1
Capitolo 1: Introduzione
1.1 ENZIMI MAO-A E MAO-B
1.1.1 Generalità
La monoammino ossidasi (o monoammino ossidoreduttasi, MAO, EC 1.4.3.4) è un flavoenzima che catalizza l’ossidazione di ammine strutturalmente diverse, inclusi alcuni neurotrasmettitori.
È un enzima di membrana, situato sulla parete mitrocondriale esterna; questa posizione, dove la MAO può essere in competizione per il consumo di ossigeno con altri enzimi (come ad esempio la citocromo-C ossidasi), può suggerire che la regolazione dell’enzima oltre ad avere effetto sulle proprietà catalitiche dello stesso, influenza anche gli enzimi limitrofi. La MAO esiste in due forme, isozimi, denominate MAO-A e MAO-B che differiscono per specificità di substrato e di inibitori.
La dimostrazione definitiva che MAO-A e MAO-B sono due proteine distinte è stata ottenuta quando, una volta isolato il cDNA che codifica le due strutture da tessuti umani, bovini e di roditore, è stata studiata la sequenza amminoacidica codificata1,2,3.
L’enzima è distribuito in vari tessuti dei mammiferi e sono state riscontrate differenze nell’attività in funzione delle specie osservate. Nell’uomo la maggior parte dei tessuti esprime entrambe le forme, ad eccezione delle piastrine dove è espressa solo la forma B4; le
2
concentrazioni maggiori si hanno nel fegato, nella placenta e nel cervello, mentre le concentrazioni minori si hanno nella milza.
Figura 1.1 - Struttura5 tridimensionale della MAO-A.
1.1.2 La struttura
L’analisi della struttura primaria, dedotta dal cDNA, mostra che i due isozimi sono formati rispettivamente da 526 (MAO-A) e 520 (MAO-B) residui amminoacidici, ed hanno un peso molecolare rispettivamente di 59 700 e 58 800; le due catene sono uguali per il 70%3,6,7,8. Studi sulla struttura secondaria della MAO-A umana e della MAO-B bovina tramite FTIR-ATR (Fourier Transfor Attenuated Total Reflection Spectroscopy) hanno mostrato l’esistenza di quattro regioni che sono presenti, sia nei due isozimi, sia nelle MAO di diverse specie animali. Queste sono:
un’unità di legame per l’ADP (residui 6-43);
un dominio putativo per il substrato (residui 178-221); un sito per il legame covalente del FAD (residui 350-458);
una regione C terminale (residui 491-511) che sembra sia necessaria per la formazione di un’α-elica trans-membrana.
L’utilizzo dello pseudo substrato-inibitore N-[2-amminoetil]-5-cloro-2-piridincarbossiammide e studi di mutazione selettiva, hanno dimostrato che i residui amminoacidici His382 e Thr158 sono essenziali per il funzionamento della MAO-B, mentre il Phe208 per la MAO-A e l’Ile199 per la MAO-B sono essenziali per la specificità di substrato9,10,11,12.
3
È stata inoltre evidenziata la presenza di due residui di cisteina nel sito attivo dell’enzima, che potrebbero essere coinvolti nel ciclo catalitico13,14.
1.1.3 Le reazioni di mono-ossigenazione enzimatica
Gli enzimi che catalizzano l’incorporazione diretta di atomi di ossigeno nella molecola di substrato sono chiamati “ossigenasi”. Il trasferimento di ossigeno al substrato può avvenire in tre differenti modi (Schema 1.1) in funzione del tipo di enzima che effettua l’ossidazione:
1. mono-ossigenasi: solo un atomo di ossigeno viene incorporato nella molecola del substrato, mentre l’altro viene ridotto per formare acqua;
2. di-ossigenasi: entrambi gli atomi della molecola di ossigeno vengono incorporati direttamente nel substrato. Questi enzimi vengono chiamati anche ossigeno-transferasi;
3. ossidasi: catalizzano solamente il trasferimento di elettroni agli atomi di ossigeno; il trasferimento può essere di 2 o 4 elettroni, portando quindi alla formazione di perossidi o ad acqua.
Sub + DonatoreH2 + O2 SubO + Donatore + H2O riciclo del cofattore
mono-ossigenasi di-ossigenasi Sub + O2 SubO2 ossidasi O2 + 2e- 2H+ H 2O2 O2 2-O2 + 4e- 4H+ 2H 2O 2O
2-Schema 1.1 - Reazioni di ossidazione enzimatiche.
