3. Materiali e metodi
3.1 Popolazione in studio
Il campione oggetto di studio è costituito da 230 individui caucasici: 115 casi affetti da tumore al colon retto sporadico e 115 controlli volontari sani. L’età media dei casi è di 66,9 anni ed è paragonabile a quella dei controlli. I due gruppi sono composti dal 57% di individui di sesso maschile. I soggetti sono stati reclutati presso l’ospedale Careggi di Firenze. Sia i casi che i controlli hanno dato l’autorizzazione ad utilizzare i loro campioni di sangue per analisi genetiche; l’approvazione per lo studio è stata approvata dal comitato etico.
3.2 Scelta degli SNPs
Sono stati scelti 108 geni candidati coinvolti nei processi infiammatori, nella riparazione dei danni al DNA, nel mantenimento della stabilità genomica, nella fisiologia dell’apparato digerente e nel metabolismo degli xenobiotici. I 364 SNPs selezionati appartengo a categorie differenti: 235 sono tag-SNPs di cui non è nota la funzione biologia (probabilmente privi di funzione biologica) ma residenti entro blocchi di LD, derivanti dal progetto HapMap; 58 sono polimorfismi probabilmente privi di attività biologica ma rappresentavano gli unici polimorfismi frequenti per un dato gene; 71 sono polimorfismi la cui azione biologica è stata studiata o di cui non è nota l’attività biologica ma che sono stati saggiati per l’associazione ad altre patologie complesse in studi di tipo caso-controllo pubblicati in letteratura. Gli SNPs selezionati sono riportati in Appendice 1
3.3 Whole genome amplification (WGA)
Si è utilizzata la tecnica WGA per ottenerne un maggior quantitativo di DNA dai campioni e per omogeneizzarne la qualità. Il principio di tale tecnica è quello di amplificare il genoma di partenza tramite l’utilizzo di random primers (piccole sequenze oligonucleotidiche casuali che fungono da innesco per la polimerasi), dNTPs e la DNA polimerasi Phi29. Il protocollo utilizzato per il “Genomiphi V2 Amplification Kit” (GE Healthcare) prevede l’utilizzo di 10 ng di DNA (in 1 µl) e di 9 µl di sample buffer. Il tutto viene riscaldato a 95°C per tre minuti e subito trasferito in ghiaccio in modo tale da mantenere il DNA denaturato. Successivamente vengono aggiunti 9 µl di reaction buffer ed 1 µl di enzima DNA polimerasi Phi 29. Il tutto viene quindi incubato a 30°C per 90 min, in questo arco di tempo i random primers esamerici si appaiano in diversi punti sul DNA e fanno da innesco per la polimerasi Phi 29 che, avendo una attività di
strand displacement, permetterà l’amplificazione isotermica di tutto il genoma secondo uno schema riportato in figura 6. Come controllo positivo, la reazione viene effettuata sul DNA del fago λ. Infine l’enzima verrà inattivato tramite un’incubazione di 10 minuti a 65°C. Per verificare che la WGA abbia avuto un esito positivo il prodotto verrà visualizzato su gel di agarosio allo 0,8 %.
Fig 6 : meccanismo di azione della WGA
3.4 Amplificazione con PCR multiplex
L’amplificazione dei frammenti di DNA recanti i polimorfismi da genotipizzare è stata ottenuta tramite multiplex PCR (Polymerase Chain Reaction), fino ad un massimo di 5 ampliconi per reazione. L’utilizzo di una PCR multiplex è richiesto al fine di minimizzare il numero di reazioni necessarie ad amplificare tutti i frammenti di interesse con conseguente economizzazione dei reagenti e del DNA e un aumento della concentrazione relativa di ciascun frammento, rispetto al totale del volume di reazione. Le sequenze dei primers impiegati sono riportate in Appendice 1. Il protocollo di amplificazione utilizzato per la PCR multiplex è il seguente: 2 µl di Taq buffer 10X, 2.4 µl di tampone contenente MgCl2 (25 mM per una concentrazione finale di 1.5 µM), 2µl di dNTPs (2mM), 1
(10µM per una concentrazione finale di 0.5µM), 0.2 µl di enzima Hotfire® Taq polimerasi (Solis Biodyne, Tartu, Estonia), un volume totale di 5,3 µl tra DNA ng (40 ng) da amplificare e H2O per un volume finale di 20 µl. I dNTPs, sono
composti da dATP, dCTP, dGTP 200 µM ciascuno, dTTP 150 µM e dUTP 50 µM. Ciò implica che gli ampliconi prodotti avranno il 25% delle timidine casualmente sostituite con gli uracili. Questo è necessario in quanto nelle fasi successive i prodotti di PCR dovranno essere frammentati a livello delle U allo scopo di agevolare la loro ibridazione con le sonde ancorate sul vetrino.
