Capitolo 1
Introduzione
L'identificazione dei possibili target di un composto chimico, dotato di attività biologica, costituisce da molto tempo un grande motivo di interesse nel processo di drug development, visti soprattutto gli ingenti sforzi compiuti nel tentare di comprimere le prime fasi di studio dello sviluppo di un nuovo farmaco. Considerando i grandi aiuti forniti dalle più moderne tecniche sperimentali, quali la chimica combinatoriale o l'high throughput screening, che permettono l'ottenimento, in tempi relativamente brevi, di un gran numero di nuove entità chimiche, l'aspettativa di trovare, tra queste, molecole potenzialmente attive dal punto di vista biologico, risulta essere, almeno agli inizi, particolarmente elevata. Nella realtà, spesso entrambe le metodiche scientifiche sopra menzionate non conducono all'ottenimento dei risultati sperati, pertanto, ultimamente, tecniche innovative in silico sono emerse e, quindi, perfezionate: il virtual screening rappresenta tra queste uno degli strumenti più ricorrenti ed utili, in quanto permette l'individuazione di composti dotati di determinate caratteristiche a partire da grandi database di molecole. In particolare, avendo a disposizione le informazioni necessarie affinché un ligando possa interagire con il rispettivo recettore, è possibile ricorrere a queste ultime per stabilire quali molecole, provenienti da una qualsiasi libreria chimica, presentano le caratteristiche necessarie affinché la medesima e specifica interazione possa essere riprodotta, facendo così delle piccole previsioni di quelli che poi risulteranno essere i corrispondenti profili farmacologici di tali composti.
1.1 Target, Off-Target
L'attività farmacologica di un composto chimico si esplica attraverso un'interazione specifica con il rispettivo target biologico macromolecolare, solitamente rappresentato da una proteina o da un acido nucleico: in seguito a tale evento, si assiste ad una modifica nell'attività di quest'ultimo, e quindi alla manifestazione dell'effetto
Nella realtà, siamo ben lontani dalla situazione, prossima all'idealità, auspicata da Paul Ehrlich, riassumibile nell'espressione "un farmaco, un unico target", in cui un farmaco viene considerato come una sorta di "proiettile magico" in grado di distinguere in maniera netta ed assoluta il proprio bersaglio molecolare, senza intaccare minimamente l'ambiente biologico circostante. La totale specificità è un obiettivo difficile da raggiungere, se non praticamente impossibile: oltre all'interazione prevista in partenza, quella per la quale il farmaco stesso è stato pensato e progettato, vengono a instaurarsi infatti molteplici interazioni secondarie, possibili cause di effetti collaterali e reazioni avverse. I bersagli alternativi con cui un farmaco sviluppa tali interazioni vengono definiti in questo contesto come off-target, vale a dire macromolecole biologiche la cui attivazione risulta inaspettata o quantomeno sgradita in seguito alla somministrazione di un particolare composto bioattivo. Ovviamente interazioni di questo tipo, definibili come off-label, sono difficili, se non impossibili, da prevedere durante le fasi di progettazione: questo si traduce nell'elevato tasso di abbandono, in genere al di sopra del 90% [1], causato dall'affioramento delle mancanze di efficacia e di sicurezza clinica nelle fasi avanzate di sperimentazione; tali fallimenti si rispecchiano inoltre nei costi richiesti in genere per lo sviluppo di un nuovo farmaco, stimabili attorno agli 800 milioni di dollari, oltre che nelle tempistiche che ne precedono l'entrata in commercio, comprese tra i 15 e i 20 anni [2].
I recettori transmembranali accoppiati a proteine G, assieme ai canali ionici, risultano essere i target primari di 9 dei 20 farmaci maggiormente prescritti in tutto il mondo a partire dal 2000 [3], il che sottolinea la loro grande importanza come bersagli terapeutici di svariate patologie. È da chiarire, comunque, che non è possibile distinguere in maniera categorica un bersaglio primario da un off-target, visto che un qualsiasi recettore, target primario di un farmaco specifico, può risultare essere la causa principale degli effetti collaterali per un altro ancora: un esempio, tra i tanti, è costituito dai recettori muscarinici, mirati, tra l'altro, per il trattamento terapeutico dell'incontinenza urinaria, e che rappresentano tuttavia i maggiori responsabili di effetti collaterali comuni di farmaci ulteriori, quali secchezza delle fauci, ritenzione urinaria e visione offuscata [4]. Alternativamente, alcune proteine possono costituire solamente un target indesiderato, e sono ritenute essere le cause principali di gravi insorgenze secondarie, come accade per il canale hERG, responsabile dei letali effetti collaterali di molti composti come l'antiistaminico terfenadina [5], ritirato successivamente dal
commercio. Altri esempi sono costituiti dal citocromo P450 o la glicoproteina P, considerati i responsabili delle variazioni di efficacia che intercorrono tra individuo e individuo, oppure delle particolari interazioni che vengono a crearsi tra farmaci somministrati contemporaneamente [6,7]. Considerando questi esempi, appare evidente il grande potenziale sfruttabile dalla possibilità di predire l'attività futura di un composto, e quindi i relativi target primari e secondari, tramite metodi in silico rapidi ed efficienti, permettendo una sottrazione ai vincoli che legano la ricerca farmaceutica sia alle lunghe e costose metodologie di sperimentazione umana che alle procedure di sorveglianza clinica dopo le rispettive entrate sul mercato. La definizione in silico del profilo farmacologico di un candidato farmaco aprirebbe quindi scenari innovativi, consentendo di prevedere, e quindi evitare, interazioni secondarie eventualmente dannose, o approfondire e potenziare quelle di farmaci già esistenti e presenti sul mercato. Questo consentirebbe anche di riproporre infine vecchi candidati per nuove indicazioni terapeutiche, percorso, questo, ampiamente intrapreso nell'ultimo periodo, sicuramente conveniente per giungere all'individuazione di un nuovo medicamento. Inoltre la cura di patologie come l'Alzheimer, varie forme di cancro o la schizofrenia, può trovare un vantaggio da un approccio consistente nel determinarne a priori i diversi target che ne stanno alla base, non correlati biologicamente tra loro: le contemporanee modulazioni di questi potrebbero portare infatti a seri benefici nei pazienti afflitti da tali gravose condizioni [8].
1.2 Identificazione del target
Attualmente, i farmaci presenti in commercio coprono solamente il 5% dell'intero proteoma dell'uomo: in altre parole, solo il 5% di quanto viene prodotto dal genoma umano costituisce effettivamente un bersaglio conclamato dei composti attivi disponibili in terapia [9]. Tale percentuale si riduce drasticamente, a valori inferiori allo 0.2%, se andiamo ad analizzare il proteoma di altri organismi, quali gli agenti patogeni M. Tubercolosis o ancora T. Brucei. Prendendo in considerazione una qualsiasi molecola di interesse, di dimensioni limitate e della quale sia nota la precisa struttura chimica, è possbile, adoperando le opportune metodologie, ricavarne informazioni riguardo i
azione in relazione alla totalità recettoriale costituita dal proteoma di un qualsiasi organismo.
Storicamente la genetica è stata ispiratrice di utilissime intuizioni biologiche, consentendo l'individuazione e la caratterizzazione della funzione di numerose proteine tramite tecniche che prevedevano l'alterazione di specifiche sequenze geniche. Uno degli approcci più utilizzati in questo senso consiste nell'evocare, ricorrendo alle opportune tecniche sperimentali, un fenotipo di interesse, procedendo quindi con l'identificazione a posteriori dei geni che ne sono direttamente responsabili e, quindi, della macromolecola che ne è coinvolta [10].
Metodi più moderni di biologia molecolare e di ingegneria genetica hanno percorso la stessa strada in direzione opposta, agendo direttamente su uno specifico gene tramite tecniche di manipolazione genica, per andare quindi ad effettuare una ricerca estesa per scovare il corrispondente fenotipo risultante [11]. Screening fenotipici costituiscono ancora oggi la fonte principale di piccole molecole farmacologicamente attive: dal momento però che il passaggio che va dall'osservazione del fenotipo particolare al corrispondente target di interesse costituisce una procedura che solamente nel 40% dei casi riscuote successo [12], sono necessarie metodologie alternative. Ad oggi, gli approcci di maggior interesse per l'identificazione di un target comprendono metodi biochimici di tipo diretto, metodi di interazione genica e strategie di deduzione computazionale [13].