Nonostante il meccanismo di ossidazione delle varie mono-ossigenazioni differisca in funzione dell’enzima e del substrato, il metodo per l’attivazione dell’ossigeno è sempre lo stesso: l’ossigeno viene attivato per riduzione a spese di un cofattore presente nell’enzima, rappresentato come “donatore” nello Schema 1.1 (generalmente NADH o NADPH).
4
Questo processo di ossidazione viene generalmente mediato da cofattori contententi metalli di transizione (generalmente Fe o Cu) o da sistemi eteroaromatici (come una flavina). Un esempio di monoossigenasi Fe-dipendente è il Citocromo P-450 (Cyt-P450) che contiene legato un gruppo Fe-eme.
Le monoossigenasi flavino-dipendenti presentano un meccanismo di ossidazione completamente diverso rispetto ad enzimi tipo Citocromo P-450. Inizialmente il NADPH riduce il sistema Enz-FAD; il FADH2 così formato viene ossidato dall’ossigeno molecolare presente in soluzione dando una specie perossidica, FAD-4a-OOH. L’anione perossido, formato per deprotonazione, può a questo punto comportarsi da nucleofilo nei confronti generalmente di un’aldeide o di un chetone; il composto tetraedrico che viene a formarsi collassa, portando al corrispettivo estere o lattone e al FAD-4a-OH che elimina H2O tornando a FAD (Schema 1.2). N H N NH N O O [Enz-FAD] NADPH NADP+ Sub N H H N NH H N O O [Enz-FADH2-Sub] O2 N H H N NH N O O O O H [Enz-FAD-4a-OOH-Sub] N H N NH N O O O H [Enz-FAD-4a-OH-SubO] H SubO + H2O
Schema 1.2 - Ciclo catalitico delle mono-ossigenasi flavino-dipendenti.
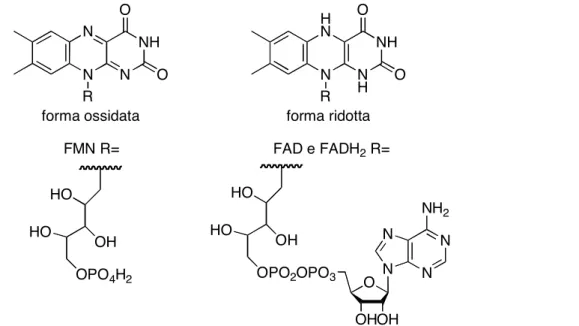
La molecola del FAD (Figura 1.2) è costituita da tre anelli condensati, che formano il cosiddetto gruppo isoalloazinico della flavina, il quale è a sua volta legato al ribitolo (zucchero a cinque atomi di carbonio) tramite l'atomo di azoto dell'anello centrale. La molecola, se presenta il gruppo OH del carbonio 5, legato al gruppo fosfato, prende il nome di Flavin Mono Nucleotide (FMN); se in posizione 5 c’è un gruppo adenosinico, allora si ha il Flavin Adenin Dinucleotide (FAD).
5
Queste molecole sono in grado di accettare 2 elettroni, sia singolarmente, formando specie ridotte semichinoniche (FADH·), sia in coppia, formando specie completamente ridotte (FADH2).
Figura 1.2 - Struttura molecolare del FMN, del FAD e del FADH2
1.1.4 Meccanismo catalitico delle MAO
La reazione catalizzata dalle MAO (A e B) è la deamminazione ossidativa di ammine 1e, 2e ed in alcuni casi 3e, come mostrato nello Schema 1.3.
Il ciclo catalitico comunemente accettato15,16 prevede due step separati. Nel primo step si ha la riduzione del FAD legato all’enzima e l’ossidazione dell’ammina con formazione dell’immina; nel secondo stadio si ha l’ossizazione del FAD da parte dell’O2 con eliminazione del perossido di idrogeno e la concomitante eliminazione dell’immina formata in precedenza che viene poi idrolizzata ad aldeide dall’acqua presente nell’ambiente di reazione.
[Enz-FAD] + Sub [Enz-FAD-Sub] [Enz-FADH2-Immina]
[Enz-FAD-Immina]
O2 H2O2
Immina
Aldeide + NH4+
Schema 1.3 – Meccanismo della deamminazione ossidativa catalizzata dalle MAO.