Il protocollo di amplificazione prevede i seguenti passaggi:
Il primo ciclo viene fatto a 95°C per 8 minuti; lo scopo di tale ciclo è quello di attivare la Taq che è inattivata chimicamente e la cui inattivazione è termolabile; la seconda fase è costituita da 19 cicli, divisi ognuno in tre passaggi, il primo dei quali (95°C per 30 secondi) ha lo scopo di denaturare il DNA. Il secondo passaggio (68°C per 30 secondi) consente l’appaiamento dei primers, in questo passaggio ad ogni ciclo la temperatura si abbassa di un grado (“touch-down PCR”). Questo fa sì che partendo da una temperatura poco permissiva e abbassandola di 1 grado ad ogni ciclo siano raggiunte le tm ottimali di ciascun
primer in modo tale da garantire un’amplificazione specifica delle sequenze bersaglio ed evitare gli aspecifici. Il terzo passaggio di questa seconda fase (72°C per 1.15 minuti) consente la sintesi del DNA. La terza fase è costituita da 30 cicli, divisi ognuno in tre passaggi: il primo a 95°C per 30 secondi per la denaturazione, il secondo a 51°C per 30 secondi per l’appaiamento dei primers. In questo caso la temperatura di 51° C è una temperatura molto permissiva che garantisce un’alta resa. Il terzo passaggio viene fatto a 72°C per 15 minuti,
serve a garantire il completamento della sintesi di ampliconi interrotti. Infine la temperatura si abbassa alla temperatura di mantenimento di 15°.
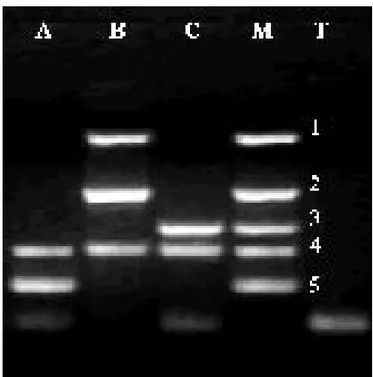
Sono stati ottenuti 324 ampliconi in un totale di 91 reazioni di amplificazione, effettuate in una placca da 96, entro lo stesso blocco del termociclizzatore. Per verificare che l’amplificazione tramite PCR abbia avuto esito positivo vengono scelti in maniera casuale 10 pozzetti della placca da 96 ed un volume di 1,5 µl di prodotto di PCR di ciascuno viene fatto correre su gel di agarosio al 2%. Nel caso in cui l’amplificazione sia avvenuta verranno visualizzate delle bande fluorescenti pari al numero di ampliconi (Fig 7).
3.5 Purificazione post-PCR
I prodotti delle varie PCR per ciascun campione vengono riuniti in un’unica provetta ed in seguito vengono purificati e concentrati tramite filtri Microcon® Millipore Y 30 che è uno dei pochissimi prodotti disponibili commercialmente in grado di permettere purificazioni di volumi di 500 µl. L’eluizione si effettua in un volume finale di 9 µl. All’eluato vengono aggiunti gli enzimi uracil N-glicosilasi (UNG; Epicentre Technologies, Madison, WI, USA) e la fosfatasi alcalina di gamberetto (sAP; GE Healthcare) (1 U di entrambi gli enzimi per ogni 15 µl di eluato, a cui aggiungiamo anche lo specifico tampone UNG dilution buffer 10x). L’enzima uracil N-glicosilasi riconosce gli uracili incorporati nei prodotti di PCR e idrolizza il legame N-glucosidico dell’Uracile con la catena del DNA producendo un sito abasico che successivamente a seguito di un trattamento ad alta temperatura diverrà il sito di rottura a singolo filamento dell’elica del DNA e questo fatto causerà la frammentazione del DNA stesso. La frammentazione agevola l’ibridazione con le sonde sul vetrino. Il ruolo dell’enzima sAP è invece quello di idrolizzare i gruppi trifosfato e eliminare così gli eventuali residui di dNTP presenti nella miscela di reazione delle PCR. La miscela contenente l’eluato, la sAP, la UNG ed il tampone per la UNG vengono sottoposte ad un’incubazione di 90 min a 37 °C che è la temperatura alla quale gli enzimi sono maggiormente attivi, cui seguono 30 minuti a 95°C che è la temperatura alla quale gli enzimi vengono inattivati ed il DNA denaturato. E’ molto importante togliere prima della fine del ciclo i campioni e metterli subito in ghiaccio in modo tale da mantenere denaturati i prodotti di PCR per poterli quindi far ibridare successivamente con le sonde dell’APEX.