I metodi biochimici sono detti diretti in quanto prevedono l'incubazione, dopo marcamento, del target con la molecola di interesse, seguita dagli opportuni saggi volti a determinarne l'affinità reciproca; pertanto, essi possono condurre all'ottenimento di preziose informazioni riguardanti i meccanismi molecolari che determinano l'efficacia o la tossicità del composto testato, essendo basati effettivamente sulle interazioni "fisiche", reali, che vengono ad instaurarsi. Il secondo approccio menzionato ricorre a induttori o soppressori genici che vanno ad agire direttamente su RNA e DNA, per andare successivamente a valutare le differenti espressioni geniche in tessuti sani e malati: organismi knock-out per geni specifici, nei quali viene inibita selettivamente l'espressione di un gene codificante per una proteina, possono quindi essere utilizzati per valutare il ruolo di un determinato target all'interno del contesto di una particolare patologia.
consentendo infatti di essere utilizzabili anche per l'identificazione di target secondari di composti noti, in aggiunta a costituire un valido appoggio per ulteriori discipline quali la proteomica e la tecnologia genetica. Sono almeno due, in questo campo, le tecniche utilizzate al fine di predire i potenziali target di una particolare molecola dotata di attività farmacologica. Un primo metodo assume che tasche recettoriali simili possano accogliere ligandi paragonabili dal punto di vista chimico e strutturale: un approccio di questo genere, definibile come structure-based, risulta utile nel creare nuove correlazioni tra target apparentemente estranei tra loro. È interessante notare, tuttavia, che farmaci chimicamente affini, appartenenti alla medesima categoria chimico-farmaceutica, spesso mostrano attività verso target dotati di sequenza amminoacidica differente a livello del rispettivo binding site, come accade per i farmaci serotoninergici, in grado di legarsi sia ai recettori metabotropici 5-HT1,2,4-7 che a quelli ionotropici
5-HT3A, dissimili tra loro sia per sequenza che per struttura [14]. Da casi come questo
emerge la necessità di correlare target farmacologici non più quindi secondo le relative caratteristiche strutturali, bensì sulla base della chimica dei rispettivi ligandi: un approccio, quindi, di tipo ligand-based, focalizzato maggiormente sul ligando, approvando il presupposto che composti chimici simili tra loro dal punto di vista strutturale possano mostrare target biologici comuni e, conseguentemente, profili farmacologici sovrapponibili. All'interno di quest'ultimo gruppo sono rintracciabili ulteriori metodologie, che fanno affidamento sulla disponibilità di complessi ligando-proteina già esistenti: queste utilizzano docking molecolare [15] o modelli farmacoforici estratti da tali complessi cristallizzati per creare dei collegamenti tra il composto analizzato e i rispettivi target macromolecolari. Le procedure di docking si rivelano essere strategie computazionali relativamente lente e soggette a seri errori che ne possono compromettere l'affidabilità e la riproducibilità, anche se sono state utilizzate con successo in diversi casi portando alla scoperta di nuovi target di molecole già conosciute [16]. Al contrario, valutare l'affinità di un set di molecole in base ad una query farmacoforica costituisce sicuramente un procedimento più veloce; inoltre, un approccio farmacoforico può essere integrato mediante il ricorso a metodologie più puramente ligand-based (ligand-centric methods) in maniera tale da estenderne l'applicabilità anche a potenziali target dei quali la struttura 3D risulta assente, pur godendo di un gran numero di ligandi conosciuti.
1.3 Strategie computazionali: stato attuale
Riportiamo nei paragrafi seguenti alcune brevi panoramiche sulle più recenti ed innovative strategie computazionali, sostanzialmente di tipo ligand-based, volte alla creazione di protocolli in silico capaci di prevedere i futuri target di un composto di particolare interesse.
1.3.1 Parallel screening
Parallel screening [17], elaborato da Langer e collaboratori, costituisce un interessante ed innovativo metodo in silico, utilizzato al fine di predire l'attività biologica di un composto, in seguito ad un virtual screening contemporaneo verso una serie di modelli farmacoforici di testata qualità.
Un sistema in grado di effettuare un parallel screening richiede un gran numero di modelli farmacoforici selettivi, uno strumento efficace in grado di generare questi in maniera veloce ed automatica ed infine una piattaforma capace di effettuare lo screening desiderato in maniera affidabile, confrontando rapidamente le caratteristiche farmacoforiche del composto analizzato con i molteplici modelli disponibili, e consentendo inoltre un'analisi, una visualizzazione ed una interpretazione corretta dei dati ottenuti.
Una metodica di questo genere permette quindi di caratterizzare, almeno in maniera virtuale, le proprietà biologiche di nuovi composti, stabilendone la sfera di azione e magari allargando quella di farmaci già noti a target non conosciuti in precedenza. Includendo gli appropriati modelli farmacoforici, è possibile inoltre fare previsioni riguardanti eventuali effetti collaterali, interazioni secondarie e possibili processi metabolici intrapresi. Al termine, per un qualsiasi composto analizzato, viene quindi stilata una vera e propria target list, traducibile come il corrispondente profilo farmacologico, nella quale sono elencate le macromolecole target dai quali cristalli provengono i modelli farmacoforici verso cui la molecola test ha mostrato maggiori affinità.
Una delle caratteristiche imprescindibili che tale sistema deve necessariamente possedere è la specificità, requisito che emerge prepotentemente lavorando su un set di composti strutturalmente o funzionalmente simili; in altre parole, tale protocollo deve
essere in grado di distinguere molecole tra loro paragonabili, in maniera tale da poter traccciare, per queste, dei profili farmacologici non sovrapponibili e ridondanti.
INPUT MOLECULE
VIRTUAL SCREENING APPROACH WITH A LARGE SET OF PHARMACOPHORE MODELS
PHARMACOPHORIC PROFILE
PHARMACOLOGIC PROFILE
ACTIVITY PROFILING
Figura 1: concetto fondamentale del Parallel screening
Langer e collaboratori sono riusciti nell'intento di mettere a punto un sistema completamente automatizzato, che quindi sfugge agli errori che possono essere commessi dall'utente, riducendo inoltre le tempistiche e gli step necessari che precedono l'ottenimento dei risultati finali.
1.3.2 Similarity Ensemble Approach (SEA)
SEA (acronimo di Similarity Ensemble Approach) [18], sviluppato da Shoichet e collaboratori, permette di correlare l'attività biologica di proteine sulla base delle caratteristiche chimiche dei rispettivi ligandi: tale metodo può essere considerato come uno degli approcci di tipo ligand-centric più significativi nello stabilire attività secondarie di composti già conosciuti.
Partendo da circa 65000 ligandi, suddivisi sulla base dei corrispondenti 246 target noti in altrettanti gruppi, sono state calcolate le similitudini che intercorrono tra i diversi set di ligandi, facendo riferimento, come termine di confronto, esclusivamente alla relativa struttura 2D dei composti che li costituiscono (tecniche di tipo fingerprint):
target, possano essere considerati simili anche se non mostrano alcun composto in comune.
Contemporaneamente è stato messo a punto un modello statistico che potesse permettere un'analisi oggettiva e una classificazione ponderata dei punteggi di similarità ottenuti. Al termine, l'attenzione è stata posta su quei composti dotati di una certa promiscuità di azione.
Un esempio particolarmente interessante è costituito dal metadone, noto agonista dei recettori µ degli oppioidi: per tale composto è emersa, tramite le particolari tecniche di fingerprint utilizzate, una similarità verso il cluster rappresentato dagli antagonisti ai recettori muscarinici M3 maggiore rispetto a quella verso il cluster di appartenenza. Confermando tale predizione, il metadone ha mostrato una Ki di 1.0 µM e quindi
confermato sperimentalmente l'attività antimuscarinica. Attività secondarie simili sono state individuate, e confermate sperimentalmente, per altri composti quali emetina e loperamide: tutte le nuove interazioni scovate giustificano inoltre i noti effetti collaterali di questi farmaci.