N N N N NH2 N N NH N O O R HO OH HO OPO4H2 FMN R= HO OH HO OPO2OPO3 O OHOH H N N NH N H O O R forma ridotta forma ossidata FAD e FADH2 R=
6
In funzione dell’andamento cinetico di questi due step si può avere un meccanismo sequenziale o a doppio-spostamento (“double-displacement”), che permettono regolazioni diverse dell’attività enzimatica. Nel caso del meccanismo a doppio spostamento15, in presenza di basse concentrazioni di ammina si ha un “effetto tampone” contro le fluttuazioni della concentrazione dell’ossigeno, mentre nel caso del meccanismo sequenziale15 il sistema è molto più sensibile a piccole variazioni della concentrazione dell’ammina.
Nonostante le differenze in selettività di substrato ed inibitori, entrambe le forme dell’enzima sono capaci di catalizzare l’ossidazione di ammine primarie, secondarie e terziarie. Il FAD covalentemente legato all’enzima è necessario per il trasferimento degli elettroni dall’azoto amminico all’ossigeno15 (Schema 1.4). In questo processo l’ammina viene convertita nello ione imminio corrispondente, e poi idrolizzata a formare l’aldeide e lo ione ammonio.
N N NH N S H3C R O O N N NH N S H3C R O O O2 + H+ H2O2 H RCH2NH2 H2O RCHO + NH4 RCH=NH2 Schema 1.4 - Ossidazione delle ammine da parte del FAD legato alla MAO.
La caratteristica strutturale principale dei substrati delle MAO è la presenza dei due idrogeni in α rispetto al gruppo amminico. È stato dimostrato17 che entrambe le forme della MAO agiscono stereospecificamente, estraendo esclusivamente l’idrogeno pro-R del gruppo metilenico prochirale del substrato amminico.
Fino ad oggi sono stati proposti tre diversi percorsi di reazione:
1. meccanismo a trasferimento di singoli elettroni (“single electron transfer”, SET)18. 2. meccanismo a trasferimento di idrogeno (“hydrogen atom transfer”, HAT)19. 3. meccanismo nucleofilo o polare20.
Il meccanismo SET (Schema 1.5) prevede l’iniziale trasferimento di un elettrone dell’azoto amminico al FAD, formando un radical-catione amminico e la specie semichinonica del FAD; il radical-catione può perdere quindi un protone, formando un radicale al carbonio, e trasferire
7
il secondo elettrone alla specie semichinonica per formare l’immina e la specie FADH2 (percorso a). R NH2 R NH2 R H C NH2 R H C NH2
FAD FAD FAD FADH
FAD FADH H -FAD FAD X R NH2 X X a b c
Schema 1.5 - Meccanismi SET e HAT.
È plausibile anche la perdita diretta, da parte del radical-catione, di un radicale idrogeno, che porta direttamente alla specie imminica (percorso b). Evidenze sperimentali che supportano questa teoria derivano da studi di inibizione con ciclopropilammina21 e ciclobutilammina22. Tuttavia non è stato ancora possibile individuare l’intermedio radical-cationico utilizzando tecniche tipo “stopped flow”, “rapid-scan” e tecniche con campi magnetici23.
Alla luce di questi risultati è stato proposto il meccanismo HAT (Schema 1.5, percorso c) che prevede l’estrazione di un radicale idrogeno dal carbonio in α, portando direttamente al radicale sul carbonio che poi evolve per dare lo ione imminio.
Il meccanismo nucleofilo o polare prevede, invece, la formazione di un addotto covalente tra il FAD e l’ammina (Schema 1.6) il cui distacco porta allo ione imminio e al FAD ridotto.
N N NH N S H3C R O O R NH2 N H N NH N S H3C R O O NH H H R N H N NH N S H3C R O O R H C NH2 +
8
1.1.5 Substrati
Le MAO catalizzano la reazione di deamminazione di ammine acicliche di varia struttura. Oltre all’ossidazione di vari neurotrasmettitori (Tabella 1.1), catalizzano anche la deamminazione di poliammidi acilate come la N-acetil-putresceina e di varie neurotossine come l’1-metil-4-fenil-1,2,3,6-tetraidropiridina (MPTP).