3.6 Preparazione dei vetrini per Micro-Array
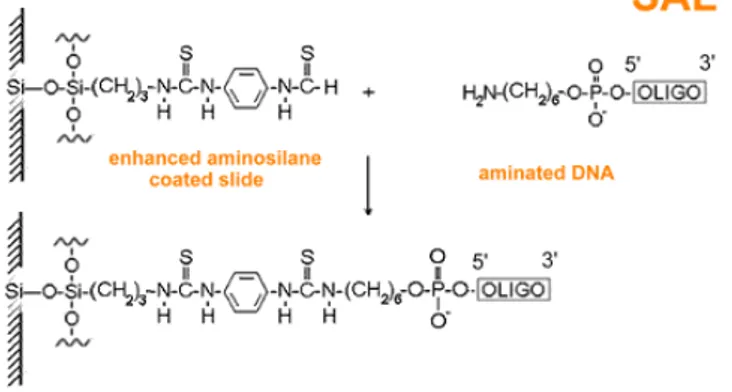
I vetrini SAL-type su cui avviene la reazione dell’APEX sono acquistati dalla Asper Biotech Ltd. (Tartu, Estonia, www.asperbio.com). La preparazione dei vetrini avviene tramite silanizzazione. Gli oligonucleotidi vengono modificati tramite un “5’–aminolinker” prodotto dalla Sigma Genosys (Sigma-Genosys Ltd, Cambridge, UK), costituito da 12 carboni (C12) e terminanti con un gruppo amminico che permette il legame covelante tra l’oligo modificato e il vetrino silanizzato (Fig 8). La sequenza degli oligonucleotidi utilizzati per l’identificazione dei polimorfismi di interesse è riportata in Appendice 2.
Fig 8: Formazione del legame covalente tra il vetrino silanizzato e la sequenza oligonucleotidica modificata con aminolinker al 5’
3.7 APEX
I vetrini necessitano di un primo lavaggio con soluzione di NaOH 100 mM per ripulirli e denaturare le sonde. Ad esso segue un lavaggio in acqua a 95°C. Sul vetrino vengono deposti 20 µl di una miscela nella quale 10 µl della miscela di reazione di purificazione vengono mescolati con 50 pmoli di ciascun terminatore della reazione di Sanger (1 µl ddATP coniugato con Cy3, 1 µl di ddGTP coniugato con Cy5, 1 µl di ddCTP coniugato con Texas Red e 1 µl di ddUTP coniugato con Fluoresceina), 4 µl di tampone di reazione 10x e 1,9 µl di enzima Thermipol (Solis Biodyne). La preparazione della miscela deve essere rapida a causa della sensibilità termica dell’enzima. Il vetrino viene messo ad incubare per 11 min a 58˚ gradi. Terminata l’incubazione il vetrino viene lavato con acqua a 95° in modo tale da eliminare gli eccessi di mix non ibridati. A questo punto viene aggiunto l’agente “Antifade” (Slowfade® Light Antifade Reagent ; Molecular Probes, Eugene, OR, USA), il cui scopo è quello di permettere la lettura dell’emissione della Fluoresceina. Il chip viene quindi coperto da un vetrino copri-oggetto ed è pronto per la lettura. La scansione avviene mediante uno scanner a quattro colori (Genorama TM-003, Asper Biotech, Tartu, Estonia) e l’acquisizione delle immagini avviene ad opera del programma Genorama Image Analysis Software (Asper Biotech, Tartu, Estonia) (Fig 9). Il programma fornisce quattro immagini corrispondenti a ciascun fluorocromo dove ognuno di essi rappresenta un diverso terminatore. Le quattro immagini vengono quindi analizzate dal software.