1.3.3 PROFILER
PROFILER [19], disponibile su richiesta degli autori, è un protocollo ibrido, in quanto combina tra loro tecniche notoriamente ligand-based con tecniche di tipo structure-based come il docking: si tratta di un metodo dinamico, in grado di scegliere per ogni possibile binomio ligando-target la strategia più appropriata da condurre, sfruttando sia i punti di forza che le debolezze di ciascuna tecnica volta a determinare il profilo farmacologico di un composto.
Il workflow seguito è piuttosto intricato: inizialmente PROFILER analizza la struttura 2D del ligando considerato, rivolgendo particolare attenzione alla relativa polarità. A seconda del numero riscontrato di accettori e donatori di legame a idrogeno viene intrapreso un percorso specifico. Se il numero calcolato è inferiore a 3, si procede con un analisi conformazionale del composto in esame: ciascun conformero è successivamente sovrapposto, tramite un software opportuno, ai vari ligandi cristallizzati con un dataset di target specifici, ed ottenendo come target potenziali quelli complessati ai ligandi verso i quali è stato raggiunto un miglior allineamento. In caso contrario, e quindi un numero di donor e/o acceptor maggiore di 3 nella molecola di
partenza, l'attenzione viene spostata sul target: in questo caso le scelte future intraprese da PROFILER saranno influenzate dal numero di volte che questo compare, complessato con ligandi tra loro differenti, all'interno del proprio set. Per altri casi specifici, sono invece considerate le caratteristiche chimiche all'interno del binding site, applicando, a seconda della polarità riscontrata, procedure di docking o ricerche di tipo farmacoforico. Qualora il target considerato sia presente in più di dieci complessi assieme a ligandi differenti, il protocollo ricorre a metodi di tipo SVM (support vector machine) per predire l'affinità o la probabilità di associazione con il ligando di input.
Applicato ad un set di 189 potenziali candidati ed utilizzando un set di 4371 proteine, PROFILER ha predetto con efficacia il target primario nel 72% dei casi.
Capitolo 2
Metodi
2.1 Fortran: breve introduzione
Il Fortran [20] è uno dei linguaggi di programmazione più comuni ed utilizzati per risolvere problemi scientifici: il suo nome deriva da FORmula TRANslation, vale a dire "traduttore di formule (o equazioni scientifiche)" in linguaggio del computer. La prima versione, denominata FORTRAN I, fu sviluppata nel 1954. In questo paragrafo sono descritte brevemente quelli che possono essere considerati gli elementi del linguaggio FORTRAN-77 [21] (quello utilizzato, e che costituisce una delle versioni più recenti) risultati fondamentali ed imprescindibili nello svolgimento di questo progetto di tesi, e che hanno permesso la costruzione di piccoli programmi necessari per la scrittura di script particolari o per l'esecuzione di determinate operazioni.
2.1.1 Istruzioni e struttura di un programma
Un programma Fortran è formato da una serie di istruzioni (dette statement) che permettono la raggiunta di un determinato obiettivo, quello per il quale lo stesso programma è stato composto. Le istruzioni possono essere eseguibili e non eseguibili: le prime esplicano le azioni che verranno svolte dal programma durante la sua esecuzione; le seconde contengono le informazioni necessarie affinché un programma possa essere eseguito correttamente.
Ciascuna linea di un programma non può contenere più di 72 caratteri: ne deriva che, se un'istruzione contiene un numero di caratteri tali per cui questo limite venga superato, sono necessari dei caratteri di continuazione. Il carattere "&" (ampersand) viene utilizzato a questo scopo. Le due istruzioni seguenti hanno quindi lo stesso significato in FORTRAN-77, e vengono lette ed eseguite nello stesso modo:
100 d=(((x-x1)**2) + ((y-y1)**2) & + ((z-z1)**2))**0.5
Una solita istruzione può continuare sino ad un massimo di 19 linee di continuazione. Il numero posto all'inizio della linea viene chiamato label (etichetta) e può essere considerato come un vero e proprio nome con cui si indica una particolare istruzione: esso può assumere un qualsiasi numero compreso tra 1 e 99999, ma non ha alcun significato temporale, quindi nessun rapporto con l'ordine con cui vengono eseguite le varie istruzioni di un programma. La label deve essere unica per l'istruzione cui viene conferita: nel caso precedente, ad esempio, ciascuna istruzione del solito programma non potrebbe essere assegnata con la label 100. Pur essendo facoltative, le label sono molto utili, in quanto tramite il loro utilizzo è possibile richiamare particolari istruzioni da altre parti del solito programma (per questo scopo si utilizza l'istruzione eseguibilie GO TO). Nelle linee di istruzione, le colonne da 1 a 5 sono dedicate all'illustrazione dell'etichetta, la colonna 6 all'eventuale segno di continuazione mentre dalla 7 alla 72 alla descrizione dell'istruzione stessa.
Sulla stessa riga di un'istruzione è possibile aggiungere delle linee di commento: in FORTRAN 77 qualsiasi linea che contiene i caratteri C o * in prima colonna viene considerata come tale. I commenti non influenzano la compilazione di un programma, né tantomeno vengono letti durante l'esecuzione dello stesso: risultano però particolarmente utili, in quanto consentono all'utente di spiegare e documentare il funzionamento di alcuni blocchi, e quindi facilitare l'orientamento all'interno del programma.
Ciascun programma Fortran deve essere composto seguendo un ordine ben preciso. La struttura comune a tutti i programmi scritti con questo linguaggio è la seguente:
1. PROGRAM nome del programma 2. Istruzioni non eseguibili.
3. Istruzioni eseguibili. 4. Stop.
Istruzione PROGRAM
L'istruzione PROGRAM è la prima istruzione di qualsiasi programma Fortran: è di tipo non eseguibile e può contenere da un minimo di 1 a un massimo di 31 caratteri, il primo dei quali costituito obbligatoriamente da una lettera dell'alfabeto.
Istruzioni non eseguibili
Le istruzioni non eseguibili, che seguono l'istruzione PROGRAM appena vista, dichiarano il nome e la tipologia delle variabili utilizzate dal programma. Possono essere di tipo:
• CHARACTER, dichiara una variabile di caratteri. Occorre specificare opportunamente il numero di caratteri che la compongono; in caso contrario, la variabile può contenere un carattere soltanto. L'espressione generale è CHARACTER*<dimension> :: nome.
• INTEGER, definisce le variabili intere (INTEGER :: nome).
• REAL, definisce le variabili reali, di virgola mobile, con o senza segno (REAL ::nome).
Inoltre, in questa sezione, è possibile assegnare il valore a variabili aggiuntive utilizzate, come il valore "3.141593" alla variabile pi o l'espressione "ipotenusa * COS(theta)" alla variabile lato. Alternativamente, è possibile inizializzare una variabile, ovvero conferirle, prima dell'esecuzione del programma, un valore iniziale che potrebbe subire dei mutamenti nel corso dello stesso.
Istruzioni eseguibili
La prima fase della sezione esecutiva consiste nello specificare i file di input e di output: questi ultimi sono costituiti dai record, che ne definiscono la struttura. Un record, definibile come una sequenza di valori o caratteri che forma l'unità di dati di un file, può essere formattato o meno: in quest'ultimo caso, la sua interpretazione dipende dal tipo di dati. I file di input e di output sono dichiarati dall'istruzione eseguibile OPEN: oltre al nome del file (indicato dalla clausola FILE) che intendiamo aprire, è
necessario specificare il numero dell'unità cui il file viene assegnato (clausola UNIT), la forma, formattata o meno, dei relativi record (clausola FORM), la clausola STATUS, che può assumere la forma 'OLD' se il file è già esistente oppure 'NEW' se intendiamo crearne uno nuovo. Le varie clausole sono separate da virgole.