I due isozimi (MAO-A e MAO-B), nonostante siano in grado di reagire con gli stessi substrati, presentano alcune specificità. Gli studi effettuati24 hanno dimostrato che le MAO sono ottimi catalizzatori per la deamminazione di ammine che presentano un gruppo arilico in posizione γ. In generale si può comunque affermare che le ammine primarie e secondarie sono ossidate indiscriminatamente da entrambi gli isozimi, mentre le ammine terziarie mostrano una selettività maggiore per una delle due isoforme.
Oltre al sito di legame per il gruppo amminico, sembra evidente la presenza nel sito attivo di una tasca idrofobica che lega il residuo aromatico del substrato. La specificità della feniletil ammina (3) per la MAO-B e della serotonina (5) per la MAO-A mostrano comunque che questo sito di legame è diverso per le due forme.
9 Tabella 1.1 - Substrati per MAO-A e MAO-B
Benzilammina NH2 1 MAO-A e MAO-B Chinuramina NH2 NH2 O 2 MAO-A e MAO-B Feniletilammina NH2 3 MAO-B Dopamina HO NH2 HO 4 MAO-A e MAO-B Serotonina N H HO NH2 5 MAO-A (±)-Adrenalina NH HO HO OH 6 MAO-A e MAO-B (±)-Nor-Adrenalina NH2 HO HO OH 7 MAO-A e MAO-B
È stato inoltre osservato che le MAO sono in grado di ossidare anche ammine alifatiche con catena medio-lunga; composti C5-C10 sono infatti ottimi substrati per questi enzimi mentre composti C1-C4 o con catene maggiori di C10 non sono buoni substrati per le MAO. Composti analoghi, ma con un gruppo metilico in α al gruppo amminico, non reagiscono, ma sono risultati inibitori reversibili e competitivi per l’enzima.
È stata osservata una differenza di reattività dei due isozimi con ammine β sostituite dimostrando che il sito attivo della MAO-A presenta una costrizione attorno a questa posizione, limitando l’accesso delle molecole sostituite. La MAO-B invece, mostrando un sito attivo più libero, risulta meno selettiva.
10
1.1.6 Inibitori della MAO-A
Lo studio degli inibitori24 della MAO (MAOIs) riveste una grande importanza, in quanto queste molecole sono state introdotte nelle terapie per la cura di disturbi neuropsichiatrici già negli ultimi cinquant’anni del secolo scorso. Il problema di queste molecole è che possono causare l’insorgere di importanti problemi cardiovascolari e presentano elevata tossicità per il fegato. Lo studio è stato ed è tuttora quindi rivolto alla ricerca di molecole meglio tollerate dall’organismo. Attualmente i MAOIs possono essere classificati in tre gruppi:
vecchi MAOIs, inibitori non selettivi ed irreversibili;
MAOIs irreversibili, inibitori selettivi che agiscono irreversibilmente sull’enzima;
MAOIs reversibili, inibitori reversibili che agiscono selettivamente sulla MAO-A (“reversible inhibitors of MAO-A”, RIMAs).
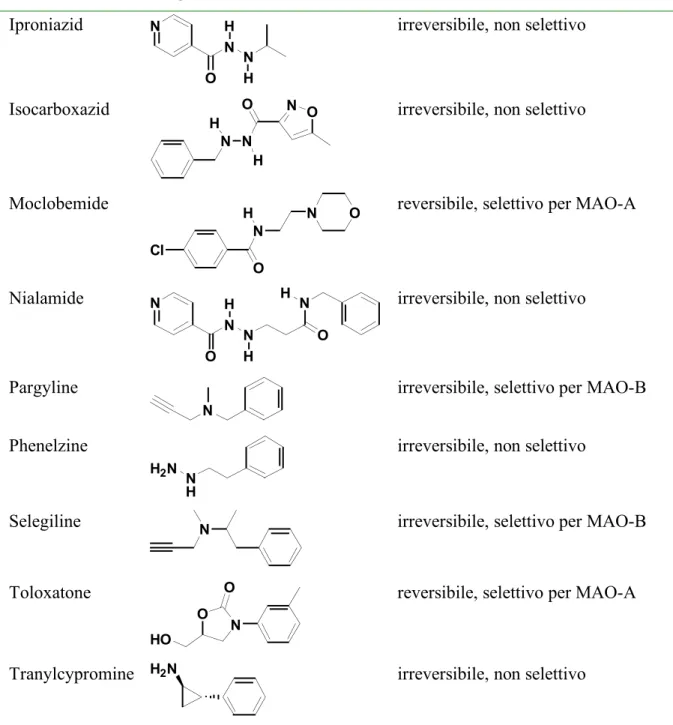
Gli inibitori disponibili in commercio ed attualmente utilizzati clinicamente per le MAO sono riportati nella Tabella 1.2. Queste molecole vengono utilizzate per il trattamento del morbo di Parkinson, depressione, fobie sociali, panico e morbo di Alzheimer.