Fig 9: griglia per l’identificazione degli SNPs su vetrino
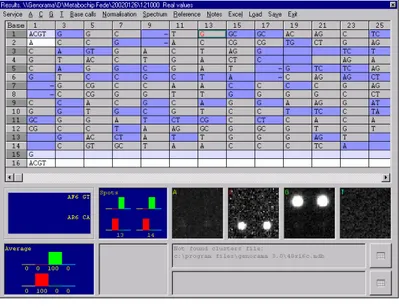
Dal momento che sui chip ciascuna sonda occupa una posizione specifica il software tramite la posizione nell’array è in grado di identificare le sonde forward e reverse di ciascun polimorfismo e di misurarne l’intensità di fluorescenza, dalla quale poi è possibile identificare la variazione genetica dello SNP in analisi e quindi determinare il genotipo dall’individuo in analisi (Fig 10). Il ruolo dell’operatore è quello di controllare che le letture dei genotipi siano stata effettuata correttamente dal software.
a)
b)
Fig 10: lettura dei genotipi da parte del programma genorama; a) in un individuo omozigote per un polimorfismo b) in un individuo eteroziogote per un polimorfismo
3.8 Analisi statistica classica
L’equilibrio di Hardy-Weinberg è stato calcolato per ciascun polimorfismo tramite il test X2 nei controlli e nei casi. I valori di P riportati sono significativi per P <0.05. Di tutti i geni esaminati sono state calcolate le frequenze genotipiche. Le frequenze alleliche calcolate sono poi state confrontate nelle due popolazioni in esame. Le associazioni tra le varianti alleliche e la patologia sono state stimate tramite un’analisi di regressione logistica multivariata, calcolando gli odds ratios (ORs) e gli intervalli di confidenza (CIs) ad essi associati (corretti per sesso ed età). L’analisi statistica è stata effettuata utilizzando i modelli dominante, codominante e recessivo. E’ stato anche effettuato il trend test per valutare la significatività del modello codominante.
3.9 Delineazione dei pathways
Una volta individuati i polimorfismi associati al CRC, i geni sono stati classificati in base alla loro funzione. Ogni gene tra quelli selezionati per il presente studio è stato posto entro uno o più pathway biologici, sulla base di una classificazione rinvenibile entro la risorsa bioinformatica ENTREZ gene (www.ncbi.nlm.nih.gov/sites/entrez) (Appendice 3).
I pathways relativi ai vari geni sono riportati in ENTREZ gene a partire da Gene Ontology e dalla Kyoto Encyclopedia of Genes and Genomes (KEGG) (Fig 11). La risorsa bioinformatica chiamata KEGG fa parte di un progetto di ricerca dei laboratori Kanheisa nei centri bioinformatica dell’Università di Kyoto e del centro Genoma Umano dell’Università di Kyoto. Una volta ottenuta la lista dei geni con i vari pathways si è creata una classificazione inversa in cui sotto i pathways sono riportati i geni che ne fanno parte (Appendice 4).
Fig 11: ricerca per ciascun gene dei pathways relativi su entrez gene
I pathways contenenti un numero di geni inferiori a tre non sono stati analizzati ed altri sono stati raggruppati sotto un unico pathway. Successivamente in ciascun pathway è stato calcolato il numero di polimorfismi totali e quelli associati al CRC.
3.10 Analisi dei pathways
Per ogni pathway biologico, di cui è ora noto il numero dei polimorfismi associati al CRC e di quelli non associati, è stata calcolata la probabilità dell’evento (n associati su k non associati), tramite il calcolo binomiale, definendo come probabilità di successo il valore soglia alpha (errore di primo tipo) pari a p=0,05 utilizzato nelle analisi statistiche preliminari. Sono stati quindi calcolati i 31 valori di probabilità corrispondenti ai 31 pathways biologici identificati. Sono stati ritenuti di interesse quei pathways che mostravano un numero di polimorfismi associati al CRC in misura maggiore rispetto all’atteso. Poichè sono stati effettuati 31 tests statistici utilizzando il calcolo binomiale, il valore soglia per ciascun calcolo binomiale è stato corretto per Bonferroni ed è risultato pari a 0.0016.
In altre parole, solamente i pathways biologici che presentavano un numero di polimorfismi associati al CRC che si discostava dall’atteso con una probabilità inferiore al 0.0016 sono stati ritenuti interessanti.