Una volta terminato il programma, tutte le unità sono chiuse automaticamente. Un'istruzione CLOSE in posizione intermedia può risultare utile qualora si desideri il riassegnamento di un'unità o di un file: ad esempio, terminata la scrittura di un file NEW, può essere necessaria la sua lettura immediata, quindi si procede con la chiusura del file stesso e la sua successiva riapertura come OLD. Per funzionare, è sufficiente specificare il numero dell'unità oggetto dell'istruzione CLOSE. Le istruzioni eseguibili OPEN e CLOSE correlano quindi le unità logiche di ingresso e uscita con i nomi dei file.
La sezione esecutiva successiva contiene le istruzioni di INPUT/OUTPUT, le quali vanno a costituire un canale di comunicazione tra il programma Fortran e il mondo esterno. Le principali sono le istruzioni READ (*,*) e WRITE (*,*), che attuano trasferimenti di dati tra file. Il primo asterisco tra parentesi occupa la posizione destinata a specificare il numero dell'unità assegnata al file oggetto dell'istruzione, il secondo mantiene il formato libero.
• L'istruzione READ (*,*) legge un valore dal file input e lo carica nella variabile specificata: nel caso in cui le variabili siano più di una, allora i valori vengono registrati nell'ordine in cui le variabili sono elencate. L'istruzione comincia con la lettura partendo dall'inizio di ogni riga dell'unità di input. L'istruzione:
read (*,*) feature
associa il primo dato che incontra nella lettura del file di input alla variabile feature.
• L'istruzione WRITE (*,*) riscrive le variabili su file interni od esterni. Nell'esempio seguente:
write (*,*) feature
scrive il valore della variabile feature nell'unità di output specificata. Alternativamente è possibile specificare direttamente il testo che vogliamo sia scritto in un dato file, ponendolo tra virgolette. Nell'esempio che segue:
il testo 'Gianluca' viene scritto nell'unità standard di output. Altre istruzioni compiono operazioni ausiliarie.
• L'istruzione BACKSPACE (unit= numero unità) torna al record precedente del file che occupa l'unità specificata.
• L'istruzione REWIND (unit= numero unità) torna al record iniziale del file.
Le istruzioni READ e WRITE in particolare possono essere eseguite mantenendo un formato libero o indirizzato da lista. La definizione del formato sostituisce il secondo asterisco nell'espressione (*,*). I dati ottenuti con il formato libero non sempre si presentano bene: spesso infatti presentano un gran numero di spazi vuoti. Per evitare questo inconveniente, è possibile rappresentare i file di output nel formato preferito dall'utente, utilizzando gli opportuni descrittori. Di seguito sono riportati quelli più utilizzati nel corso di questa esperienza di programmazione:
• I: controlla il formato dei valori interi, dichiarati dall'istruzione INTEGER nella sezione non esecutiva del programma. La sua formula generale è rIw, dove: r= fattore di ripetizione, specifica il numero di volte che deve essere utilizzato un descrittore di formato; w=larghezza di campo, specifica il numero di caratteri da utilizzare per quel dato.
• F: controlla il formato dei valori reali, definiti dall'istruzione REAL. La sua formula generale è rFw.d, dove: d= indica il numero delle cifre decimali a destra della virgola nei numeri reali di input ed output; r e w mantengono il significato visto prima.
• A: controlla il formato dei testi di input ed output. La sua forma generale è rAw.
• X: ignora all'interno dei campi di input quei dati che non vogliono esser letti (nell'istruzione READ) oppure introduce un numero di spazi pari a n nel buffer di output (nell'istruzione WRITE); è di formula generale nX.
Tutte le istruzioni eseguibili possono essere compiute in maniera condizionata utilizzando il blocco IF: solo se viene rispettata una condizione, introdotta dallo stesso IF, allora le istruzioni successive, facenti parte del blocco ed introdotte da THEN, saranno lette, e quindi eseguite, dal programma. Il costrutto termina in corrispondenza della voce ENDIF: una volta eseguite le istruzioni contenute nel blocco, il programma passa alla prima istruzione che segue l'ENDIF.
Termine del programma
L'istruzione STOP blocca l'esecuzione del programma. Può o meno precedere l'END finale: in questo caso può essere omessa.
L'istruzione END indica al compilatore che non ci sono più istruzioni da compilare all'interno del programma. Con essa si chiude il programma Fortran.
2.1.2 Compilazione del programma
Terminata la scrittura di un programma Fortran, si procede con la sua compilazione, vale a dire con la sua traduzione in un linguaggio che risulti comprensibile per il computer. I file contenenti il codice sorgente vengono salvati come <nomeFile>.f e la compilazione viene definita con la preparazione di un file eseguibile; il comando utilizzato è f77. Lo script utilizzato è il seguente:
f77 -i <nomeFile>.f -o <nomeFile>
Dove: -i= indica il nome del file contenete il programma che vuole essere compilato; -o= specifica il nome del file eseguibile che viene creato.
Adesso è possibile eseguire il programma:
./<nomeFile>
2.2 ROCS
ROCS, acronimo di Rapid Overlay of Chemical Structures, è un software creato per effettuare ricerche su grandi database di molecole 3D tramite l'utilizzo di un metodo di sovrapposizione che permette di scovare composti non intuitivamente simili, che possono successivamente essere analizzati e sottoposti alle fasi posteriori di un processo di drug discovery.
1-2 molecole per secondo); al contrario, ROCS è in grado di sovrapporre dai 600 fino agli 800 conformeri per secondo. A questo si aggiunge la possibilità di dividere un solito calcolo su più processori, traendo un vantaggio massimo dal lavoro contemporaneo di più macchine e rendendo possibile lo screening di database di decine di milioni di composti a tempi ragionevoli.
Per le sue ricerche, ROCS utilizza un metodo di sovrapposizione di tipo shape-based: le molecole sono allineate attraverso un processo di ottimizzazione del corpo rigido che massimizza il volume condiviso: nel fare ciò, il programma considera solamente gli heavy atoms dei ligandi e ignora gli idrogeni. Dal momento che in questo contesto forma e volume sono due entità correlate tra loro, la suddetta procedura di massimizzazione del volume condiviso si rivela un utile strumento per raggiungere informazioni riguardo la somiglianza delle forme.
Sebbene il progamma consista principalmente in un metodo shape-based, l'utente può includere nel processo di sovrapposizione e di analisi della somiglianza una sua personale considerazione della chimica, in modo da facilitare l'identificazione di quei composti che sono simili sia dal punto di vista della forma che dal punto di vista elettrostatico. La possibilità di unire uno screening riguardante la forma a uno specifico per la chimica, il tutto frazionabile su più macchine, rende ROCS un software incredibilmente potente per la ricerca di composti all'interno di grandi database.
Pur essendo storicamente uno strumento utilizzabile esclusivamente da command line, la release 3.0 ha incluso un visualizzatore grafico e un editore di query, denominato vROCS.
2.2.1 Teoria
Parlando di ROCS, e quindi del suo metodo di lavoro, deve essere ben chiaro il significato della parola "forma": in questo contesto si afferma che due entità hanno la stessa forma se il loro volume corrisponde perfettamente. Al contrario, più la forma differisce e più il volume occupato dalle stesse si allontana dal corrispondere. Il volume è invece definito come un campo scalare, ovvero una funzione che ha un singolo valore numerico in ogni punto dello spazio:
La funzione del volume viene inoltre definita come funzione caratteristica. Dobbiamo aggiungere inoltre che due oggetti non possono avere la stessa forma se i loro volumi sono differenti; contemporaneamente, possono però avere lo stesso volume ma forma diversa. A questo punto possiamo scrivere una precisa definizione matematica di similitudine della forma considerando il seguente integrale:
S1= ∫ |f (x, y, z) – g (x, y, z)| dV
dove f e g sono due diverse funzioni caratteristiche. Quando l'integrale assume un valore pari a zero allora f e g rappresentano esattamente la stessa funzione e corrispondono quindi alla stessa forma. Al contrario, più il valore dell'integrale si allontana da zero, più differenti sono le forme rappresentate da f e g.
L'integrale rappresenta quindi una quantità metrica tra le due grandezze f e g: il termine metrico viene usato raramente per riferirsi alla forma, ma qui assume un preciso significato matematico, cioè una distanza che:
• è sempre positiva;
• assume valore zero solo quando due entità sono identiche;
• rispetta la disuguaglianza triangolare, la quale afferma che se un'entità A dista x da un'entità B, e B dista y da C, allora la distanza tra A e C è compresa tra |x-y| e |x+y|.