11
Tabella 1.2 - Elenco degli inibitori delle MAO utilizzati attualmente.
Iproniazid N N N O H H
irreversibile, non selettivo
Isocarboxazid
N N H H
O N O irreversibile, non selettivo
Moclobemide
Cl
N O
H N O reversibile, selettivo per MAO-A
Nialamide O N N N O N H H
H irreversibile, non selettivo
Pargyline
N
irreversibile, selettivo per MAO-B
Phenelzine
N H H2N
irreversibile, non selettivo
Selegiline N irreversibile, selettivo per MAO-B
Toloxatone
O N O
HO
reversibile, selettivo per MAO-A
12
1.2 ENZIMI IMMOBILIZZATI
1.2.1 Introduzione
Negli ultimi anni l’uso di enzimi come catalizzatori per la sintesi di prodotti otticamente attivi è diventato un’alternativa attraente a metodologie chimiche di sintesi asimmetrica. Numerose applicazioni sono riportate nella letteratura e utilizzate in campo industriale25. Il vantaggio dell’uso di biocatalizzatori risiede nel fatto che le reazioni possono essere condotte in condizioni blande, con elevate rese e ridotta formazione di sottoprodotti, evitando l’uso di catalizzatori metallici, spesso tossici o difficilmente smaltibili. Per quanto riguarda la produzione di enantiomeri puri, le reazioni enzimatiche minimizzano problemi di isomerizzazione, racemizzazione, epimerizzazione e riarrangiamenti che possono invece avvenire durante i processi chimici. Gli enzimi inoltre grazie alla loro attività in vivo possono essere molto utili in studi farmacologici e bioanalitici.
Tuttavia l’uso di enzimi isolati può presentare alcuni svantaggi come ridotta stabilità dell’enzima nelle condizioni di reazione (solvente organico, pH, temperatura), difficile recupero dei prodotti o dell’enzima e costo iniziale elevato.
Molti di questi inconvenienti possono essere efficacemente superati mediante l’immobilizzazione dell’enzima su un’opportuna matrice insolubile25,26. In questo modo il recupero ed il successivo riutilizzo dell’enzima al termine della reazione viene notevolmente semplificato. In secondo luogo, l’immobilizzazione generalmente conduce ad un significativo aumento della stabilità dell’enzima.
La struttura e la superficie del supporto hanno un ruolo fondamentale nel mantenimento della struttura terziaria dell’enzima e quindi delle sue proprietà. Modificazioni di questa struttura infatti influiscono sulla stabilità termica, sull’attività catalitica e sulla cinetica (modificando la costante di Michaelis-Menten, Km, e la velocità massima, Vmax), possono inoltre causare una
variazione del profilo di attività enzimatica in funzione del pH e della temperatura.
La scelta quindi sia del supporto, sia del metodo d’immobilizzazione variano in funzione dell’enzima e non possono essere individuati dei metodi “universali”.
Le principali tecniche di immobilizzazione di enzimi sono rappresentate nella Figura 1.4 e saranno brevemente descritte nei paragrafi successivi.
13 Adsorbimento Legame covalente Incapsulazione Intrappolamento Reticolazione
Figura 1.3 - Principali tecniche di immobilizzazione di enzimi.
1.2.1.1 Adsorbimento
Il metodo più semplice per immobilizzare un enzima è quello di farlo adsorbire su un supporto macroscopico insolubile in acqua25,27. I materiali generalmente utilizzati come supporti sono sia di tipo organico che inorganico (carbone attivato, ossido di alluminio, celite, cellulosa, vetro e resine sintetiche). Le forze che tengono l’enzima legato al supporto sono forze di Van der Waals, interazioni ioniche e legami ad idrogeno.
