale condizione di carenza di
ossigeno estesa all’intero ambiente, con situazioni ipossiche od anossiche a livello del sedimento (quantomeno durante il periodo estivo). Simili misurazioni non sono state effettuate per quanto riguarda lo Stagno 2; tuttavia, il sedimento ivi raccolto presenta uno strato RPD (Redox Potential Discontinuity layer, cfr. Sezione 1: Introduzione) appena al di sotto della superficie del sedimento (1-2 cm). Una tale altezza dello strato di discontinuità, analoga anche nello Stagno 1, indica scarsa o nulla ossigenazione del sedimento sottostante: è pertanto ipotizzabile che lo Stagno 2 presenti condizioni di ossigeno disciolto paragonabili a quelle dello Stagno 1.
Durante il periodo di studio sono stati effettuati tre campionamenti, in date scelte casualmente in un arco di tempo di otto mesi (cfr. Tabella 2.1). Le repliche, consistenti in campioni di 5-10 cm3 di sedimento coperti da circa 30 ml di acqua, sono state prelevate in stazioni distribuite casualmente lungo una delle due sponde (E/W) di entrambi gli stagni costieri. La profondità di prelievo del sedimento è stata fissata a circa 2-3 cm dalla superficie del sedimento stesso. La distanza dalla sponda e la profondità della colonna d’acqua
1Dati provenienti rispettivamente da misurazioni effettuate in 3 stazioni scelte in maniera casuale
lungo la sponda E, e da 5 stazioni sulla medesima sponda. Le misure non sono state replicate. Tali dati, non utilizzabili a fini statistici, forniscono tuttavia una indicazione sulla quantità di ossigeno disciolto a livello del sedimento e nella colonna d’acqua durante il periodo estivo.
sovrastante la zona di prelievo risultano variabili da replica a replica; tali misure non superano in nessun caso i valori di 2 e 0,7 m rispettivamente.
I campioni così prelevati sono stati conservati a temperatura ambiente, non sigillati. Nel caso del campionamento C2, sono state prelevate 2 repliche gemelle per ogni stazione. Di tali repliche, l’una è stata mantenuta in condizioni di contatto con l’aria (tappo leggermente sollevato), mentre l’altra è stata tenuta sigillata per due mesi (successivamente a tale data, i campioni sono stati aperti per consentire il prelievo delle cellule; sono stati tuttavia sigillati nuovamente dopo ogni osservazione).
I campioni non sono stati arricchiti con sostanze nutrienti di alcun tipo. Nel caso del campionamento C1, i campioni erano stati inizialmente arricchiti mediante l’aggiunta, direttamente nel contenitore, di mezzo chicco di riso bollito (il riso, decomponendosi, rilascia sostanze amilacee che favoriscono la crescita di batteri, utilizzati come cibo da alcuni protozoi ciliati). Tale arricchimento ha però favorito l’incremento di alcune delle specie batterivore presenti (in part. Paramecium calkinsi), selezionando negativamente altre specie (in part. Frontonia salmastra) ed, in generale, alterando marcatamente la biodiversità originaria presente nel campione. Considerando gli scopi del presente lavoro, tali arricchimenti sono stati in seguito evitati.
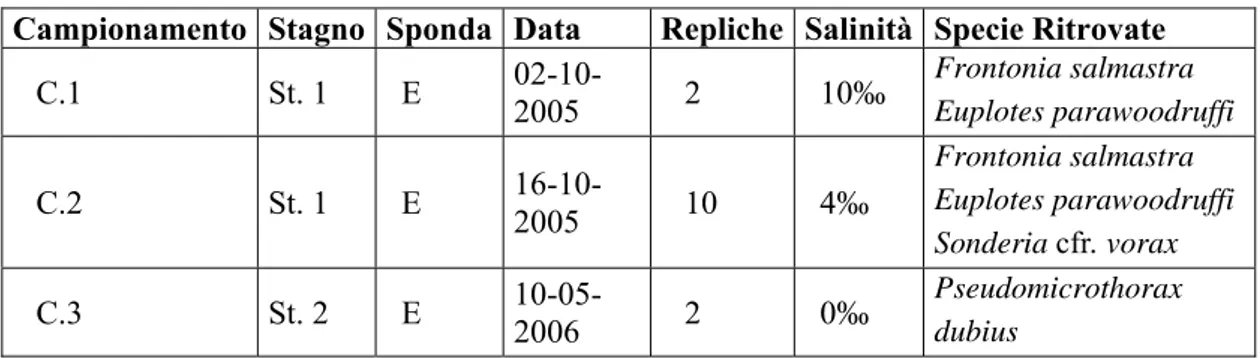
Campionamento Stagno Sponda Data Repliche Salinità Specie Ritrovate
C.1 St. 1 E 02-10-2005 2 10‰ Frontonia salmastra Euplotes parawoodruffi C.2 St. 1 E 16-10-2005 10 4‰ Frontonia salmastra Euplotes parawoodruffi Sonderia cfr. vorax C.3 St. 2 E 10-05-2006 2 0‰ Pseudomicrothorax dubius
Tabella 2.1: Schema dei campionamenti effettuati nel corso del presente lavoro. La misura di salinità riportata è la moda calcolata su tutte le repliche Nell’ultima colonna sono riportate alcune delle specie di ciliati reperite nel campionamento e successivamente utilizzate nel presente lavoro.
2.2: Isolamento delle cellule e tentativi di coltura
Le cellule sono state individuate osservando il campione al microscopio binoculare; sono quindi state prelevate singolarmente tramite micropipetta capillare e poste in salierine o vetrini a tre depressioni contenenti acqua di mare artificiale. L’acqua artificiale utilizzata è stata preparata con sali “Red Sea Salt” sciolti in acqua distillata autoclavata; la salinità finale della soluzione è pari a quella del campione di provenienza dei ciliati. Nel caso di specie particolarmente delicate o necessitanti di condizioni del mezzo difficilmente replicabili (ex: specie microaerofile) è stata utilizzata acqua salmastra prelevata dallo stesso campione, filtrata attraverso un filtro sterile da 0,2 μm onde eliminarne il particellato più grossolano ed
abbatterne la carica batterica. Gli organismi isolati sono stati conservati in camere umide poste in cella termostatata a 19±1°C, con alternanza luce-buio di 12 ore.
Sono stati effettuati tentativi di messa in coltura su tutte le specie isolate. Alcune specie sono state riconosciute, sulla base dei caratteri morfologici, come appartenenti a gruppi le cui condizioni generali di allevamento sono già note (es. ciliati del gen. Euplotes). Per tali specie si è proceduto come segue: sono state isolate alcune cellule (6-12); ciascun individuo è stato posto in vetrini a tre depressioni, e cibato con il nutrimento più opportuno per il gruppo in questione. All’aumentare del numero di cloni, la coltura è stata via via trasferita in contenitori più capienti. E’ stato possibile ottenere in tal modo colture monoclonali stabili, composte da numerosi individui.
Per gli organismi il cui protocollo di allevamento fosse sconosciuto, si è tentato di individuarlo, isolando alcuni individui (non meno di 15) o riunendoli in popolazioni composte da non più di una decina di cellule. Sono quindi stati divise in gruppi, e differenti tipi di nutrimento sono stati somministrati a ciascuno di essi: alghe verdi (Dunaliella salina), alghe brune (Pheodactylum tricornutum), cianobatteri (Oscillatoria formosa), flagellati eterotrofi (Chilomonas spp.) grani di riso bollito. Una volta individuato il cibo ottimale per l’organismo in questione, questo è stato esteso a tutte le cellule isolate. Ne sono state somministrate diverse quantità, a differenti cadenze temporali, per ciascun gruppo di cellule, in modo da stimare la quantità di cibo ed i tempi di somministrazione più opportuni. Sono state ottenute in questo modo alcune colture (originate da singola cellula o da popolazione) stabili per brevi periodi (uno-due mesi): i protocolli di allevamento individuati sono ancora approssimativi, e necessitano di ulteriori revisioni per consentire il mantenimento delle colture per periodi più lunghi.
Alcuni organismi (in part. Sonderia cfr. vorax) sono risultati non coltivabili nelle condizioni di allevamento testate. E’ stato tuttavia possibile mantenere per lungo tempo popolazioni vitali all’interno del campione di origine, cercando di riprodurvi le condizioni ottimali per la specie in questione (nel caso di Sonderia cfr. vorax, specie microaerofila obbligata, i campioni sono stati sigillati per circa 72 giorni, ed in seguito aperti il meno possibile ed a intervalli di tempo non più brevi di sette giorni: in tal modo la popolazione si è mantenuta stabile per oltre nove mesi. Simili risultati sono stati ottenuti mantenedo comunque una condizione di anossia dello strato di sedimento del campione).
Sono di seguito riportati i protocolli per la preparazione delle colture di organismi autotrofi utilizzate nel presente lavoro:
- Dunaliella salina. Alga verde. Coltura al 5‰.
Mezzo di coltura: 500 ml di acqua deionizzata autoclavata e 100 ml di acqua di mare artificiale (salinità 33‰), con l’aggiunta del mezzo di Walne (1966), specifico per
organismi fotosintetici, in ragione di 1 ml per litro, e del complesso vitaminico B1 –
BB6 – B12 (0,1 ml di soluzione vitaminica commerciale Benexol [Roche] per litro).
®
Mantenimento: in cella termostatata a 19±1°C, con alternanza luce-buio di 12 ore; lo scambio gassoso viene fornito da pompe per l’insufflazione di aria microfiltrata. Una volta staccata dalla pompa, la coltura resta utilizzabile per quattro-cinque giorni. - Pheodactylum tricornutum. Alga bruna. Coltura al 5‰.