Il tipo di metrica descritto da S1 viene indicato come metrica L1. Un altro tipo di metrica è la S2:
S2 =√ ∫ [f (x, y, z) – g (x, y, z)]2 dv
Questo integrale rappresenta quello standard utilizzato per definire l'uguaglianza di forma. Elevando ambo i membri dell'uguaglianza alla seconda otteniamo:
S22 = ∫ f (x, y, z)2 dV + ∫ g (x, y, z)2 dV – 2 ∫ f(x, y, z) g (x, y, z) dV
Essa può essere riscritta come:
Sf,g = If + Ig – 2Of,g
I termini I rappresentano la sovrapposizione self-volume delle entità (molecole, in questo caso), il termine O invece è la sovrapposizione tra le due funzioni: mentre i due termini I sono indipendenti dall'orientamento, il termine O varia al variare di quest'ultimo. Trovare l'orientamento che massimizza il valore di O e che allo stesso tempo minimizzi Sf,g equivale a trovare la miglior sovrapposizione tra le due entità.
Il valore noto come coefficiente di Tanimoto può essere ottenuto ricombinando i termini I e O secondo l'equazione:
Tanimotof,g =
Of , g If +Ig Of , g
In maniera similare è possibile definire un'alternativa misura della forma, utilizzando il coefficiente di Tversky, la cui equazione base è la seguente:
Tverskyf,g =
Of , g
α If +β Ig
Normalmente, α + β = 1. Il calcolo secondo Tversky dipende da quale self-volume acquisisce per primo il fattore α: ROCS effettua due calcoli, il primo in cui è la query ad acquisire α, e il secondo dove invece lo stesso pre-fattore viene assegnato alla molecola del database. Il valore del coefficiente di Tversky può assumere valori maggiori di 1, dal momento che il volume condiviso Of,g può essere maggiore del self volume If (o Ig) di una molecola.
In aggiunta allo shape-based, ROCS può come opzione considerare anche la "chimica" (Color) durante la sovrapposizione delle molecole, in maniera tale da poter identificare durante lo screening di un database tutte quelle molecole simili sia nella forma che nelle caratteristiche chimiche. I color atoms sono descritti come gaussiane e visibili in vROCS come sfere colorate.
Il programma è fornito di due color force fields, Implicit Mills Dean (di default) e Explicit Mills Dean, i quali sono descritti in color force field files (*.cff) localizzati
all'interno della ROCS data directory: questi sono utilizzati per identificare le similitudini chimiche tra la query e le molecole del database, e quindi per rifinire la sovrapposizione basata esclusivamente sullo shape.
2.2.2 Preparazione dei file di input
File del Database
L'utilizzo più comune di ROCS consiste nel sovrapporre una grande raccolta di composti (dbase file) su una molecola di riferimento, denominata query. Il formato più comune per il database è il multi-conformer file .OEBinary, creato dal programma OpenEye OMEGA; alternativamente è possibile utilizzare SDF, MOL2 e PDB come formati alternativi.
Un altro tipo di file consentito è il dbase file: un file il cui nome termina per .list o .lst viene letto come una lista di file di molecole, una per linea.
File Query
Il secondo file richiesto, necessario affinché una corsa di ROCS possa essere effettuata, è il file query, il quale può contenere una o più molecole su ciascuna delle quali verrà allineata volta volta una molecola del database. Aggiungendo il comando -mcquery allo script si sceglie di trattare il file query come una libreria di più conformeri di un solito composto: in questo caso il software allineerà ciascuna molecola del database ad ogni conformero del file query, riconsegnando comunque un unico file di output, contenente il miglior allineamento scaturito da tutte le sovrapposizioni possibili.
Tipologie alternative sono lo shape query, che comprende diversi elementi di forma, incluse molecole, griglie e feature di tipo color, e le griglie, utilizzabili anch'esse come file di riferimento.
2.2.3 Interfaccia della Command Line
Una descrizione dell'interfaccia della Command Line può essere ottenuta eseguendo ROCS specificando l'opzione help:
prompt> ROCS –help
In questo modo genera il seguente output:
Help functions
ROCS --help simple : get a list of simple parameters ROCS --help all : get a complete list of parameters ROCS --help <parameter> : get a detailed help on parameter
ROCS --help html : create an html help file for this program
Il calcolo di ROCS può quindi essere lanciato da shell, digitando il comando desiderato, come nell'esempio:
rocs -query <queryfile> -dbase <dbasefile>
Dove: -query=indica il file di riferimento sul quale effettuare la sovrapposizione; -dbase=specifica il nome del file da utilizzare come database.
2.2.4 Parametri di input e output
Oltre ai già noti files di query e database, richiesti per eseguire un calcolo di ROCS, sono disponibili ulteriori parametri per rifinire e migliorare il risultato del programma, tutti visibili eseguendo ROCS e aggiungendo l'opzione help come visto in precedenza.
#InputOptions :
• -param <paramfile> : definisce il file dei parametri. Esso contiene una vera e propria lista di tutti i parametri che possono essere utilizzati, invece che
riportarli ciascuno sulla Command Line. In aggiunta, il file dei parametri scritto in seguito a un qualsiasi calcolo di ROCS può essere utilizzato in successive esecuzioni dello stesso programma, tramite il comando:
rocs -param <paramFile>
• -mcquery : tratta la query come una libreria di conformeri di un solito composto. Di default, questo parametro è posto come
falso.-• scdbase : non tratta due conformeri contigui del file del database come fossero generati da una solita molecola.
#OutputOptions :
• -prefix <name> : prefisso utilizzato per nominare tutti i file di output; di default è utilizzato il prefisso ROCS.
• -besthits <N> : numero massimo di hit che vengono riportati nel file di output. Ovviamente tutte le molecole presenti nella hitlist dovranno aver superato il valore di cutoff, se questo è stato specificato, e ivi saranno tenute o meno a seconda dello score specificato alla voce rankby. Di default, N è stabilito 500.
• -cutoff <F> : parametro che stabilisce se una particolare sovrapposizione è buona abbastanza da essere inclusa nella hitlist. F= -1.0 di default.
• -rankby <score> : definisce lo score da utilizzare per classificare gli hit. Le varie possibilità includono: TanimotoCombo; ShapeTanimoto (tanimoto); ColorTanimoto; ComboScore (combo); ScaledColor; RefTverskyCombo; RefTversky; RefColorTversky; FitTverskyCombo; FitTversky; FitColorTversky; Overlap. Di default gli hit sono classificati tramite la TanimotoCombo.
• -maxconfs <N>: numero massimo di sovrapposizioni da ottenere per ogni confronto tra query e molecole del database.
#HitsOutputOptions :
• -outputquery : posiziona la query come prima struttura all'interno del file di output. True di default.
• -oformat <extension> : specifica il formato del file di output. Di default, viene mantenuto il formato SDF.
2.2.5 Report file
Oltre al file hits.sdf, contenente gli hit del database che meglio si sovrappongono alla query opportunamente classificati, un calcolo eseguito con ROCS fornisce come file di output un file report (*.rpt), simile ad una tabella con su specificati i diversi campi.
Name è il nome del database di molecole. Se il database contiene più conformeri
per una solita molecola, allora l'indice dello specifico conformero segue il nome della molecola.
ShapeQuery è il nome della query, cui i corrispondenti hit sono stati allineati.
Anche in questo caso, se il riferimento è costituito da più conformeri l'indice dello specifico conformero segue il nome della molecola.
Rank è la classificazione numerica della hitlist basata sullo score scelto di
applicare. Se non sono state effettuate modifiche, gli hit sono classificati in base al punteggio di Tanimoto.
TanimotoCombo fornisce un punteggio che tiene conto sia dello shape che del
color, entrambi "secondo Tanimoto". Può assumere tutti i valori compresi tra 0 e 2.
ShapeTanimoto fornisce il punteggio di Shape Tanimoto, compreso tra 0 e 1.