Questo tipo d’immobilizzazione, oltre alla velocità del processo, presenta il vantaggio di non interferire eccessivamente con la struttura terziaria dell’enzima e quindi con le sue caratteristiche, causando solo una bassa perdita dell’attività catalitica grazie alla mancanza di modificazioni chimiche dell’enzima o del supporto.
Tuttavia, proprio a causa delle interazioni deboli, si ha facilmente il distacco del biocatalizzatore dal supporto a causa di variazioni di pH, concentrazione del substrato, temperatura, forza ionica o qualsiasi altra condizione che possa far variare la conformazione dell’enzima. Altri svantaggi di questo metodo di immobilizzazione possono derivare dall’“overloading” di enzima sul supporto, che può causare una diminuzione dell’attività catalitica derivata dall’assenza di spazio sufficiente tra le molecole di enzima ed il supporto.
14
1.2.1.2 Legame covalente
A differenza dell’adsorbimento, la formazione di un legame covalente tra la matrice ed il biocatalizzatore lega stabilmente l’enzima al supporto impedendone quindi il rilascio. Il legame che viene a formarsi però può causare modificazioni strutturali all’enzima e quindi la perdita di attività catalitica.
Per effettuare il legame vengono normalmente coinvolti gruppi nucleofili presenti sull’enzima come ad esempio gruppi ε-amminici della catena laterale della lisina o il gruppo amminico terminale della catena polipeptidica. Un requisito importante è che non vengano interessati nel legame i gruppi funzionali del sito attivo.
La procedura di immobilizzazione prevede generalmente due stadi: (i) l’attivazione del supporto tramite reazioni che inseriscono un gruppo spaziatore reattivo e (ii) l’attacco dell’enzima. Come supporti possono essere utilizzati sia composti inorganici che polimeri naturali o sintetici.
1.2.1.3 Reticolazione
L’unione di più unità enzimatiche attraverso la formazione di legami covalenti viene chiamata reticolazione25,27: ne risulta un aggregato insolubile ad elevato peso molecolare. Le molecole di enzima possono essere reticolate tra di loro o co-reticolate utilizzando una proteina di “riempimento” inattiva come l’albumina. Per questo tipo di immobilizzazione vengono normalmente utilizzati come agenti aggreganti α,ω-glutaraldeide, esametilendiisocianato o isotiocianato.
Anche questo metodo di immobilizzazione è abbastanza semplice, ma gli aggregati che si ottengono sono spesso gelatinosi e quindi non possono essere utilizzati come impaccamento per bioreattori. Inoltre l’attività catalitica dell’enzima può essere limitata a causa della complessa struttura che viene a crearsi, che può impedire l’accesso al sito attivo da parte del substrato.
1.2.1.4 Intrappolamento
Gli enzimi possono essere fisicamente “intrappolati” in una matrice macroscopica. Le molecole di enzima sono libere in soluzione ma ristrette nei movimenti dalla struttura della matrice: per assicurare l’attività è necessario che il substrato ed il prodotto possano
15
attraversare tale struttura. L’intrappolamento può essere effettuato facendo formare un gel od un polimero in presenza dell’enzima.
1.2.1.5 Incapsulazione
Questo metodo è simile all’intrappolamento e prevede l’inclusione dell’enzima in una membrana semipermeabile che può essere sia di tipo biologico che sintetico25,27. Le piccole molecole, come i substrati ed i prodotti, possono facilmente diffondere attraverso i pori della membrana che però non lasciano passare l’enzima. In questo caso il supporto può essere preparato nella forma più conveniente (sfere, fibre, fogli, cilindri, etc) in funzione del tipo di processo nel quale verrà utilizzato.