Mezzo di coltura: 500 ml di acqua deionizzata autoclavata e 100 ml di acqua di mare sintetica (salinità 33‰), con l’aggiunta del mezzo di Walne (1966) in ragione di 1 ml per litro, e del complesso vitaminico B1 – B6 – B12 (0,1 ml di soluzione vitaminica
commerciale Benexol® [Roche] per litro).
Mantenimento: in cella termostatata a 19±1°C, con alternanza luce-buio di 12 ore; lo scambio gassoso viene fornito da pompe per l’insufflazione di aria microfiltrata. E’ consigliabile utilizzare le alghe subito dopo aver staccato la coltura dall’insufflatore, in quanto esse tendono a depositarsi sul fondo e morire in breve tempo.
- Oscillatoria formosa. Cianobatterio.
Mezzo di coltura (Eisler, 2006; pers. comm.): sono richieste tre differenti soluzioni:
Soil Medium
Soil-Stock Solution 50 ml
NaNO3 2% in H2O 1 ml
Na2HPO4 2% in H2O 1 ml
H2O distillata 1 l
Cyanophora Medium (stock solution /1)
Ca(NO3) 2 x 4 H2O 28,78 g
KNO3 x 7 H2O 5,10 g
MgSO4 x 4 H2O 10,24 g
K2HPO4 x 3 H2O 9,82 g
Biotina BioChemika 14400 (Fluka) 20 mg
Complesso vitaminico B1–B6–B12 Benexol® (Roche) 12,5 mg
H2O distillata 250 ml
Cyanophora Medium (stock solution /2)
Titriplex III (C10H14N2Na2O8: EDTA disodico) 375 mg
FeCl3 x 6 H2O 50 mg
MnCl2 x 4 H2O 20 mg
ZnCl2 2,5 mg
H2MoO4 2 mg
H2O distillata 250 ml
La soluzione finale di Cyanophora Medium è preparata aggiungendo 3 ml di stock
Il mezzo di coltura finale per O. formosa è preparato miscelando 1 parte di Soil
Medium per 1 parte di Cyanophora Medium.
Mantenimento: in cella termostatata a 19±1°C, con alternanza luce-buio di 12 ore, in fiasche chiuse ma non sigillate. Non è richiesta insufflazione di aria. La coltura si mantiene stabile per alcuni mesi dopo l’inoculo.
2.3: Caratterizzazione morfologica degli organismi
Nel presente lavoro sono state utilizzate procedure volte ad evidenziare alcuni dettagli morfologici dei ciliati oggetto di studio, importanti dal punto di vista tassonomico, al fine di determinare il genere e, ove possibile, la specie di appartenenza. Sono stati inoltre realizzati preparati per l’osservazione al microscopio elettronico a scansione (S.E.M.) od a trasmissione (T.E.M.), al fine di visualizzare, rispettivamente, i dettagli della morfologia esterna delle cellule e l’ultrastruttura interna, nonché la presenza, la collocazione e l’aspetto dei batteri simbionti eventualmente presenti.
Sono di seguito riportate le descrizioni delle tecniche impiegate ed i protocolli utilizzati.
- Colorazione nucleare di Feulgen
Questa procedura, che prende il nome dal biochimico tedesco Robert Joachim Feulgen (1884-1955), permette di evidenziare l’apparato nucleare delle cellule. Nel caso di protozoi ciliati, essa consente di valutare il numero, l’aspetto e la posizione del macronucleo e dei micronuclei presenti nella cellula. Tali caratteristiche sono tassonomicamente significative e rendono possibile una identificazione più precisa dell’organismo in questione.
Protocollo utilizzato:
1. Concentrazione delle cellule su vetrino portaoggetti.
2. Fissazione con miscela Sanfelice aggiunta in rapporto 1:1 col mezzo (5’).
3. Rimozione del fissativo tramite aspirazione con micropipetta; adesione delle cellule al vetrino con 1-2 gocce di alcole etilico (EtOH) al 95% in H2O.
4. Reidratazione dei campioni tramite passaggi in soluzioni di etanolo a concentrazione decrescente secondo il seguente schema:
- EtOH 95% (10’) - EtOH 90% (10’) - EtOH 70% (10’)
- EtOH 50% (10’) - EtOH 30% (10’)
5. Passaggio in H2O distillata (10’).
6. Passaggio in soluzione di HCl 1N a 60° C per 1h.
7. Breve sciacquo in H2O distillata (T ambiente).
8. Passaggio in reattivo di Schiff a 4°C per 30’; la reazione deve avvenire al buio. 9. Risciacquo dei vetrini sotto acqua di fonte corrente (durata variabile; di solito, 5’-10’
risultano sufficienti).
10. Due passaggi in H2O distillata (5’ ciascuno).
11. Disidratazione progressiva dei preparati tramite passaggi in soluzioni di EtOH a concentrazione crescente secondo il seguente schema:
- EtOH 30% (10’) - EtOH 50% (10’) - EtOH 70% (10’) - EtOH 90% (10’) - EtOH 95% (10’) - EtOH assoluto (10’) - EtOH assoluto (10’)
12. Asciugatura dei vetrini e montaggio con resina Euparal.
Note al protocollo:
(a) Composizione del fissativo Sanfelice (punto 2):
Acido cromico 1% 7 parti
Formalina commerciale Assoluta 7 parti Acido acetico glaciale Assoluto 4 parti La miscela deve essere conservata al riparo dalla luce.
(b) E’ possibile abbreviare i passaggi di reidratazione e disidratazione (punti 4 e 11) sostituendo ai passaggi in EtOH 70%, 90% e 95% un unico passaggio di 10’ in EtOH 80%.
(c) La durata e la temperatura riportate per l’idrolisi acida (punto 6) risultano adeguate se si lavora con strutture cellulari particolarmente resistenti. Viceversa, in simili condizioni le cellule più delicate possono danneggiarsi. In tali casi è opportuno effettuare un’idrolisi più blanda (30’ a 60° C oppure 60’ a T ambiente).
(d) Il reattivo di Schiff contiene fucsina basica e metabisolfito di sodio: durante il passaggio di colorazione (punto 8), la fucsina va a legarsi con un gruppo aldeidico del deossipentoso fosfato presente nelle catene degli acidi nucleici, precedentemente liberato e reso disponibile durante l’idrolisi acida (punto 6). Il materiale nucleare assume pertanto una colorazione rosso magenta. Il reattivo è fotosensibile, e deve essere conservato al buio a 4°C. Può essere impiegato per più colorazioni. Risultati analoghi sono ottenibili utilizzando una soluzione molto concentrata di fucsina basica.
(e) Durante il risciacquo sotto il getto di acqua corrente (punto 9) può accadere che parte delle cellule si distacchi dal vetrino e vada perduta. Nei casi in cui l’analisi venga condotta su un numero di cellule basso (situazione frequente in caso di ciliati non coltivabili), il passaggio può essere modificato nel seguente modo: si pongano i vetrini in una giara riempita di acqua corrente sino all’orlo, indirizzando il getto d’acqua sulla sua superficie; si lasci tracimare l’acqua dalla giara per 10’.
In alternativa, è possibile sciacquare manualmente i vetrini, immergendoli in acqua di fonte ed agitandoli delicatamente.
(f) La resina Euparal (punto 12), una volta solidificata, possiede un indice di rifrazione simile a quello del vetro: ciò riduce il numero di diottrie che la luce dovrà attraversare durante l’osservazione del preparato al microscopio. Tale resina è tossica: il montaggio deve essere eseguito sotto cappa aspirante.
- Impregnazione argentica (Corliss, 1953)
L’impregnazione argentica permette di visualizzare nei dettagli, mediante osservazione al microscopio ottico, la struttura e la disposizione della ciliatura somatica ed orale della cellula. Tale carattere è uno dei marcatori morfologici più significativi per quanto riguarda la sistematica dei protozoi ciliati. La tecnica non permette di visualizzare altre strutture cellulari quali l’apparato nucleare.
Protocollo utilizzato:
1. Concentrazione delle cellule in salierina o depressione.
2. Fissazione con miscela di Champy aggiunta in rapporto 1:1 col mezzo (5’). 3. Postfissazione in fluido Da Fano (12h).
4. Sciacquo in H2O distillata (3-5 passaggi da 5’-10’).
5. Concentrazione delle cellule su vetrino portaoggetti.
6. Aggiunta di una goccia di gelatina liquida in rapporto 1:1 col mezzo contenente le cellule; la gelatina deve essere spalmata lungo la superficie del vetrino. Collocare il vetrino in una camera umida.
7. Passaggio per 5’ a 4°C.
8. Passaggio in AgNO3 (3%) a 4°C per 30’.
9. Sciacquo in H2O distillata a 4°C (1-2 passaggi, pochi secondi).
10. Collocare i vetrini in acqua a 0°C; porre sotto lampada a UV per circa 1h [cfr. Note
al protocollo, (g)].
11. Passaggio in alcole etilico 70% a 4°C (10’-15’). 12. Passaggio in alcole etilico 96% a T ambiente (10’).
13. Passaggio in alcole isopropilico assoluto a T ambiente (10’). 14. Montaggio con resina Euparal.
Note al protocollo
(a) Composizione della miscela di Champy (punto 2):
CrO3 1% 7 parti
K2Cr2O7 3% 7 parti
OsO4 2% 4 parti
La miscela deve essere conservata al buio e a 4°C; si mantiene stabile per alcune settimane.
(b) Composizione del fluido Da Fano (Foissner, 1993; punto 3):
Formalina commerciale 37% 100 ml
Co(NO3) 2 x 6 H2O 10 g
NaCl 10 g
H2O distillata 900 ml
La miscela è stabile per anni.
La durata del passaggio in Da Fano (punto 3) può essere prolungata a piacere.