ColorTanimoto fornisce il punteggio di Color Tanimoto, anch'esso compreso tra
0 e 1.
FitTverskyCombo fornisce un punteggio che tiene conto sia dello shape che del
color (entrambi secondo Tversky), unendo la FitTversky con la FitColorTversky. Può assumere tutti i valori compresi tra 0 e 2.
FitTversky fornisce il valore di shape secondo l'equazione di Tversky (vedi
2.2.1 Teoria), assegnando a β un valore di 0.95. Precedentemente questo score era noto come Tversky(d).
FitColorTversky calcola il valore di color sempre secondo l'equazione di
Tversky, assumendo β=0.95.
RefTverskyCombo fornisce un punteggio che tiene conto sia dello shape che
del color (entrambi secondo Tversky), unendo la RefTversky con la RefColorTversky. Può assumere tutti i valori compresi tra 0 e 2.
RefTversky fornisce il valore di shape secondo l'equazione di Tversky (vedi
2.2.1 Teoria), assegnando, questa volta, a α un valore di 0.95. Questo punteggio era precedentemente chiamato Tversky(q).
RefColorTversky calcola il valore di color sempre secondo l'equazione di
Tversky, assumendo α=0.95.
ScaledColor calcola un valore graduato del colore, condiderando il punteggio
reale ottenuto da un hit e dividendolo per il punteggio di color ottenuto dalla query contro se stessa.
ComboScore unisce la ShapeTanimoto con la ScaledColor, fornendo un valore
che tiene conto sia dello shape che del color, e che può assumere tutti i valori compresi tra 0 e 2.
SubTan è definita prendendo in considerazione il file di riferimento e la
molecola del database alla fine della sovrapposizione e rimuovendo tutti gli atomi della molecola del database che distano maggiormente di 1.5 Å da qualsiasi atomo della query. A questo punto si calcola lo ShapeTanimoto per queste due strutture e il punteggio ottenuto è salvato proprio come SubTan.
Overlap consiste nel valore assoluto del volume sovrapposto tra la query e la
molecola del database. Questo parametro è utilizzato quando è una griglia ad essere utilizzata come query.
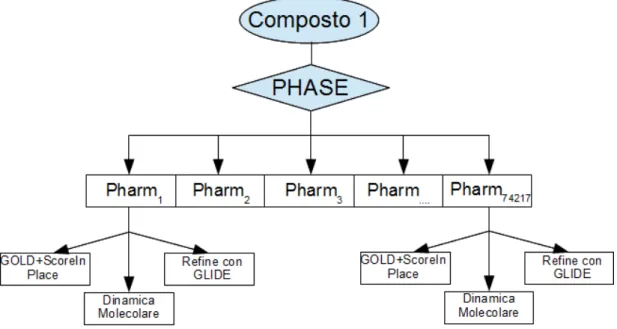
2.3 PHASE
PHASE [22] è un software molto versatile utilizzato per la preparazione di modelli farmacoforici, per l'allineamento di strutture, per la predizione dell'attività e per la ricerca delle strutture 3D all'interno di un database. Dato un insieme di molecole con elevata affinità per una particolare proteina bersaglio, PHASE utilizza un algoritmo di campionamento conformazionale e una serie di tecniche di classificazione per individuare elementi comuni e generare così un farmacoforo, vale a dire un arrangiamento spaziale di caratteristiche chimiche comuni a due o più ligandi attivi, con il quale si propone di spiegare le interazioni chiave coinvolte nel legame ligando-proteina. Ogni ipotesi è accompagnata da una serie di conformazioni allineate che suggeriscono il modo in cui le molecole interagiscono con il sito di legame; inoltre il programma permette la possibilità di combinare una data ipotesi con dati noti relativi all'attività per creare un modello 3D-QSAR. Il modello così generato può essere utilizzato in combinazione con l'ipotesi per estrarre da un database di molecole 3D quelle che hanno più probabilità di essere fortemente attive nei confronti del bersaglio.
PHASE può anche essere utilizzato come programma di allineamento molecolare in concomitanza con software di terze parti per lo sviluppo di modelli 3D-QSAR. Questo programma è integrato nella suite SCHRODINGER, di cui fa parte anche il programma di visualizzazione grafica MAESTRO.
2.3.1 Funzionalità di PHASE
PHASE dispone delle seguenti quattro funzionalità, ciascuna delle quali supportata graficamente da un pannello di MAESTRO:
• costruzione di modelli farmacoforici da un insieme di ligandi;
• preparazione di un database 3D che include informazioni utili alla costruzione di un farmacoforo;
• ricerca in un database di molecole generiche delle caratteristiche descritte da un'ipotesi farmacoforica.
Qualsiasi via si scelga di percorrere, il lavoro di ricerca prevede sempre la creazione di un modello farmacoforico di partenza. Questa operazione può essere schematizzata nelle seguenti fasi:
• creazione dei ligandi, minimizzazione e conversione in 3D;
• ricerca e definizione dei siti farmacoforici in comune nei ligandi;
• creazione dell'ipotesi;
• sviluppo dell'ipotesi, con l'aggiunta, per esempio, del volume escluso;
• costruzione ed esame di eventuali modelli 3D-QSAR.
La trattazione di questa tesi si è incentrata sulla creazione di modelli farmacoforici partendo da un unico ligando, complessato con il rispettivo target.
Per sviluppare un modello farmacoforico è sempre necessario assicurarsi di disporre della struttura 3D della molecola di partenza, nella conformazione attiva e con giusta carica e chiralità. I potenziali punti farmacoforici sono individuati utilizzando una serie di modelli, chiamati feature, che rappresentano i vari gruppi funzionali, i quali possono essere definiti, modificati e creati ex novo, per far sì che rappresentino le particolari caratteristiche ricercate.
Inoltre è possibile aggiungere al modello alcune opzioni che aiutano a renderlo più selettivo, come il volume escluso, cioè la porzione di spazio che non deve essere occupata da atomi del ligando; la maschera, che impone la presenza di alcune feature; la tolleranza, espressa in Angstrom [Å], che indica la distanza dalla quale si possono trovare i punto farmacoforici delle molecole da esaminare rispetto alla posizione di riferimento. Se si crea un'ipotesi da un recettore noto o complesso recettore-ligando, è possibile utilizzare lo stesso recettore per generare automaticamente i volumi esclusi: questa, tra l'altro, è l'unica opzione aggiuntiva utilizzata in questo lavoro, con lo scopo di incrementare la selettività di ciascun farmacoforo creato.
2.3.2 Creazione dell'ipotesi farmacoforica
Feature farmacoforiche
Una volta a disposizione la struttura 3D di un composto, dal quale si vuole trarre il farmacoforo, si procede con l'utilizzare l'insieme di funzioni caratteristiche di ogni gruppo funzionale, le cosiddette feature, per creare una rappresentazione tridimensionale delle parti essenziali dell'attività della molecola.
Una serie di feature predefinite è proposta dal programma:
• accettore di legame ad idrogeno (A);
• donatore di legame ad idrogeno (D);
• gruppo idrofobo (H);
• gruppo con carica negativa (N);
• gruppo con carica positiva (P);
• anello aromatico (R).
Ogni feature è definita da un insieme di gruppi funzionali con determinate caratteristiche e da una geometria che le conferisce le proprietà fisiche. La geometria della feature può essere di tre tipi:
• puntiforme: il sito si trova su un singolo atomo;
• vettoriale: il sito si trova su un singolo atomo, ma sono specificati anche uno o più vettori provenienti dall'atomo stesso;
• gruppo: il sito si trova nel baricentro di un gruppo di atomi. Per gli anelli aromatici al sito è assegnata una direzione definita da un vettore che è normale al piano dell'anello.
Nel caso le feature predefinite non coprano tutto lo spazio chimico necessario alla realizzazione del farmacoforo, è possibile integrarle con nuovi gruppi funzionali, oppure crearne di nuove.
Creazione dell'ipotesi
Nello sviluppo del modello farmacoforico le ipotesi sono generate da un insieme di molecole attive tenendo conto di caratteristiche comuni e caratteristiche escluse non
comuni. Il processo non tiene conto direttamente delle conoscenze circa il legame delle molecole al recettore.