1.2.2 IMERs (“Immmobilized Enzyme Reactors”)
L’uso di reattori contenenti enzimi immobilizzati (“Immobilized Enzyme Reactors”, IMERs) è stato recentemente sviluppato ed è stata dimostrata l’importanza di questa metodologia in vari campi della ricerca28. Gli enzimi infatti possono essere immobilizzati su una fase stazionaria ed utilizzati per impaccare colonne per HPLC che possono essere impiegate come bioreattori permettendo di condurre reazioni enantioselettive in modo estremamente efficiente, riciclando ripetutamente l’enzima. I bioreattori sono stati inoltre utilizzati con successo anche per studi di carattere prettamente biochimico, che hanno riguardato, ad esempio, la separazione e la successiva identificazione di metaboliti in studi sul metabolismo dei farmaci,29 nonché l’identificazione di nuovi inibitori selettivi di specifiche attività enzimatiche. Inoltre i sistemi così preparati hanno trovato vasta applicazione anche per la preparazione di fasi stazionarie chirali per HPLC, impiegate nella cromatografia per affinità.30 Questo approccio oltre a non richiedere elevate quantità di proteina, ne aumenta la stabilità, rendendo allo stesso tempo possibile il suo riutilizzo fintanto che questa rimane attiva; vi è inoltre la possibilità di immobilizzare enzimi in sequenza, permettendo quindi l’ottenimento di processi “multistep” con elevata specificità e selettività.
L’immobilizzazione per la realizzazione di IMERs può essere ottenuta in due modi: “in situ” o “in batch”. La tecnica “in batch” prevede l’immobilizzazione dell’enzima sul supporto in un recipiente di reazione e poi l’impaccamento della fase stazionaria così realizzata nella colonna; la tecnica “in situ” prevede invece l’immobilizzazione diretta dell’enzima in una colonna precedentemente impaccata.
16
È stato dimostrato31,32 che la metodologia “in situ” è quella ottimale per ottenere risultati migliori in termini di quantità di enzima legato al supporto e ritenzione di attività catalitica. Gli IMERs possono essere utilizzati come semplici bioreattori se montati in un sistema costituito da una pompa per HPLC ed un detector (Schema 1.7).
POMPA HPLC IMER DETECTOR
scarico
Schema 1.7 –IMER inserito in uno strumento HPLC.
Con questo sistema si possono effettuare reazioni nell’IMER e raccogliere i prodotti di reazione come eluati della colonna. La possibilità di inserire anche un detector permette un’analisi preliminare, qualitativa o quantitativa in funzione del detector e delle condizioni, all’uscita del bioreattore.
Gli IMERs, inoltre, una volta integrati in un sistema cromatografico HPLC, permettono la messa a punto di metodi analitici ad elevata accuratezza e riproducibilità33,34,35,36, consentendo anche l’analisi di matrici complesse.
Sono però da evidenziare alcuni problemi dovuti al fatto che le condizioni sperimentali in cui si può operare sono ristrette: (i) è necessario lavorare in uno specifico range di flussi, in funzione del tipo di immobilizzazione dell’enzima e del tipo di colonna utilizzato, (ii) il pH e l’eluente devono essere compatibili con l’enzima oltre che con la fase stazionaria. L’IMER inoltre non è una colonna cromatografica vera e propria, di conseguenza i picchi cromatografici che si ottengono non sono propriamente risolti sia a causa di interazioni non specifiche, sia per la cinetica di reazione tra il substrato e l’enzima.
Per ovviare a questi problemi sono stati ideati dei sistemi accoppiati, separati da una
“switching valve”. In questo modo l’IMER ed il sistema HPLC possono lavorare in
17
1.3 BIBLIOGRAFIA DEL CAPITOLO 1
1 Bach A. W. J., Lan N. C., Johnson D. L., Abell C. W., Benmbenek M. E., Kwan S. W., Seeburg P. H., Shih J. C., Proc Nat1. Acad. Sci. U.S.A., 1988, 85, 4934-4938.
2 Hsu Y. P. P, Weyler W., Chen S., Sims K. B., Rinehart W. B., Utterback M., Powell J. F., Breakefield X. O. J. Neurochem., 1988, 51, 1321-1324.
3 Powell J. F., Hsu Y. P. P., Weyler W., Chen S., Salach J., Andrilopoulos K., Mallet J., Breakefield X. O. Biochem. J., 1989, 259, 407-413.
4 Biochem. Pharmacol., 1989, 38, 901-905 5 2BXS, Protein Data Bank
6 Kuwahara T., Takamoto S., Ito A., Agric. Biol. Chem., 1990, 54, 253-257.
7 Ito A., Kuwahara T., Inadome S., Sagara Y. Biochem. Biophys. Res. Commun., 1988, 157, 970-976.
8 Chen K., Wu H. F., Shih J. C. J. Neurochem., 1993, 61, 187-190.
9 Cesura A. M., Gottowik J., Lahm H. W., Eur. J. Biochem, 1996, 236, 996-1002.
10 Cesura A. M., Gottowik J., Lang G., Malherbe P., DaPrada M. J. Neural Transm. (Suppl.), 1998, 52, 189-200.
11 Veselovsky A. V., Ivanov A. S., Medvedev A. E., Biochemistry (Moscow), 1998, 63, 1441-1446.
12 Tsugeno Y., Ito A. J. Biol. Chem., 1997, 272, 14033-14036. 13 Wu H. F., Chen K., Shih J. C. Mol. Pharmacol., 1993, 43, 888
-893.
14 Zhong, B., Silverman R. B. J. Am. Chem. Soc., 1997, 119, 6690
-6691.
15 Tipton K. F., Boyce S., O’Sullivan J., Davey G. P., Healy J. Curr. Med. Chem., 2004, 11, 1965-1982.
16 Edmondson D. E., Mattevi A., Binda C. , Li M., Hubàlek F. Curr. Med. Chem., 2004, 11, 1983-1993.
17 Yu P. H., Davis A. B. Int. J. Biochem., 1988, 20, 1197-1201
18 (a) Silverman R. B. Acc. Chem. Res., 1995, 28, 335-342. (b) Silverman, R. B., Hoffman, S. J., Catus W. B. J. Am. Chem. Soc., 1980, 102, 7126-7128.
18
20 (a) Kim J. M., Hoegy S. E., Mariano P. S. J. Am. Chem. Soc., 1995, 117, 100-105. (b) Kim J. M., Bogdon M. A., Mariano P. S. J. Am. Chem. Soc., 1993, 115, 10591-10595.
21(a) Silverman R. B. Biochem. Soc.Trans., 1991, 19, 201-206. (b) Silverman R. B., Cesarone J. M., Lu X. J. Am. Chem. Soc., 1993, 115, 4955-4961.
22 Silverman R. B., Zhou J. P., Eaton P. E., J. Am. Chem. Soc., 1993, 115, 8841-8842. 23 Miller R. J., Edmondson D. E., Grissom C. J. Am. Chem. Soc., 1995, 117, 7830-7831. 24 Kalagutkar A. S., Dalvie D. K., Castagnola N. Jr., Taylor T. J. Chem. Res. Toxicol., 2001,
9, 1140.
25 Faber K. Biotransformation in Organic Chemistry II Ed.. Sprinter, Germany, Berlin, 1995. 26 Linqiu Cao Carrier-Bound Immobilized Enzymes, Wiley-VCH. Weinheim, 2005.
27 Bickerstaff G. F. Immobilization od Enzymes and Cells, Bickerstaff G. F. ed., Umana Press, Totowa, New Jersey, 1997.
28 (a) Jadaul P., Wainer I. W. Chirality 1990, 2, 32-37; (b) Bertucci C., Petri A., Felix G., Perini B., Salvadori P. Tetrahedron: Asymmetry 1999, 10, 4455-4462; (c) Petri A., Gambicorti T., Salvadori P. J. Mol. Catal. B: Enzym. 2004, 27, 103-106.
29 (a) Sotolongo V., Johnoson D. V., Wahnon D., Wainer I. W. Chirality 1999, 11, 39-45; (b) Pasternyk M., Ducharme M. P., Vescorps V., Felix G., Wainer I. W. J. Chromatogr. A
1998, 828, 135-140.
30(a) Felix G., Descorpos V. Chromatographia 1999, 49, 595-605; (b) Domenici E., Bertucci C., Salvadori P., Félix G., Cahagne I., Motellier S., Wainer I. W. Chromatographia 1990,
29, 170-176; (c) Erlandsson P., Marle I., Hanson L., Isaksson R., Petterson C., Petterson G. J. Am. Chem. Soc. 1990, 112, 4573-4574.
31 Massolini G., Calleri E., De Lorenzi E., Pregnolato M., Terreni M., Felix G., Gandini C., J.
Chromatogr. A, 2001, 921, 147.
32 Félix G., Liu M. Bio-Sciences, 1989, 8, 2-6. 33 Klibanov A. M., Science 219 (1983) 722.
34 Moo-Young M. (Ed.), Bioreactor Immobilized Enzymes and Cells: Fundamental And
Application, Elsevier Applied Science, London,UK, 1988, Chapter IV.
35 D’Souza F. S., Curr. Sci. 77 (1999) 69.