(c) La gelatina commerciale (CarloErba) va preparata alla concentrazione del 3% in H2O con l’aggiunta di 0,005 g di NaCl ogni 20 ml di H2O (punto 6). Deve essere
usata calda. Prima di gocciare la gelatina sul mezzo contenente le cellule, riscaldare il vetrino appoggiandolo sulla retina spargifiamma del bunsen. Spalmare la gelatina sul vetrino usando una pasteur scaldata, senza esercitare una pressione eccessiva, in modo da creare uno strato non troppo sottile e non troppo spesso.
(d) Utilizzare come camera umida (punto 6) delle capsule Petri coperte; rivestire il fondo della capsula con carta da filtro inumidita per impedire alla gelatina di disseccarsi. Porre il tutto in frigorifero per 5’.
(e) Durante questa fase, il nitrato d’argento si lega ai cinetosomi della cellula. La successiva irradiazione con UV causerà la precipitazione dello stesso nitrato d’argento, che assumerà un colore brunastro: le basi delle ciglia saranno pertanto evidenziate come corpuscoli marroni, mentre il citoplasma della cellula resterà traslucido.
La soluzione di AgNO3 3% in H2O (punto 8) va preparata in anticipo e conservata a
4°C in modo da poterla utilizzare fredda. Terminati i 30’, può essere recuperata e utilizzata di nuovo. Conservare al buio e a 4°C. Se appare di colore marroncino non è più utilizzabile.
(f) L’acqua distillata (punto 9) deve essere posta con alcune ore di anticipo a 4°C, in modo che sia fredda al momento dell’uso.
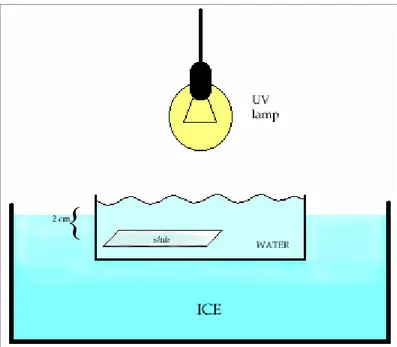
(g) La distanza fra la lampada a UV ed i vetrini (punto 10) deve essere di circa 20-25 cm. I vetrini devono essere collocati in un contenitore trasparente o metallico (o, in alternativa, rivestito d’alluminio), possibilmente non di plastica e non scuro; devono essere ricoperti da uno strato di acqua di circa 2 cm (comunque, mai inferiore ad 1 cm.). Il contenitore deve essere appoggiato o incassato in un supporto di ghiaccio (cfr. Figura 2.1). La durata dell’esposizione può variare in funzione della potenza della lampada.
Durante questa fase, non è infrequente che la gelatina si stacchi dal vetrino. Ciò comporta la perdita delle cellule ivi incluse. Tale distacco può essere causato da più fattori (distanza e/o potenza della lampada UV inadeguati, tracce di fluido Da Fano attorno alle cellule, precipitazione incontrollata del nitrato d’argento, scarsa qualità della gelatina): la buona riuscita di tale passaggio richiede esperienza.
(h) La resina Euparal (punto 14), una volta solidificata, possiede un indice di rifrazione simile a quello del vetro: ciò riduce il numero di diottrie che la luce dovrà attraversare durante l’osservazione del preparato al microscopio. A causa della tossicità della resina, il montaggio deve essere eseguito sotto cappa aspirante.
Figura 2.1: Schema della collocazione dei vetrini sotto lampada UV per l’impregnazione argentica. La distanza fra lampada e supporto (non indicata) deve essere di circa 20-25 cm. Lo strato di acqua che ricopre i vetrini non deve essere di spessore inferiore a 1 cm. L’acqua stessa deve mantenersi fredda onde evitare il distacco della gelatina: a tale scopo è opportuno incassare il contenitore in un supporto di ghiaccio come illustrato.
{
- Realizzazione di preparati per S.E.M.
Il microscopio elettronico a scansione (S.E.M.), utilizzando gli elettroni anziché i fotoni per generare un’immagine, consente una risoluzione ed un ingrandimento migliori rispetto ad un microscopio ottico, in virtù della loro minor lunghezza d’onda (10˙000 volte più corta di quella fotonica). Gli elettroni, emessi nel vuoto da un filamento di tungsteno incandescente, vengono accelerati e convogliati verso il campione; le cellule, opportunamente trattate e ricoperte da una lamina di metallo pesante (oro), ricevono tale fascio; gli elettroni secondari che si liberano al suo passaggio vengono quindi raccolti e convogliati per produrre un’immagine tridimensionale su di un tubo a raggi catodici. Questa tecnica permette di ottenere immagini chiare della morfologia esterna degli organismi trattati, anche se non consente l’osservazione di preparati viventi; inoltre, le procedure di fissazione possono alterare l’aspetto delle cellule.
Protocollo utilizzato:
1. Concentrazione delle cellule in salierina o depressione.
2. Fissazione con OsO4 2% in H2O distillata, aggiunto in rapporto 1:1 col mezzo (10’).
3. Sciacquo in H2O distillata (5’-10’).
4. Trasferimento delle cellule su vetrini coprioggetti precedentemente trattati con una soluzione di polilisina 0,01 mg/ml in H2O.
5. Disidratazione dei preparati tramite passaggi in soluzioni di EtOH a concentrazione crescente secondo il seguente schema:
- EtOH 30% (10’) - EtOH 50% (10’) - EtOH 70% (10’) - EtOH 90% (10’) - EtOH 95% (10’) - EtOH assoluto (15’) - EtOH assoluto (15’) 6. Critical Point Drying con CO2.
7. Montaggio dei campioni su appositi supporti. 8. Metallizzazione con sottile (20 nm) strato di oro.
Note al protocollo:
(a) Per gli organismi marini, è spesso più utile diluire il tetrossido d’osmio in acqua di mare sintetica a salinità 33‰, onde evitare che lo shock osmotico possa danneggiare la cellula. Le specie oggetto di studio sono adattate a salinità del 5-10‰: è stato pertanto deciso di utilizzare ugualmente OsO4 diluito in acqua distillata.
(b) La soluzione di polilisina (punto 5) è ottenuta diluendo 1:10 la polilisina commerciale 0,1 mg/ml in H2O (Sigma-Aldrich Co.). Tale soluzione viene gocciata
su vetrini coprioggetto sgrassati con alcole, ed in genere tagliati in modo da adattarsi alla misura degli alloggiamenti di un tamburo portavetrini (i vetrini rimarranno all’interno di tale supporto durante i passaggi successivi, sino alla doratura). La goccia viene quindi lasciata evaporare in stufa a 60°C per 30°. La polilisina si deposita sulla superficie del vetrino, favorendo l’adesione delle cellule allo stesso. (c) Scopo del Critical Point Drying (punto 6) è rimuovere i residui di etanolo ed
essiccare il preparato prima che questo venga sottoposto a metallizzazione: ciò è ottenuto mediante numerosi passaggi in fluido di transizione (CO2 liquida: Costante
Critica = 31,1°C; 1072 P.S.I.), sino alla completa rimozione dell’etanolo. Tali passaggi vengono effettuati in un apposito dispositivo (Critical Point Dryer). Una volta eliminati i residui di alcole, la temperatura dell’apparecchio viene progressivamente aumentata finché la tensione superficiale dell’anidride carbonica non diviene molto bassa o nulla: una volta raggiunto tale “punto critico” si verifica il fenomeno della “continuità di stato”: il passaggio della CO2 da liquido a gas avviene
contatto con alcun menisco in tensione (durante l’essiccazione, gli effetti della tensione superficiale sono la principale fonte di danneggiamento dei preparati, causando la deformazione od il collasso delle strutture. Tale metodo consente di ridurre al minimo gli effetti di queste forze sui preparati microbiologici più delicati). L’anidride carbonica, per le sue caratteristiche fisicochimiche, è uno dei fluidi di transizione più utilizzati: non essendo ben miscibile con l’acqua, è tuttavia necessario procedere preventivamente alla sostituzione dell’acqua presente nel campione con un altro fluido miscibile con CO2 (“fluido intermedio”: punto 5).
- Realizzazione di preparati per T.E.M.
Il microscopio elettronico a trasmissione (T.E.M.) è impiegato per indagare l’ultrastruttura della cellula. Il preparato, incluso in una resina epossidica, viene tagliato in sezioni sottili (300-500 Å) tramite un ultramicrotomo. Tali sezioni, opportunamente contrastate con metalli pesanti, vengono sottoposte al fascio di elettroni dello strumento: la diffrazione più o meno grande di questi ultimi all’attraversamento della sezione, determinata dagli elementi che costituiscono gli organuli e/o i componenti cellulari dell’organismo, impressionerà differentemente una lastra fotografica posta al disotto di essa e fornirà un’immagine delle strutture interne della cellula. Nel corso del presente lavoro, tale tecnica è stata ugualmente utilizzata per la visualizzazione delle cellule batteriche eventualmente presenti all’interno di cellule di protozoi ciliati o sulla loro superficie; è stato in tal caso applicato un protocollo di fissazione apposito. Le informazioni così ottenute vengono impiegate per l’identificazione dell’ospite e dei simbionti, nei casi in cui essi presentino peculiarità morfologiche particolarmente rilevanti.
Sono stati utilizzati due differenti metodi di preparazione del campione, volti a garantire una fissazione ottimale rispettivamente delle strutture cellulari dell’ospite e dei batteri simbionti. Protocollo utilizzato/1 (indicato per visualizzare la struttura delle cellule di ciliati):
1. Concentrazione delle cellule in salierina o depressione.
2. Fissazione con OsO4 2% in H2O distillata, aggiunto in rapporto 1:1 col mezzo (10’).
3. Postfissazione in glutaraldeide 2,5% in tampone cacodilato 0,2 M (10’).
4. Disidratazione delle cellule tramite passaggi in soluzioni di EtOH a concentrazione crescente secondo il seguente schema:
- EtOH 30% (10’) - EtOH 50% (10’) - EtOH 70% (10’)
- EtOH 90% (10’) - EtOH 95% (10’) - EtOH assoluto (15’) - EtOH assoluto (15’)
5. Trasferimento delle cellule in acetone assoluto (30’).
6. Aggiunta di resina epossidica liquida in rapporto 1:1 con l’acetone (12h).
7. Trasferimento delle cellule in resina epossidica assoluta.
8. Polimerizzazione della resina contenente le cellule (in stufa a 60°C sino ad avvenuta polimerizzazione).
9. Taglio manuale del blocchetto di resina contenente le cellule e montaggio dello stesso su supporto per ultramicrotomo.
10. Taglio del blocchetto mediante ultramicrotomo in sezioni sottili (300-500 Å); collocazione delle sezioni su retini.
11. Contrasto delle sezioni mediante immersione in acetato di uranile e citrato di piombo.
12. Osservazione al microscopio.
Note al protocollo/1:
(a) Per l’utilizzo di osmio tetrossido in H2O distillata (punto 2), cfr. Note al protocollo
di realizzazione di preparati per S.E.M., (b).
(b) La postfissazione in glutaraldeide (OHC-[CH2]3-CHO; punto 3) è necessaria per
fissare la componente proteica della cellula (i gruppi aldeidici condensano con le estremità amminiche a formare azometine); l’osmio, ossidandosi, fissa invece prevalentemente la componente lipidica (es. membrane cellulari).
(c) Il passaggio in miscela di acetone e resina (punto 6) è necessario per consentire la permeazione dell’intera cellula alla resina (una miscela contenente la sola resina sarebbe troppo viscosa per penetrare correttamente in tutti i comparti cellulari). Durante tale passaggio, è opportuno collocare le cellule al centro di una salierina riempita per metà di acetone, e percolare la resina lungo i bordi della salierina stessa, fino a quando questa non sia completamente riempita. In tal modo si evita che le cellule vengano trascinate dalla resina in posizione periferica e non si impregnino correttamente.
Protocollo utilizzato/2 (indicato per visualizzare la struttura delle cellule batteriche) 1. Concentrazione delle cellule in salierina o depressione.
2. Fissazione con miscela fissativa [cfr. Note al protocollo, (a)], aggiunta in rapporto 1:1 col mezzo (1h30’).
3. Sciacquo in soluzione tampone [cfr. Note al protocollo, (b)] (pochi minuti).
4. Rimozione del tampone e postfissazione con OsO4 1,5% in soluzione tampone [cfr.
Note al protocollo, (b)] (1h).
5. Disidratazione delle cellule tramite passaggi in soluzioni di EtOH a concentrazione crescente secondo il seguente schema
- EtOH 30% (10’) - EtOH 50% (10’) - EtOH 70% (10’) - EtOH 90% (10’) - EtOH assoluto (15’) - EtOH assoluto (15’)
6. Procedere con il punto 5 del Protocollo/1 sino al termine.
Note al protocollo/2:
(a) Composizione della miscela di fissazione:
Glutaraldeide 2,5% Sol. tampone 2 parti Paraformaldeide 8% H2O distillata 4 parti
Sol. tampone 6 parti
H2O distillata 8 parti
La miscela non può essere conservata per lunghi periodi.
L’aggiunta di paraformaldeide aumenta la permeabilità delle membrane cellulari (l’impiego della sola glutaraladeide può talvolta mantenere intatta la semipermeabilità naturale delle membrane stesse). La soluzione viene preparata sciogliendo 1g di paraformaldeide commerciale in 12,5 ml di acqua. E’ necessario aggiungere una goccia di NaOH per consentire la completa solubilizzazione della polvere.
(b) La soluzione tampone è composta da tampone fosfato pH 7,2 e saccarosio (70 mg/ml). L’aggiunta del saccarosio limita il rischio di shock osmotico durante la
procedura di fissazione. La soluzione si degrada rapidamente: non è possibile conservarla a lungo.
2.4: Caratterizzazione molecolare degli organismi
Nel presente lavoro è stata effettuata la caratterizzazione del gene codificante per l’rRNA della subunità piccola del ribosoma (rispettivamente, 16S nei procarioti e 18S negli eucarioti) degli organismi oggetto di studio. Le ragioni della scelta di tale marcatore molecolare sono spiegate nella Sezione 1: Introduzione.
Vengono di seguito riportate le descrizioni delle procedure impiegate nella estrazione, amplificazione e sequenziamento del DNA, tanto dell’ospite che dei simbionti, nonché la procedura di ibridazione in situ utilizzata.
-Estrazione di DNA standard
Protocollo utilizzato (Wisotzkey, 1990; modificato):
1. Centrifugare le cellule (in Eppendorf) a 12000 x g (5’); eliminare il surnatante. 2. Risospendere il pellet in Saline-EDTA (volume consigliato: 500 μl).
3. Aggiungere 20 μl di lisozima 10 mg/ml in TrisHCl 50 mM pH=8 ed incubare a 37°C (30’).
4. Aggiungere 10 μl di RNasi A 0,5 mg/ml in SSC 2x ed incubare a 37°C (15’). 5. Aggiungere 10 μl di Proteinasi K ed incubare a 37°C (60’).
6. Aggiungere 40 μl di Sodio Dodecilsolfato 25% in H2O ed incubare a 65°C (10’)
onde completare la lisi cellulare ed inattivare le DNasi.
7. Aggiungere 180 μl di Sodio Acetato 5 M pH=5,5 e 745 μl di Chisom, agitare delicatamente.
8. Centrifugare a 12000 x g (5’, 4°C).
9. Recuperare la fase superiore (contenente il DNA).
10. Aggiungere 2 voll. EtOH assoluto. Dopo breve agitazione, incubare a 0°C (10’). 11. Centrifugare a 12000 x g (10’, 4°C); eliminare l’etanolo per svuotamento. 12. Lavare il pellet con EtOH 70%.
14. Far asciugare il pellet.
15. Risospendere il DNA in un appropriato volume di acqua sterile.
Note al protocollo:
(a) Composizione Saline-EDTA:
NaCl 0,15 M
EDTA 0,01 M
Il pH deve essere pari a 8,0. (b) Composizione Chisom:
Cloroformio 24 parti
Alcole Isoamilico 1 parte (c) Composizione SSC 20x
NaCl 3 M
TriSodio Citrato 0,3 M Il pH deve essere pari a 7,0.
La soluzione deve essere diluita 1:10 in H2O al momento dell’uso.
(d) L’aggiunta di RNasi durante le fasi di lisi cellulare (punto 4), se il contenuto in RNA non è alto, può essere saltata.
(e) Il trattamento con Proteinasi K (punto 5) può essere prolungato overnight.
-Procedura di estrazione di DNA da micelio applicata a campioni di cellule fissate.
E’ stato utilizzato il kit “Nucleospin® Plant” (Macherey-Nagel) per ottimizzare l’estrazione
di DNA nei casi in cui questa venisse fatta a partire da un basso numero di cellule (10-20). Reagenti e protocollo (CTAB) sono concepiti per ottimizzare la resa nelle estrazioni da tessuto di funghi; sono stati comunque ottenuti buoni risultati utilizzandoli nelle estrazioni del DNA di protozoi ciliati e dei loro batteri simbionti.
Protocollo utilizzato (CTAB method; modificato):
1. Centrifugare le cellule (fissate in EtOH 70%) a 11000 x g (12000 rpm). Rimuovere il surnatante per rovesciamento; attendere l’avvenuta essiccazione dei residui di alcole. 2. Risospendere il pellet in 200 μl di Buffer C1. Rendere omogeneo il campione
3. Aggiungere ulteriori 100 μl di Buffer C1. Continuare ad omogenare il campione (5’-10’).
4. Aggiungere 100 μl di cloroformio. Porre su Vortex (10’’).
5. Centrifugare a 11000 x g (5’-10’) in modo da separare le due fasi. Recuperare la fase acquosa soprastante (contenente il DNA).
6. Incubare a 60°C (30’).
7. Caricare l’intero volume di lisato in una colonnina NucleoSpin Filter Column. Centrifugare per 5’ a < 11000 x g. Recuperare l’eluato. La colonnina è monouso. 8. Trasferire 300 μl di eluato in una Eppendorf da 1,5 ml. Aggiungere 300 μl di Buffer
C4 e 200 μl di EtOH assoluto. Porre su Vortex (30’’).
9. Caricare la miscela in una colonnina NucleoSpin Plant Column. Centrifugare per 5’ a 11000 x g. Scartare l’eluato.
10. Aggiungere 400 μl di Buffer CW. Centrifugare per 1’ a 11000 x g. Scartare l’eluato. 11. Aggiungere 700 μl di Buffer CW. Centrifugare per 1’ a 11000 x g. Scartare l’eluato. 12. Aggiungere ulteriori 200μl di Buffer CW. Centrifugare a 11000 x g per 2’ onde
rimuovere eventuali residui di etanolo ed asciugare la membrana silicea della colonnina.
13. Porre la colonnina in una nuova Eppendorf da 1,5 μl. Pipettarvi 100 μl di Buffer CE (preriscaldato a 70°C). Incubare a T ambiente per 5’. Centrifugare a 11000 x g (1’) onde eluire il DNA legato alla membrana silicea.
14. Recuperare l’eluato (conservare a –20°C). La colonnina è monouso.
Note al protocollo:
(a) L’omogeneizzazione del campione (punti 2-3) viene ottenuta aspirando ed estrudendo ripetutamente la soluzione di lisi contenente le cellule mediante una siringa ipodermica sterile da 5 ml. L’uso del Vortex durante questa fase, previsto nel protocollo originale CTAB, è stato ridotto od evitato in quanto le sollecitazioni impresse dallo strumento possono frammentare grandi macromolecole quali il DNA genomico.
(b) Il tampone di lisi (punti 2-3) contiene sali caotropici, agenti denaturanti e detergenti che emulsionano le membrane ed inattivano le componenti cellulari.
(c) Il passaggio in colonna descrittto al punto 7 serve a rimuovere i detriti cellulari (polisaccaridi, frammenti cellulari particolati) tuttora presenti nella soluzione.
(d) Il tampone C4 (punto 8) ottimizza la fase di binding del DNA alla matrice silicea contenuta nella Plant Column. Può essere premiscelato all’etanolo secondo i dosaggi riportati. Il tampone C4 non può essere conservato per lunghi periodi: deve essere preparato al momento dell’uso miscelando i tamponi C2 e C3 (forniti nel kit Nucleospin® Plant) in ragione di 4 parti a 1.
(e) L’eluizione del DNA può essere ottimizzata pipettando 50 μl di tampone iposalino di eluizione (CE), preriscaldato a 70°C, sulla matrice, lasciando incubare a 70°C per 3’, aggiungendo i restanti 50 μl, incubando ulteriormente per altri 2’ e procedendo quindi con la centrifugazione. Si è fatto ricorso a tale procedura in caso di estrazioni di DNA genomico da un basso numero di cellule (es. 12).
-Amplificazione del 18S rDNA tramite Reazione a Catena della Polimerasi (Mullis, 1984)
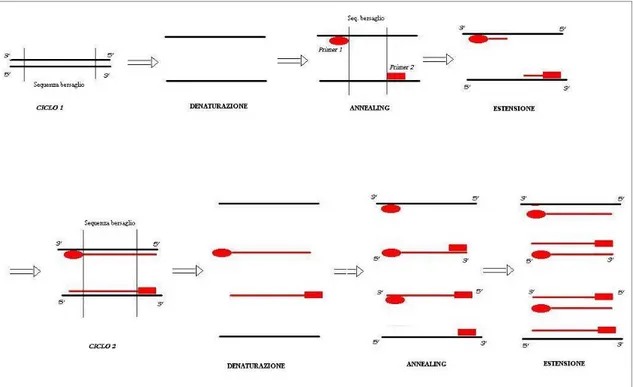
Questa tecnica consente di produrre un notevole numero di copie di una sequenza specifica di DNA (cfr. Figura 2.2). E’ infatti possibile indirizzare l’enzima termostabile Taq DNA polimerasi (estratto dal batterio Thermus aquaticus) in modo da sintetizzare esclusivamente una regione del genoma compresa fra due sequenze nucleotidiche note, i cui oligonucleotidi complementari possono essere usati come inneschi (primer) per avviare la reazione stessa. Il
primer che presenta la stessa sequenza del filamento codificante per l’RNA è detto
“forward”, mentre l’altro, che quindi ha quella del filamento opposto, “reverse”. Tali inneschi, una volta denaturata la doppia elica del filamento di DNA che si desidera amplificare (stampo), andranno ad appaiarsi (fase di annealing) alle sequenze loro complementari, situate rispettivamente a monte ed a valle della regione bersaglio: poiché la
Taq polimerasi procederà a sintetizzare un nuovo filamento, in direzione 5’-3’,
complementare a ciascuno dei due stampi, verranno prodotti due nuovi filamenti di DNA che copriranno l’intera zona bersaglio delimitata dai primer. Grazie alla termostabilità dell’enzima, il processo può essere ciclicizzato: è possibile cioè scaldare la miscela sino ad ottenere la separazione dei filamenti di nuova sintesi dallo stampo originale. Abbassando nuovamente la temperatura, gli inneschi si appaieranno sia allo stampo originale che ai due nuovi filamenti (che includono la sequenza complementare al primer opposto), ed il ciclo successivo inizierà servendosi di quattro stampi. Virtualmente, poiché ogni stampo dà luogo a due molecole di nuova sintesi, il risultato netto di una reazione di PCR di n cicli condotta a partire da una sola molecola di DNA stampo sarà di 2n frammenti di DNA amplificato.
Figura 2.2: La reazione a catena della polimerasi (2 cicli). Il filamento-stampo viene denaturato; i
primer individuano i rispettivi siti di legame e vi si appaiano; la Taq polimerasi sintetizza nuovi
filamenti di DNA complementari alla sequenza bersaglio. Con la successiva denaturazione, i filamenti di nuova sintesi (che includono la sequenza complementare al primer opposto) funzionano come stampo per il ciclo successivo.
Composizione standard della miscela di reazione: 15. H2O sterile.
16. Tampone di pH specifico per la polimerasi utilizzata (concentrazione finale 1x). 17. MgCl2 50 μM (concentrazione finale variabile; gen. 1,5 μM).
18. Deossinucleotidi trifosfati (dNTP) 2,5 mM (concentrazione finale variabile; gen. 0,25 mM).
19. Primer forward 100 μM (concentrazione finale 10 μM). 20. Primer reverse 100 μM (concentrazione finale 10 μM).
21. Enzima Taq DNA polimerasi (concentrazione finale 1u/50 ml). 22. DNA stampo.
Note:
(a) Ove il rischio di amplificazione aspecifica da contaminazione fosse basso (es. nelle reazioni condotte con inneschi specifici e/o ad alte condizioni di stringenza) è stata impiegata acqua distillata autoclavata preparata manualmente in laboratorio. Viceversa, per reazioni ad alto rischio di contaminazione e amplificazione aspecifica
(es. utilizzando primer universali e/o basse condizioni di stringenza) si è fatto ricorso ad acqua ultrapura commerciale (Fluka).
(b) Il cloruro di magnesio è un cofattore dell’enzima: lo ione Mg2+ va infatti a
stabilizzare i polianioni presenti nella miscela di reazione. Modificando la concentrazione finale di tale ione, è possibile alterare le condizioni di reazione. Alcuni dei tamponi impiegati contengono già MgCl2 in soluzione: non è stato in tal
caso possibile modificarne la concentrazione.
(c) I deossinucleotidi trifosfati (dNTP) comporranno le molecole di nuova sintesi che costituiscono il prodotto di reazione. La concentrazione finale deve essere quindi proporzionata alla lunghezza del tratto di genoma che si desidera amplificare e del numero di cicli di amplificazione previsto.
(d) Nel presente lavoro sono stati utilizzati differenti combinazioni di primer forward e
reverse, a seconda del tipo di reazione effettuata (cfr. Tabella 2.2). Nel caso in cui il
gruppo tassonomico (es. “Classe”) di appartenenza dell’organismo oggetto di amplificazione fosse noto, e di tale gruppo fossero disponibili, nei database molecolari, sequenze relative al 18S-16S rDNA, e’ stato possibile disegnare od impiegare inneschi già disegnati in modo da appaiarsi a regioni di DNA, fiancheggianti la sequenza bersaglio o interne alla sequenza stessa, molto conservate all’interno del gruppo bersaglio e presentanti quante più differenze possibili con le regione omologhe degli organismi appartenenti ad altri gruppi. Simili inneschi “specifici”, combinati con temperature di appaiamento medio-alte, rendono possibile amplificare selettivamente e in misura maggiore il gene dell’organismo bersaglio; il DNA di organismi appartenenti ad altri gruppi ed eventualmente presente nella miscela di reazione risulterà sfavorito dalla minore affinità con gli inneschi, e sarà amplificato in quantità nettamente inferiori.
Nei casi in cui la collocazione tassonomica dell’organismo bersaglio fosse ignota e/o non fossero disponibili dati molecolari al riguardo, è stato utilizzato un approccio diametralmente opposto: sono stati impiegati primer disegnati su regioni altamente conservate nel più alto numero possibile di gruppi tassonomici. Nel caso in cui la sequenza bersaglio presenti uno o più nucleotidi variabili, è stata impiegata nella reazione una miscela di primer (“primer degenerato”): le molecole componenti tale miscela sono sintetizzate in modo da presentare in parte l’uno, in parte l’altro nucleotide complementare a quello presente nella posizione variabile. In questo modo, in sede di reazione, vi sarà comunque appaiamento da parte di una delle due versioni. Queste “degenerazioni” possono essere introdotte più volte nella sequenza dell’innesco, ed alternare due, tre o tutti i nucleotidi. Tali primer, utilizzati a condizioni blande di stringenza, consentono l’amplificazione in egual misura di tutte
le sequenze di DNA bersaglio presenti nella miscela di reazione. In tali condizioni, l’amplicone del gene dell’organismo in esame (se si è nella condizione in cui questo prevale quantitativamente rispetto ad altri geni eventualmente presenti), comporrà gran parte del prodotto di reazione: un successivo clonaggio, o, più raramente, il sequenziamento diretto del prodotto stesso permetterà di risalire alla sequenza. Le combinazioni di inneschi risultate efficaci per l’amplificazione degli organismi in esame sono riportati nei capitoli relativi a ciascuno di essi (cfr. sezione successiva).
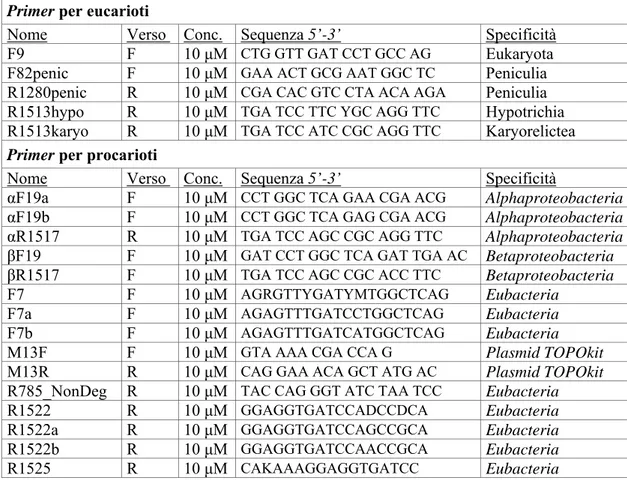
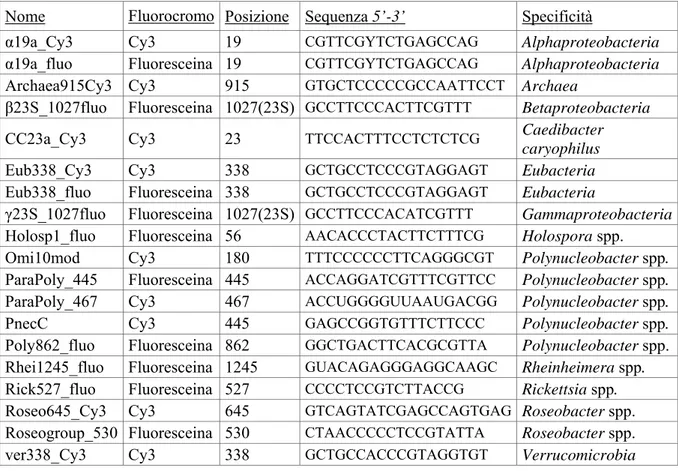
Primer per eucarioti
Nome Verso Conc. Sequenza 5’-3’ Specificità F9 F 10 μM CTG GTT GAT CCT GCC AG Eukaryota F82penic F 10 μM GAA ACT GCG AAT GGC TC Peniculia R1280penic R 10 μM CGA CAC GTC CTA ACA AGA Peniculia R1513hypo R 10 μM TGA TCC TTC YGC AGG TTC Hypotrichia R1513karyo R 10 μM TGA TCC ATC CGC AGG TTC Karyorelictea
Primer per procarioti
Nome Verso Conc. Sequenza 5’-3’ Specificità
αF19a F 10 μM CCT GGC TCA GAA CGA ACG Alphaproteobacteria
αF19b F 10 μM CCT GGC TCA GAG CGA ACG Alphaproteobacteria
αR1517 R 10 μM TGA TCC AGC CGC AGG TTC Alphaproteobacteria
βF19 F 10 μM GAT CCT GGC TCA GAT TGA AC Betaproteobacteria
βR1517 F 10 μM TGA TCC AGC CGC ACC TTC Betaproteobacteria
F7 F 10 μM AGRGTTYGATYMTGGCTCAG Eubacteria
F7a F 10 μM AGAGTTTGATCCTGGCTCAG Eubacteria
F7b F 10 μM AGAGTTTGATCATGGCTCAG Eubacteria
M13F F 10 μM GTA AAA CGA CCA G Plasmid TOPOkit
M13R R 10 μM CAG GAA ACA GCT ATG AC Plasmid TOPOkit
R785_NonDeg R 10 μM TAC CAG GGT ATC TAA TCC Eubacteria
R1522 R 10 μM GGAGGTGATCCADCCDCA Eubacteria
R1522a R 10 μM GGAGGTGATCCAGCCGCA Eubacteria
R1522b R 10 μM GGAGGTGATCCAACCGCA Eubacteria
R1525 R 10 μM CAKAAAGGAGGTGATCC Eubacteria
Tabella 2.2: Quadro riassuntivo dei primer utilizzati nel presente lavoro. La concentrazione riportata è quella effettiva di impiego. La specificità indica il gruppo tassonomico sulla base del quale l’innesco è stato disegnato.
(e) La maggior parte degli enzimi Taq DNA polimerasi attualmente in commercio viene solitamente ingegnerizzata (Taq ricombinanti) al fine di aumentarne l’efficienza catalitica e/o di evitare amplificazioni aspecifiche durante le prime fasi della reazione. Nel presente studio sono state impiegate le seguenti Taq polimerasi: TaKaRa Ex Taq (TaKaRa Biomedicals); Taq DNA Polymerase (Promega); EUROBIOTAQ ADN POLYMERASE
(Laboratoires EUROBIO); Platinum Taq (Invitrogen). Risultati positivi sono
stati ottenuti unicamente con la TaKaRa Ex Taq e la Platinum Taq.
(f) Nelle reazioni di PCR, il DNA rappresenta la fonte principale di contaminazione. È pertanto consigliabile preparare separatamente la quantità di miscela necessaria per l’amplificazione di tutti i campioni in esame
(Master Mix), utilizzando una provetta Eppendorf da 1,5 ml: tale miscela sarà quindi aliquotata in miniprovette Eppendorf da 0,2 ml, e soltanto all’interno di ciascuna di esse sarà aggiunto il DNA stampo. È inoltre consigliabile conteggiare, nella preparazione della Mix, un controllo positivo (una aliquota contenente DNA di provata amplificabilità alle condizioni di reazione adottate: l’assenza di prodotto finale in questa aliquota indica il fallimento dell’intera reazione) e di un controllo negativo (“bianco”: una aliquota della Master Mix a cui non viene aggiunto alcun DNA stampo: la presenza di prodotto finale in questa aliquota indica una contaminazione da DNA avvenuta all’interno della Master Mix, e quindi estesa all’intera reazione).
(g) La preparazione della miscela di reazione deve avvenire in ghiaccio, impiegando reagenti preraffreddati, in modo da evitare amplificazioni aspecifiche durante l’allestimento della reazione.
Ciclo di PCR (bassa stringenza):
1. Preparazione della miscela di reazione; suddivisione in aliquote; inserimento delle Eppendorf da 0,2 ml nel termociclatore.
2. Denaturazione del DNA genomico (94°C, 3’). 3. Ciclo (ripetuto 35 volte):
• Denaturazione delle molecole di nuova sintesi (94°C, 30’’) • Annealing dei primer (30-50°C, 30’’).
• Sintesi del nuovo filamento (72°C, tempo variabile; per la sintesi della molecola del 16-18S rDNA si richiede circa 1,5’).
4. Estensione finale a 72°C per 7’. 5. Raffreddamento a 8°C (∞).
Note:
(a) Nel presente lavoro è stato utilizzato il termociclatore Primes 96 plus (MWG BIOTECH. AG).
(b) La denaturazione iniziale (punto 2) è più lunga in quanto è necessario separare i filamenti dell’intero DNA genomico. Viceversa, durante il ciclo iterato occorre denaturare solo molecole di nuova sintesi, lunghe al massimo alcune Kbasi: i tempi necessari risultanto pertanto più brevi.
(c) Nel caso in cui la quantità di DNA stampo iniziale sia scarsa (es. amplificazione da poche cellule, caso frequente se si lavora con ciliati difficilmente coltivabili) è possibile aumentare il numero di cicli (punto 3), in modo da ottenere maggiori quantità di prodotto. Tale procedura comporta però alcuni svantaggi: aumenta la probabilità che la polimerasi, specialmente se sprovvista dell’attività “correttore di bozze”, incorpori nucleotidi sbagliati durante la sintesi; l’enzima è soggetto a degradazione dopo un determinato numero di cicli; il rischio di amplificazioni aspecifiche, infine, è tanto più alto quanto più si prolunga la durata della reazione. Durante questo lavoro, è stato a volte aumentato il numero di cicli sino ad un massimo di 45; nei casi in cui il prodotto non sia risultato ugualmente sufficiente, abbiamo proceduto con altre tecniche (es. eminesting).
(d) La temperatura di annealing può essere variata in relazione alla specificità di reazione richiesta: una temperatura relativamente bassa facilita l’appaiamento degli inneschi, ma può rendere stabili anche gli eventuali appaiamenti effettuati su sequenze non perfettamente complementari (mismatch), determinando così una amplificazione aspecifica. Viceversa, mantenere la temperatura troppo alta sfavorisce termodinamicamente l’appaiamento dei primer con conseguente bassa resa di prodotto finale; l’alta temperatura può inoltre far sì che i primer, soprattutto se non esattamente complementari, non riescano a mantenersi appaiati alle regioni bersaglio, e non si abbia pertanto amplificazione. In un ciclo standard di PCR a bassa stringenza con primer universali è stata solitamente impostata una temperatura di annealing di 50°C.
(e) L’estensione finale prolungata dà modo all’enzima di completare la sintesi di eventuali frammenti lasciati incompiuti.
Ciclo di PCR touchdown (amplificazione quantitativa della sequenza bersaglio: Don et al., 1991; modificato):
1. Preparazione della miscela di reazione; suddivisione in aliquote; inserimento delle Eppendorf da 0,2 ml nel termociclatore.
2. Denaturazione del DNA genomico (94°C, 3’). 3. Ciclo (ripetuto 5 volte):
• Denaturazione delle molecole di nuova sintesi (94°C, 30’’) • Annealing dei primer (63°C, 30’’).
4. Ciclo (ripetuto 10 volte):
• Denaturazione delle molecole di nuova sintesi (94°C, 30’’) • Annealing dei primer (57°C, 30’’).
• Sintesi del nuovo filamento (72°C, 2’). 5. Ciclo (ripetuto 25 volte):
• Denaturazione delle molecole di nuova sintesi (94°C, 30’’) • Annealing dei primer (63°C, 30’’).
• Sintesi del nuovo filamento (72°C, 2’). 6. Estensione finale a 72°C per 7’.
7. Raffreddamento a 8°C (∞).
Note:
(a) Nel presente lavoro è stato utilizzato il termociclatore Primes 96 plus (MWG BIOTECH. AG).
(b) La denaturazione iniziale (punto 2) è più lunga in quanto è necessario separare i filamenti dell’intero DNA genomico. Viceversa, durante il ciclo iterato occorre denaturare solo molecole di nuova sintesi, lunghe al massimo alcune Kbasi: i tempi necessari risultano pertanto più brevi.
(c) Il programma di reazione riportato (punti 3-4-5) prevede inizialmente alcuni cicli ad alta stringenza, nei quali, virtualmente, viene amplificato soltanto la sequenza bersaglio, che diviene pertanto quantitativamente dominante. I successivi cicli, a stringenza progressivamente più bassa, comportano l’amplificazione collaterale del DNA non-bersaglio; tuttavia, la sequenza bersaglio, ora quantitativamente dominante, viene prodotta in un numero di copie maggiore, e potrà essere in seguito determinata per sequenziamento diretto o clonaggio. Tale tecnica consente di selezionare positivamente i geni di un organismo bersaglio da un campione contenente DNA di altri organismi. La procedura è stata da me utilizzata con primer specifici per organismi (in genere procarioti simbionti) bersaglio già parzialmente determinati dal punto di vista tassonomico, tramite procedure di ibridazione in situ o grazie all’identificazione di caratteristiche morfologiche tipiche dell’organismo o del gruppo tassonomico in questione. Può essere utilizzata anche con inneschi universali. Non è conveniente impiegare primer degenerati in questo tipo di reazione, poiché l’alta temperatura di annealing favorisce selettivamente le
degenerazioni in G e C (formanti 3 legami) rispetto a quelle in A-T (formanti due legami e conseguentemente meno stabili): i geni contenuti nel campione non sono pertanto amplificati nelle stesse proporzioni e la sequenza bersaglio potrebbe risultare poco rappresentata nel prodotto finale.
(d) L’estensione finale prolungata dà modo all’enzima di completare la sintesi di eventuali frammenti lasciati incompiuti.
-Purificazione del prodotto di reazione
Al termine di una reazione di PCR, all’interno della eppendorf da 0,2 ml estratta dal termociclatore saranno presenti, oltre al DNA stampo originale ed alle copie della sequenza bersaglio ottenute, anche frammenti oligonucleotidici indesiderati quali amplificati parziali, inneschi appaiatisi tra di loro (dimeri dei primer), dNTP e soprattutto primer inutilizzati. Questi prodotti secondari a basso peso molecolare costituiscono un rumore di fondo che può compromettere l’esito di una seconda reazione di PCR e/o della reazione di sequenziamento. Per selezionare positivamente i prodotti principali, l’intero volume della miscela di reazione viene purificato dai frammenti oligonucleotidici mediante passaggio attraverso colonnine di resina commerciali Quantum Prep® (BioRad).
Protocollo utilizzato (PCR Kleen Spin Column):
1. Risospendere la resina contenuta nella colonnina agitandola su Vortex (5’’)
2. Rimuovere il tappo e la chiusura inferiore della colonnina; collocare la stessa in un tubo da 2 ml.
3. Centrifugare la colonnina a 735 x g (3100 rpm) per eliminarne il liquido preservativo (1’).
4. Collocare la colonnina in una Eppendorf sterile da 1,5 ml. Pipettare l’intero volume di amplificato sulla parte più alta della resina.
5. Centrifugare a 735 x g (3100 rpm) (2’).
6. Raccogliere l’eluato (contenente il DNA). La colonnina è monouso.
-Procedura di eminest su DNA amplificato
Come ricordato, la replicazione del DNA tramite PCR può comportare errori sperimentali. La probabilità di incorrere in tali errori aumenta proporzionalmente al numero di cicli di reazione impiegati per ottenere una quantità di prodotto sufficiente. Una possibile soluzione consiste nel dare inizio alla PCR con un alto numero di molecole-stampo nella miscela di
reazione: in questo modo, pochi cicli saranno sufficienti a produrre grandi quantità di amplificato, e la sintesi complessiva sarà minore. Ove ciò non fosse possibile (situazione frequente in caso di organismi non coltivabili) occorre procedere diversamente. Dopo una prima reazione di PCR, il prodotto di reazione, opportunamente purificato, viene impiegato come DNA stampo per eseguire una seconda reazione. Gli inneschi utilizzati in questa seconda reazione, a differenza dei precedenti, non costeggiano la zona da amplificare, ma sono interni alla prima coppia di primer: vengono detti “primer annidati” (nested primers), ed il processo prende il nome di nesting. Per evitare la perdita delle zone del gene rimaste all’esterno dei primer annidati, è possibile eseguire una “emi-nested PCR reaction”: si preparano due diverse miscele di reazione, in ciascuna delle quali si utilizza uno degli inneschi “esterni” impiegati precedentemente, mentre sul filamento opposto si impiega un
primer “annidato”. Ogni reazione amplificherà una parte della sequenza bersaglio, che sarà
ricostruita successivamente, sequenziando e sovrapponendo le sequenze dei prodotti di entrambe le reazioni.
Questo procedimento consente di amplificare ulteriormente il prodotto di una prima reazione non più proseguibile; inoltre, utilizzando primer interni, più specifici, riduce significativamente la possibilità di amplificare sequenze indesiderate.
Il protocollo utilizzato per queste reazioni non differisce da quello per una normale PCR (vedi); essendo però i due tratti da sintetizzare più brevi che nel caso precedente, la durata della fase di estensione viene accorciata di conseguenza.
-Visualizzazione del prodotto di reazione mediante corsa elettroforetica su gel
L’esito della reazione di PCR può essere osservato caricando parte del prodotto su gel di agarosio colorato con bromuro d’etidio (intercalante delle basi del DNA) e sottoponendolo a corsa elettroforetica: le molecole polianioniche del DNA migreranno dal polo negativo a quello positivo in relazione di proporzionalità inversa al proprio peso molecolare, ed i frammenti amplificati, essendo delle stesse dimensioni, tenderanno a formare una banda ad una determinata altezza sul gel. Per stimare la lunghezza (in paia basi) delle molecole componenti tale banda, viene fatta correre in parallelo una miscela di frammenti di DNA di lunghezza nota (ladder): la lunghezza della banda incognita potrà essere stimata dal confronto con il pattern di bande, di lunghezza nota, generato dal ladder. Similmente è possibile estrapolare un valore approssimativo della quantità (in ng) di DNA ottenuto, paragonando l’intensità della banda incognita con quella delle bande del ladder, la cui concentrazione è nota.
Materiale utilizzato:
- Tampone di caricamento: Blu di bromofenolo (1,2 μl ogni 5 μl di campione). - Ladder GeneRulerTM DNA Ladder Mix (Fermentas) (3 μl per corsa).
- Apparecchio elettroforetico a corsa orizzontale (voltaggio variabile).
- Transilluminatore Spectroline mod. TR-312A Transilluminator 312 Ultraviolet.
Note:
(1) Tampone TBE 1x:
Tris Base 0,89 M Acido Borico 0,89 M
EDTA 0,02 M
Il pH della soluzione deve essere compreso fra 8,3 e 8,4 (2) Soluzione di Blu di bromofenolo:
Bromofenolo 0,25% Xilene cianolo 0,25%
Glicerolo idrato 30%
-Inserzione di geni amplificati in vettori di clonaggio
Il sequenziamento diretto di un prodotto di reazione di PCR è possibile soltanto nel caso in cui tale prodotto sia costituito dall’amplificato del gene di un unico organismo, oppure nel caso vi sia una forte dominanza della sequenza genica dell’organismo bersaglio rispetto a quelle di altri organismi eventualmente presenti. Nel corso del presente lavoro, durante la caratterizzazione del 16S rDNA di batteri simbionti, si è talvolta verificato che il prodotto di reazione fosse costituito da due o più ampliconi, in proporzione variabile, provenienti l’uno dal gene dell’organismo bersaglio (generalmente più rappresentato) e l’altro/gli altri da batteri contaminanti o da eventuali simbionti secondari presenti nello stesso ciliato ospite. Il cromatogramma ottenuto dal sequenziamento di tale prodotto mostra, per ogni nucleotide della sequenza, un picco principale ed uno o più sottopicchi, di altezza variabile, derivanti da nucleotidi presenti su altre sequenze ed ugualmente letti dal sequenziatore. Poiché non è possibile assegnare con certezza ciascuno dei nucleotidi visualizzati alle diverse sequenze di origine, esse risultano confuse fra loro. In questi casi si è fatto ricorso a procedure di clonaggio al fine di separare ciascuna delle sequenze presenti nella miscela originaria e poter procedere alla loro caratterizzazione. I geni amplificati sono stati clonati in vettori plasmidici; tali vettori sono stati utilizzati per trasformare cellule di Escherichia coli appositamente trattate (rese competenti) per poter incorporare il plasmide stesso. Poiché ogni plasmide contiene un unico gene, ciascun batterio trasformato dà luogo ad una colonia i cui
cloni contengono unicamente la copia del gene inserito nel plasmide originale. Estraendo i plasmidi dai cloni di un sufficiente numero di colonie, e sequenziando l’inserto in essi contenuto mediante primer appositi, è possibile caratterizzare singolarmente ciascuna delle sequenze presenti nel prodotto di partenza.
Le sequenze ottenute mediante tale procedimento risultano di qualità migliore rispetto a quelle derivanti dal sequenziamento diretto di prodotti di PCR in quanto il DNA-stampo utilizzato per la reazione di sequenziamento è privo dei sottoprodotti oligonucleotidici (in part. di residui di primer) che possono disturbare la reazione stessa. Tuttavia, tale DNA-stampo rappresenta la copia fedele del frammento originario inserito nel plasmide, il quale è frutto di amplificazione tramite PCR. Poiché l’enzima Taq DNA polimerasi è suscettibile di errori durante l’incorporazione di nucleotidi nei frammenti di nuova sintesi, è possibile che in essi siano contenuti nucleotidi diversi da quelli effettivamente presenti nella sequenza genica stampo. La frequenza di tali errori varia a seconda del tipo di enzima utilizzato; essendo generalmente molto bassa, le sequenze contenenti errori sono poco rappresentate nel prodotto finale: sequenziando tale prodotto, i nucleotidi “sbagliati” non risultano in quantità sufficiente da generare ambiguità nell’interpretazione del cromatogramma in uscita. Se però una molecola contenente errori viene clonata, l’errore sarà replicato nel 100% dei cloni prodotti, poiché il batterio duplicherà fedelmente la sequenza contenuta nel plasmide ad ogni divisione (gli errori commessi in vivo dalla polimerasi del batterio sono supposti trascurabili in quanto più rari di quelli commessi in vitro e poco rappresentati nel prodotto finale). Per ovviare a tale problema, sono stati sequeziati gli inserti presenti in almeno tre colonie trasformate con il medesimo amplicone (per la tecnica utilizzata nello screening dei cloni, cfr. Controllo della correttezza della ricombinazione e digestione del DNA con endonucleasi
di restrizione, presente sezione), ed è stata ottenuta una sequenza consensus: la probabilità
che il medesimo nucleotide sia stato inserito erroneamente in più di due molecole è infatti estremamente bassa.
Un ulteriore problema derivante dall’impiego di procedure di amplificazione-clonaggio è quello del possibile ottenimento di sequenze chimeriche. Ciò si verifica quando nel campione sottoposto a PCR sono presenti sequenze geniche stampo provenienti da più organismi. In tale situazione, può accadere che una molecola di nuova sintesi si distacchi dal filamento stampo prima che la Taq polimerasi abbia coperto l’intera lunghezza del gene bersaglio. Durante la successiva fase di annealing, tale molecola parzialmente amplificata può nuovamente appaiarsi con un DNA stampo appartenente al medesimo organismo: in tal caso, la sintesi viene completata correttamente. Poiché nella miscela è presente il DNA genomico di più organismi, può tuttavia accadere che essa possa appaiarsi con la regione, parzialmente complementare, del medesimo gene di un organismo diverso da quello originale. Anche in questo caso, il doppio filamento funge da innesco per l’enzima e la
sintesi viene condotta a termine: la molecola così ottenuta conterrà parte della sequenza genica del primo organismo e parte del secondo. Durante i successivi cicli di reazione, essa verà amplificata al pari dei filamenti “corretti”. Tali molecole ibride (“chimere”, dal nome del mostro mitologico composto da parti di animali differenti) possono formarsi a partire dalla commistione da due, tre o più geni di diversi organismi. La loro rappresentatività nel prodotto finale di reazione è in genere bassa, e, in caso di sequenziamento diretto del prodotto stesso, non creano ambiguità nell’interpretazione del cromatogramma ottenuto. Viceversa, se una molecola chimerica viene clonata, si ottiene la sequenza di un gene, in realtà inesistente, che mostra una collocazione filogenetica peculiare e che può essere interpretato come la sequenza genica di una nuova specie. Tale interpretazione può risultare avvalorata nel caso in cui si utilizzi la sequenza per disegnarvi specifiche sonde oligonucleotidiche a fluorescenza: esse, infatti, marcheranno positivamente uno degli organismi componenti la chimera, e precisamente quello il cui gene è servito da stampo per la regione nella quale è stata disegnata la sonda: in tal caso è possibile commettere l’errore di associare all’organismo stesso la sequenza chimerica (ove la sonda venisse disegnata esattamente nella regione in cui le sequenze di due organismi diversi si congiungono, essa potrebbe effettivamente non legarsi ad alcuno dei due, ed in tal caso la chimera sarebbe riconoscibile come tale; le probabilità che ciò avvenga sono però molto basse). L’esistenza di tali artefatti è stata riconosciuta in tempi relativamente recenti (Amann et al., 1995); il problema sembra essere più frequente di quanto supposto in passato. L’identificazione delle chimere non è semplice. Nello svolgimento del presente lavoro, è stato tenuta presente la possibilità di ottenere delle chimere. La procedura utilizzata per la loro identificazione è stata la seguente: le sequenze ottenute da procedure di clonaggio sono state inserite in un albero filogenetico; l’attenzione è stata focalizzata su quelle presentanti una collocazione filogenetica inusuale (ad esempio, molto distanti dal gruppo filogeneticamente più prossimo). Le sequenze in questione sono state rimosse dall’albero e divise in due parti: è stata quindi testata la collocazione filogenetica di ciascuna delle due metà (rispettivamente,
5’ e 3’). In tal modo è stato possibile scoprire alcune sequenze chimeriche, nonché
identificare, almeno parzialmente, gli organismi che le componevano. Il metodo utilizzato è empirico, ma è risultato efficace nell’individuazione di chimere composte da due organismi le cui sequenze vi siano rappresentate per un numero di nucleotidi tale da consentirne l’identificazione.
Nel presente lavoro, sono stati utilizzati vettori plasmidici pCR®2.1-TOPO® (Invitrogen).
Tutti i vettori della classe TOPO® vengono forniti linearizzati, dotati di un singolo
nucleoside timidina, non appaiato (overhanged), all’estremità 3’ di ciascun filamento e legati covalentemente all’enzima topoisomerasi I, purificato da Vaccinia sp. (“activated” vector). Durante la reazione di inserzione, il segmento di DNA genomico si lega alle estremità del
vettore grazie all’appaiamento con la deossitimidina (le Taq DNA polimerasi possiedono una attività trasferasi terminale, indipendente dallo stampo, che aggiunge un singolo nucleoside deossiadenosina a ciascuna estremità 3’ del filamento di nuova sintesi: tutti i prodotti di PCR presentano perciò tale nucleoside overhanged alle estremità di ciascun filamento). L’enzima topoisomerasi si lega alla doppia elica e taglia lo scheletro fosfodiestereo della molecola immediatamente dopo il sito target 5’-CCCTT su entrambi i filamenti. L’energia derivante dalla rottura del ponte fosfodiestereo viene conservata in forma di legame covalente fra il fosfato 3’ ed il residuo tirosinico Tyr-274 dell’enzima. All’inserimento dell’amplicone, la reazione procede in senso opposto, riformando il ponte fosfodiestereo e liberando l’enzima. Utilizzando vettori TOPO® non è quindi necessario ricorrere a reazioni di ligation, o
all’inserimento di apposite sequenze terminali nel prodotto di PCR prima del clonaggio. Il plasmide pCR®2.1-TOPO® è lungo 3931 pb. Il sito multiplo di clonaggio (MCS) è
fiancheggiato dal promotore per la RNA polimerasi T7 ed è inserito nel gene dell’operone del lattosio, codificante per l’enzima β-galattosidasi, che conferisce alle cellule la capacità di metabolizzare il galattosio: l’inserzione del frammento nel sito di clonaggio interrompe tale operone, rendendo le cellule incapaci di sintetizzare β-galattosidasi. Il plasmide contiene inoltre i geni per la resistenza all’ampicillina ed alla kanamicina: ciò consente di creare terreni di coltura selettivi per cellule trasformate (cfr. Trasformazione e selezione delle
cellule competenti, presente sezione).
La reazione di ricombinazione del vettore è eseguita come riportato: il tampone salino utilizzato (“Salt Solution”: 200 mM NaCl, 10 mM MgCl2) è aggiunto nella quantità standard
riportata nel protocollo TOPO TA Cloning® (Invitrogen).
L’amplificato perde rapidamente il nucleoside adenosina overhanged, situato all’estremità 3’ di ciascun filamento, che, appaiandosi con il nucleoside timidina presente sul filamento del vettore, consente l’aggancio e conseguentemente l’inserimento dell’amplicone nel vettore stesso. Lavorando con prodotti di PCR conservati da più di tre giorni, si è fatto perciò ricorso a procedure volte ad aggiungere nuovamente una deossiadenosina in posizione 3’ su ciascun filamento dell’amplificato (A-tailing): è stato impiegato un enzima DNA polimerasi a bassa attività correttrice di bozze (TaKaRa rTAQ) per catalizzare l’inserimento di deossiadenosina trifosfato (dATP) alle estremità del frammento.
Composizione della miscela per la ricombinazione del vettore: - Amplificato (eventualmente sottoposto ad A-tailing) (2 μl). - Salt Solution (0,5 μl).
- Vettore (0,5 μl). Procedura per A-tailing:
1. Miscela di reazione: - Amplificato (6,4 μl).
- Tampone 10x TaKaRa Buffer (1 μl). - MgCl2 25 mM (0,6 μl).
- dATP 2 mM (1 μl). - TaKaRa rTAQ (1 μl).
2. Incubare a 70°C (30’) in termociclatore Primes 96 plus (MWG BIOTECH. AG).
-Trasformazione e selezione delle cellule competenti
Una volta ottenuti i plasmidi ricombinanti, essi vengono impiegati per la trasformazione di cellule competenti di E. coli del ceppo DH5α (Invitrogen). Tali cellule sono Lac- e sensibili ad ampicillina e kanamicina. Una volta trasformate con plasmidi pCR®2.1-TOPO® (cfr.
Inserimento di geni amplificati su vettori di clonaggio, presente sezione) esse acquisiscono
resistenza agli antibiotici e possono essere coltivate su terreni selettivi (es. LB-agar-ampicillina). L’aggiunta di X-GAL (5-Br-4-Cl-3-indolil-β-D-galattopiranoside: substrato cromogeno per la β-galattosidasi) permetterà inoltre di distinguere le colonie originanti da cellule correttamente trasformate dalle quelle di batteri trasformati con plasmidi non ricombinanti.
Protocollo utilizzato (TOPO TA Cloning®):
1. Prelevare una aliquota (ca. 200 μl) di cellule competenti; velocemente aggiungere 3 μl della miscela contenente il vettore.
2. Porre in ghiaccio per 20’.
3. Shock termico: trascorsi i 20’, passare rapidamente le cellule a 42°C (30’’), quindi porre nuovamente in ghiaccio.
4. Aggiungere 200 μl di terreno di coltura SOC Medium commerciale. 5. Collocare a 37°C per 75’ in agitazione.
6. Piastrare rispettivamente 50, 75 e 100 μl del mezzo contenente le cellule in capsule Petri con terreno solido LB Medium con l’aggiunta di ampicillina 50 mg/ml in H2O
(1 ml/l) e X-GAL 40 mg/ml in DMSO (1 ml/l). 7. Incubare le piastre a 37°C per 12h.