PHASE fornisce i mezzi per mettere a frutto la conoscenza sul tipo di legame di un particolare ligando con il recettore, dando la possibilità di costruire un'ipotesi da una singola molecola. Per questa molecola, PHASE rileva tutti i possibili siti che aderiscono alle feature farmacoforiche e lascia all'utente la possibilità di selezionare i siti da includere nell'ipotesi. Partendo da una struttura di riferimento si possono seguire due metodi per la creazione dell'ipotesi:
• Ligand-based ipotesi: è possibile utilizzare un insieme di feature proposte da PHASE che identificano tutti i possibili punti farmacoforici e selezionare quindi quelli desiderati;
• FreeStyle ipotesi: è possibile inserire le caratteristiche farmacoforiche a volontà nell'area di lavoro, in relazione ad un ligando di riferimento. Il ligando di riferimento è preso solo come guida ed è eliminato una volta che l'ipotesi è stata creata.
Per questo lavoro, le ipotesi sono sempre state create a partire da un singolo ligando, e scegliendo tra tutte le feature proposte dal programma quelle considerate fondamentali per l'interazione con il rispettivo recettore, e quindi per l'attività.
Seguendo la procedura totalmente Ligand-based, si procede come segue:
1. Dall'interfaccia di MAESTRO selezionare Applications/Phase/Create Hypotesis, aprendo il pannello corrispondente;
2. Selezionare il ligando desiderato, si apre il pannello New Hypotesis nel quale sono elencati tutti i gruppi farmacoforici presenti nella molecola;
3. Selezionare le feature desiderate e quindi premere Add per aggiungerle; infine premere OK;
4. Selezionare Export per salvare l'ipotesi
L'ipotesi così creata sarà composta, al termine, da quattro file con le seguenti estensioni: .mae il file apribile con MAESTRO che contiene il nome degli atomi ed i riferimenti agli altri file; .def che contiene le definizioni delle varie tipologie di feature; .tab che riporta alcune regole da seguire nel caso si vogliano modificare i vari dati; .xyz che contiene le coordinate dell'ipotesi. Quest'ultimo in particolare può essere considerato il file fondamentale dell'ipotesi e di seguito ne è riportato un esempio:
0 A 15.4670 25.7140 5.4150 1 D 15.4944 24.7447 5.4387 2 N 16.9360 24.0285 3.5030 3 N 12.2775 25.5830 6.6845 4 N 14.2550 28.1265 4.0895
Nella prima colonna è presente un numero progressivo assegnato alle varie feature dell'ipotesi, nella seconda il codice di ogni feature e nella terza le relative coordinate spaziali x, y, z.
Aggiunta del volume escluso all'ipotesi
Se nel set di molecole iniziali, utili per la costruzione del faramcoforo, sono presenti anche composti inattivi, è possibile aggiungere all'ipotesi regioni di spazio in cui non dovrebbero essere presenti gli atomi di qualsiasi molecola attiva. Questi volumi esclusi sono considerati come zone morte e nel momento in cui si cercano le corrispondenze nel database vengono esclusi tutti i ligandi che posizionano i propri atomi in tali spazi.
Inoltre è possibile aggiungere i volumi esclusi ad un ipotesi basandosi sulla struttura del recettore, qualora esso sia disponibile, come accade nel nostro caso di studio. Per generare il volume escluso può essere utilizzato il seguente comando:
$SCHRODINGER/utilities/create_xvolReceptor -hypo $PATH/ipotesi – receptor $PATH/recettore.pdb -buff 2 -limit 5
Dove -receptor= percorso del recettore; -hypo= percorso dell'ipotesi, ove se ne specifica solamente il prefisso (ricordiamo che l'ipotesi è sempre costituita da quattro file, di formato diverso); -buff= distanza in Angstrom tra il volume escluso e la superficie data dalle forze di Van der Waals del ligando di riferimento; -limit= limita lo spessore del guscio creato, ignorando tutti gli atomi del recettore che sono più lontani del valore impostato.
2.3.3 Ricerca delle corrispondenze all'ipotesi
Nel momento in cui l'ipotesi è stata creata e si dispone di un database 3D, si può procedere alla ricerca delle molecole potenzialmente attive, in quanto affini al modello appena creato. Il processo di ricerca è generalmente eseguito in due fasi: ricerca e recupero.
Nel primo passo si procede a trovare nel database tutte le disposizioni geometriche delle molecole che possiedono i punti farmacoforici richiesti, dopo di che si misurano tutte le distanze tra i vari punti. Ad esempio, per l'ipotesi ADRR che contiene un accettore (A), un donatore (D), e due anelli aromatici (i due R), il programma misurerà le sei distanze: dAD, dDR1, dDR2, dAR1, dAR2 e dR1R2. A questo passaggio segue la scansione del database per trovare le molecole in cui le sei distanze sono sufficientemente vicine a quelle dell'ipotesi. Rispettata questa caratteristica, ad ogni molecola viene abbinato un file.
In fase di recupero, il file è utilizzato come una tabella di ricerca in modo da trovare rapidamente i conformeri che si allineano meglio all'ipotesi. Le molecole così ottenute possono proseguire alla fase successiva: sono quindi prima ordinate in base al loro fitness score, ovvero in base alla qualità di adesione al farmacoforo, poi filtrate in base all'occupazione dei volumi esclusi. Il processo è articolato in queste due fasi, in modo da evitare di ripetere l'intera procedura di ricerca se si desidera cambiare i parametri.
Un'ipotesi potrebbe avere più caratteristiche che sono effettivamente necessarie per il legame, ma potrebbe esserci una certa incertezza su quali siano quelle effettivamente utili. Quindi è possibile richiedere che solo un certo numero di funzioni trovi corrispondenza nelle molecole in esame: questa funzione è detta corrispondenza parziale. Per procedere alla ricerca nei database commerciali sono stati utilizzati i seguenti comandi:
$SCHRODINGER/utilities/phasedb_findmatches -setup <file> -db $PATH_db/database -hypo $PATH_hypo/ipotesi -mode find+fetch+flex -flexAmideOption trans -minSites N
(anche in questo caso, si specifica il nome dell'ipotesi senza estensioni); -mode find+fetch+flex= consente di eseguire una ricerca più recupero seguita da una rifinitura fatta generando conformeri aggiuntivi; -flexAmideOption trans= blocca tutti i legami ammidici in posizione trans; -minSites N= numero minimo di feature da ricercare; -maxHits M= numero massimo di risultati proposti. Questo comando genera un file di partenza contenenti tutti i parametri di ricerca e la posizione dei file.
A questo punto, può iniziare la ricerca vera e propria, lanciando da shell il seguente comando:
$SCHRODINGER/utilities/phase_dbSearchStart_File -NO_CHECKPOINT
Dove -NO CHECKPOINT impedisce la creazione dei file di controllo.
2.3.4 Ricerca dei file con phase_gridSearch
Quando lavoriamo con una gran quantità di strutture salvate in formato SDF possiamo utilizzare l'opzione phase_gridSearch per cercare corrispondenze all'ipotesi all'interno di un database. Per il suo utilizzo è necessario creare una lista di file, uno per riga, per i quali è da ricercare la corrispondenza ad una data ipotesi farmacoforica: il nome dei file può includere il loro indirizzo assoluto e il loro formato può essere solamente di tipo SDF, standard o compresso.
Il calcolo eseguito può essere diviso in più processori: i file oggetto della ricerca sono distribuiti sui diversi processori e quindi aggiunti, uno per volta, ad un file temporaneo, uno per CPU. Quando un numero prestabilito di file è stato aggiunto, il file temporaneo viene chiuso e compresso con gzip, e quindi uno nuovo viene aperto per quel solito processore: dividendo il calcolo su più CPU è conveniente mantenere basso il numero di strutture per file temporaneo, in maniera da non appesantire, e quindi rallentare, la procedura. La distribuzione dei file avviene sul processore locale, i file temporanei vengono quindi immagazzinati nella cartella di lavoro, della quale è opportuno assicurarsi la presenza di spazio libero sufficiente per l'esecuzione dei calcoli.
La ricerca dei file può avvenire, a questo punto, tramite l'utilizzo del seguente comando:
phase_gridSearch structFileList <filename> -hypoID <hypoID> -minSites M -scoreInPlace true -maxHits N jobname -WAIT
Dove -structFileList= nome del file contenente la lista delle strutture in formato SDF; -hipoID= prefisso utilizzato per indicare tutti i file dell'ipotesi farmacoforica che intendiamo adoperare per la ricerca; -minSites= numero minimo dei siti del farmacoforo che devono essere mantenuti; -scoreInPlace true= non si generano conformeri durante la ricerca e il risultato di fitness viene calcolato direttamente sulla pose di input, senza che questa venga allineata al farmacoforo; -maxHits= numero totale di strutture risultato dell'analisi, che andranno a comporre il file jobname-hits.mae; jobname= nome del lavoro che comparirà nei file di output.
2.4 Omega 2
Omega 2 è un programma utilizzato per effettuare velocemente analisi conformazionale di ligandi: è costituito da due componenti principali, ovvero un modellatore di frammenti molecolari e un campionatore dei torsionali. Il primo componente può essere trascurato, qualora le strutture fossero importate dall'esterno, come accade nel corso di questo studio.
Tale programma inizia il processo di analisi dei torsionali esaminando il grafico molecolare e determinando i legami che possono essere liberamente ruotati. La selezione finale si basa sulla distanza RMS degli heavy atoms e gli atomi di idrogeno campionati, mentre quelli non campionati non influenzano la distanza RMS. Ad ogni legame rotabile viene quindi assegnata una lista di possibili angoli diedri: l'attuale meccanismo per questa assegnazione si basa sulla corrispondenza SMART; esistono tuttavia ulteriori strategie che hanno basi sia sperimentali che teoriche.
Una profonda ed esaustiva ricerca dei torsionali viene quindi eseguita su ciascun frammento e i conformeri risultanti sono ordinati in una lista secondo la loro energia. A questo punto le strutture molecolari nella loro interezza vengono costruite combinando tra loro i frammenti a più bassa energia.
delle energie dei frammenti supera la finestra energetica della struttura a più bassa energia. I conformeri migliori, trovati durante il campionamento dei torsionali, sono ordinati in base alla loro energia: per essere accettati nel raggruppamento finale, ciascuno di essi deve avere una distanza RMS nei confronti di ciascun altro membro del medesimo gruppo superiore al valore di cutoff definito in partenza dall'utente.
Per lanciare il calcolo di OMEGA 2 è necessario digitare da shell il comando come nell'esempio:
omega -in <inputFile> -out <outputFile> -ewindow 25 -maxconfs 100 -maxtime 600 -rms 0.2 -strictatomtyping false
Dove -in= specifica il percorso del file di input; -out= specifica il percorso del file di output; -ewindow= imposta il valore di finestra energetica, espressa in kcal/mol, da utilizzare come criterio per accettare o rifiutare conformeri: un conformero qualsiasi è accettato se possiede un'energia tensionale inferiore alla somma tra la finestra energetica e l'energia del conformero corrispondente al minimo globale; -maxconfs= specifica il numero massimo di conformeri da generare (200 come valore di default); -maxtime= limita a 600 secondi il limite massimo da utilizzare per la generazione dei conformeri; -rms= valore minimo di Root Main Square al di sotto del quale due conformeri sono considerati come duplicati (il valore di default è 0.8); -strictatomtyping false= è permesso l'utilizzo di un atom type simile per uno non espressamente descritto nel force field di utilizzo (MMFF94).
L'aggiunta dell'opzione -param permette la creazione di un file contenente tutti i parametri di controllo, il cui nome sarà dato dalla combinazione del prefisso di base con l'estensione .param: tale file può essere utilizzato nei successivi lanci del programma con l'opzione -param.
2.5 GOLD
GOLD (Genetic Optimization for Ligand Docking) è un algoritmo genetico che permette il docking flessibile ed automatico di un set di composti all'interno del sito di binding di una proteina. Le molecole analizzate vengono posizionate nel sito
recettoriale, individuato all'interno della proteina dal programma, e le varie soluzioni vengono valutate sulla base del punteggio ottenuto a seconda della scoring function utilizzata: le migliori vengono ottenute come file di output al termine della procedura. Le scoring function delle quali si può usufruire sono molteplici: GoldScore, ChemScore, ASP (Astex Statistical Potential) e CHEMPLP (Piecewise Linear Potential). Le stesse sono inoltre modificabili, in modo da implementare la precisione del calcolo.
Nella suite di programmi, oltre a GOLD, sono compresi due software aggiuntivi: Hermes e GoldMine.
• Hermes è il visualizzatore grafico di GOLD e può essere utilizzato per facilitare la preparazione dei file di input, la visualizzazione dei risultati di docking e il calcolo dei descrittori;
• GoldMine è uno strumento per l'analisi e la post-processazione dei risultati di docking.
•
2.5.1 Preparazione dei file di input
Preparazione della proteina
La procedura di preparazione può coinvolgere sia una proteina singola che un complesso cristallografico proteina-ligando: solitamente quest'ultimo caso è preferibile, in quanto risulta più facile rivelare il sito di legame. Alternativamente, è pure possibile utilizzare come file di input esclusivamente i siti amminoacidici presenti nel sito attivo: in questo caso però dobbiamo essere sicuri che tali amminoacidi siano completi. Solitamente si includono tutti i residui che rientrano all'interno di un raggio di 5.0 Å dalla superficie della cavità accessibile al solvente.
In ogni caso, la proteina che è stata scelta deve contenere tutti gli idrogeni, inclusi quelli che definiscono il corretto stato di ionizzazione e tautomerico, e presentare un corretto bond-order: sulla base di queste informazioni, GOLD deduce automaticamente gli atom type che costituiscono il file di input. Nel caso in cui GOLD non riesca a definire un atom type particolare, questo viene allora sostituito da un dummy atom (letteralmente, "atomo fantoccio"), indicato come Du: in questo caso un messaggio di Warning viene riportato nel file gold_protein.log. La presenza dei dummy
che questi non vengono trattati né come donatori né come accettori di legame a idrogeno.
Qualora sia presente uno ione metallico all'interno del file di input, è necessario accertarsi che questo non sia legato direttamente agli atomi costituenti la proteina o molecole di acqua, in quanto i legami che lo coinvolgono vengono ridefiniti automaticamente dal programma; allo stesso tempo, affinché GOLD possa predirne la corretta geometria di coordinazione, lo ione deve essere coordinato ad almeno due atomi della proteina o a due molecole di acqua.
Sempre nel file di input è possibile scegliere se mantenere le molecole di acqua, e quindi tenere conto di queste, o almeno di quelle ritenute importanti ai fini dell'interazione tra ligando e recettore, durante il calcolo di docking; il programma, dopo averle individuate, determina quelle necessarie per il legame con la proteina, quindi decide automaticamente se lasciarle libere o vincolate durante la procedura di calcolo. L'acqua deve essere specificata in un file separato, in formato MOL2, e quindi caricata separatamente o estratta direttamente dal complesso. I seguenti parametri definiscono l'utilizzo delle molecole di acqua:
• On, utilizza l'acqua durante il docking;
• Off, non utilizza l'acqua durante il docking;
• Toggle, lascia la scelta a GOLD.
L'orientamento degli atomi di idrogeno delle molecole di acqua può essere ottimizzato dal programma durante il docking. Le opzioni disponibili al riguardo sono:
• Spin, Gold ottimizza automaticamente l'orientamento degli atomi di idrogeno;
• Trans Spin, si specifica un valore di distanza entro il quale far girare e traslare le molecole di acqua, compreso tra 0 e 2 Å;
• Fix, utilizza l'orientamento indicato nel file di input.
Definizione del sito di legame
Solo gli atomi inclusi nel sito di legame sono considerati durante la procedura di docking. La definizione del sito recettoriale della proteina deve quindi essere grande abbastanza da poter contenere ogni possibile binding pose del ligando e da contenere tutti i residui amminoacidici che potrebbero essere coinvolti nel legame con lo stesso. Questo passaggio può essere svolto in diversi modi: