FARMACI CHE AGISCONO SULLE FUNZIONI MITOCONDRIALI
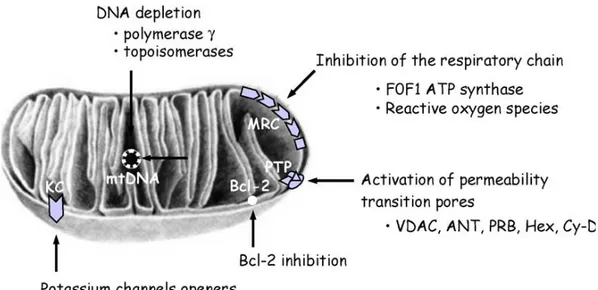
Il ruolo centrale dei mitocondri nella morte cellulare programmata ha indirizzato la ricerca verso agenti chemioterapici in grado di provocare la morte di cellule cancerose [33]. La maggior parte dei farmaci anticancro convenzionali sfrutta indirettamente i mitocondri per svolgere la sua azione citotossica, per esempio attraverso l'attivazione multipla delle vie che coinvolgono la proteina p53 o la morte dei recettori. Quindi, bersagliare direttamente le funzioni mitocondriali potrebbe essere di significativa rilevanza terapeutica, dal momento che la crescita rapida e continua delle cellule tumorali è altamente energia-dipendente, che le cellule tumorali spesso sviluppano farmaco-resistenza, causando resistenza nei confronti dei segnali pro-apoptotici. Di conseguenza, oggi i mitocondri rappresentano i potenziali bersagli per la terapia anti-cancro, e sono stati proposti vari approcci per interferire con le loro funzioni all'interno delle cellule tumorali [34] (figura 8).
Figura 8. Siti d'azione dei farmaci che agiscono sui mitocondri.
Questi organuli cellulari, oltre ad offrire un'ulteriore strategia per la cura del cancro, rappresentano il bersaglio anche di agenti usati o sviluppati per il trattamento di altre patologie, in particolare quelle neurodegenerative e cardiovascolari, come pure il diabete e certe infezioni virali.
Lo scopo principale di questa tesi è quello di dare un prospetto delle diverse categorie di farmaci che interagiscono con i mitocondri.
(acronimo di mitocondri e cancro), alterano i mitocondri e provocano l'apoptosi, che, almeno in alcuni casi, è selettiva per le cellule tumorali.
I mitocani sono stati classificati in sette gruppi diversi, in base alla funzione mitocondriale che vanno a colpire; se una di queste funzioni viene inibita all'interno della cellula tumorale, la sopravvivenza di quest'ultima viene compromessa [35-37].
Composti attivi sulla biosintesi del DNA mitocondriale (mtDNA)
Si tratta di un gruppo di mitocani identificato di recente. Sebbene il ruolo dei mitocondri nella carcinogenesi sia ancora poco chiaro, mutazioni del mtDNA, dalla mutazione di una singola base ad un’estesa delezione, sono state evidenziate in diversi tumori.
L’analisi dell’intero genoma mitocondriale ha evidenziato che su dieci linee cellulari del tumore colon-rettale, sette mostrano mutazioni che non si riscontrano nel tessuto normale da cui il tumore è derivato (mutazione somatica).
Fino ad oggi, nessuna particolare mutazione del mtDNA è stata correlata ad uno specifico tipo di tumore. Le mutazioni possono influenzare la velocità con cui il mtDNA si replica, e le cellule che non esprimono il gene del mtDNA possono diventare più sensibili all’induzione dell’apoptosi. Comunque, in molte linee cellulari tumorali, mutazioni del mtDNA contribuiscono alla chemioresistenza.
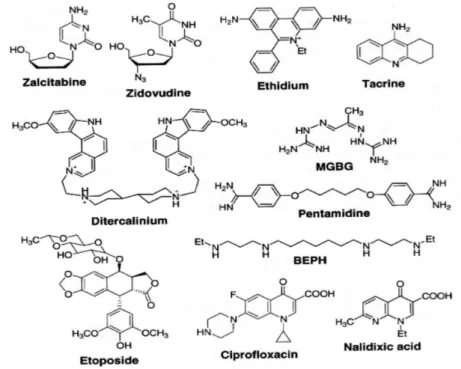
La selettività d’azione verso il mtDna o la sua deplezione possono costituire un’interessante modalità di inibizione della proliferazione o della sopravvivenza delle cellule tumorali. Le caratteristiche del mtDNA, che non è protetto dagli istoni e possiede una limitata capacità di riparo, lo rendono un bersaglio interessante per agenti farmacologici. Un ampio gruppo di molecole strutturalmente diverse sono in grado di indurre diminuzione selettiva dei livelli di mtDNA, che è stata correlata alla citotossicità (figura 9). Per esempio, gli analoghi nucleosidici antivirali come 3’-azido-3’deossitimidina (AZT, zidovudina) o 2’-3’-dideossicitidina (ddC, zalcitabina), usati nella cura dell’immunodeficienza umana (HIV) e nell’epatite B, provocano il rallentamento del metabolismo del mtDNA, con conseguente riduzione dei livelli cellulari di mtDNA e citotossicità.
Figura 9. Farmaci che inducono la deplezione del DNA mitocondriale.
Alcuni di questi analoghi nucleosidici riducono la sintesi del mtDNA attraverso l’inibizione della γ- polimerasi, che media la replicazione del mtDNA; tale inibizione avviene attraverso l'interruzione dell’allungamento del filamento del DNA, per mancanza dei gruppi ossidrilici all’estremità 3’. Anche se questi analoghi nucleosidici hanno mostrato in vitro la capacità di causare deplezione del mtDNA nelle cellule di mammifero, portando alla perdita della crescita cellulare, non è ancora chiaro il ruolo dei mitocondri nel meccanismo d’azione di questi farmaci.
In aggiunta all’indebolimento della funzione della γ-polimerasi, la zidovudina è anche in grado di inibire la telomerasi, l’attività del trasportatore mitocondriale dell’ADP/ATP e gli enzimi della fosforilazione ossidativa. Poiché l’azione dell’AZT non è specificamente rivolta ai mitocondri, il ruolo di questi organelli nell’azione del farmaco è difficile da stabilire con precisione.
Numerosi studi hanno dimostrato che alcuni agenti intercalanti, tra cui il bromuro di etidio o il derivato antitumorale dimerico ditercalinio, possono causare la deplezione anche di mtDNA in colture cellulari di mammifero. Queste cellule, chiamate ρ0 , mostrano una
respirazione carente, ma possono essere mantenute attraverso la sola glicolisi. Il meccanismo attraverso il quale questi agenti intercalanti inibiscono la sintesi del mtDNA, e non del DNA nucleare, non è del tutto chiaro, ma è possibile che questi si accumulino
preferenzialmente nei mitocondri piuttosto che nel nucleo, e che possano agire, come recentemente è stato dimostrato per il ditercalinio, attraverso l'interruzione della replicazione del mtDNA, mediante interazione con la γ-polimerasi mitocondriale ed elicasi Twinckle specificamente mitocondriale.
Il metabolismo del mtDNA può essere colpito anche attraverso gli inibitori delle Topoisomerasi.
Entrambi i tipi, I e II, sono stati identificati nei mitocondri, e alcuni inibitori delle topoisomerasi si sono dimostrati capaci di inibire gli enzimi mitocondriali.
Gli antibatterici 4-chinolonici, come l’acido nalidixico o la ciprofloxacina, causano deplezione del mtDNA, perdita della respirazione mitocondriale, aumento della glicolisi e crescita ritardata.
Il derivato della epipodofillotossina teniposide (VM-26) , inibisce la topoisomerasi II estratta dai mitocondri, e induce la rottura del mtDNA. In maniera analoga, il farmaco antitumorale etoposide (VP-16), inibitore della topoisomerasi II, ha mostrato di agire a livello mitocondriale; il trattamento con basse concentrazioni di etoposide (10 µM) provoca danni al DNA nucleare che alterano i mitocondri attraverso l’attivazione della caspasi 2 e il rilascio di citocromo c; al contrario, a concentrazioni più alte (50 µM) il rilascio di citocromo c è caspasi-indipendente, e sembra correlato alla formazione dei pori di transizione di permeabilità (PTP).
Anche il cisplatino (CDDP) agisce a livello del DNA mitocondriale. Esso si lega in maniera molto più selettiva al mtDNA che al DNA nucleare, provocando l'inibizione dell'enzima NADH-ubiquinone reduttasi e la diminuzione della sintesi di ATP. In seguito alla somministrazione di un'unica dose di farmaco nelle cavie, si è potuta osservare la formazione di addotti farmaco-mtDNA in quantità maggiore rispetto a quelli che si formano con il DNA genomico; elevate quantità di addotti cisplatino-mtDNA possono danneggiare le funzioni di quest'ultimo, fino a causare la morte della cellula. Poiché CDDP lega sia il DNA nucleare che quello mitocondriale, entrambi i meccanismi d'azione possono contribuire all'attività antitumorale di questo composto.
In questo gruppo viene inserita anche la vitamina K3 (menadione).
Il suo effetto sulla coagulazione del sangue e la sua utilità terapeutica sono riconosciuti da tempo, ed è stato valutato il suo utilizzo anche come potenziale agente antitumorale.
Vitamina K3
Sebbene non agisca direttamente sul DNA mitocondriale, essa inibisce in maniera specifica la DNA polimerasi γ (pol γ), l'enzima mitocondriale responsabile della replicazione del mtDNA. Uno studio recente mostra che a concentrazioni di 30 µM, la vitamina K3 inibisce l'attività della pol γ dell'80% senza avere alcun effetto sulle altre DNA polimerasi, e induce alterazioni mitocondriali, produzione di ROS e apoptosi. Questo composto induce anche l'apoptosi nei mitocondri di cellule di tumore mammario. Studi più recenti hanno dimostrato la sua azione inibitoria sulle cellule pancreatiche cancerose.
Composti attivi sulla catena respiratoria
Gli ultimi studi sulla catena di trasporto elettronico hanno permesso la preparazione di farmaci efficaci e selettivi.
Il flusso di elettroni dal NADH all'ossigeno attraverso complesso I, ubiquinone, complesso III, citocromo c, complesso IV ( e attraverso il complesso II dall'ossidazione del succinato) è stato identificato con l'aiuto di inibitori specifici, come il rotenone per il complesso I, il malonato per il complesso II, il mixotiazolo e l'antimicina A per il complesso III, il cianuro per il complesso IV e l'oligomicina o i protonofori, o come il p-clorofenilidrazone di carbonilcianuro (CICCP) per inibire l'ATP-sintasi. E' interessante notare che la 2-metossi-antimicina perde la sua azione inibitoria sul trasporto elettronico, ma si lega direttamente alle proteine Bcl-XL /Bcl-2 per indurre l'apoptosi.
Il rotenone, che è stato usato inizialmente per studiare il trasporto elettronico verso l'ossigeno nella catena respiratoria mitocondriale, inibisce il complesso I in modo non-competitivo, e aumenta la produzione di ROS inducendo l'apoptosi nelle cellule con leucemia primaria. Questo composto non altera in maniera significativa la diffusione o la crescita delle cellule di linfoma B, suggerendo che l'ATP prodotto all'esterno dei mitocondri
potrebbe essere sufficiente per mantenere vitali le cellule di questo tipo di linfoma. Si è osservato che il trattamento cronico con rotenone potrebbe stimolare un aumento della produzione di superossido, portando all'alterazione dei mitocondri. I risultati degli effetti del rotenone sui livelli cellulari di ROS sono contrastanti, avendo osservato sia un aumento che una diminuzione della concentrazione di ROS nelle cellule trattate con il farmaco. Non si conoscono le ragioni di questi opposti risultati. Alla variazione della quantità di ROS potrebbero contribuire differenze a livello del genoma cellulare, una diversa funzionalità mitocondriale e la quantità di farmaco impiegata durante le sperimentazioni. Nonostante il rotenone sia un potente e specifico inibitore del complesso I, e venga comunemente usato, la sua utilità come agente antitumorale non è ancora chiara, sebbene il suo potenziale chemopreventivo sia stato dimostrato.
Nel corso degli anni sono stati identificati potenti inibitori sia sintetici che di derivazione naturale, dotati di proprietà farmacologiche interessanti, ad esempio maggiore stabilità e più specificità in determinati tessuti (figura 10).
Figura 10. Farmaci che agiscono sulla catena respiratoria, producendo ROS.
È il caso di un gruppo di polifenoli fotochimici attivi sulla F0-F1-ATP sintasi. Lo stilbene
naturale fitoalexina piceatannolo ha dimostrato di essere un inibitore della F0-F1-ATP
sintetasi più potente (IC50~8-9 µM), o degli estrogeni naturali come il 17α-estradiolo e il
17β-estradiolo (IC50>50 µM). Sia il piceatannolo che il resveratrolo inibiscono il complesso
F1 dell'enzima, mentre i due estrogeni bersagliano preferenzialmente il complesso F0.
L'inibizione della F0-F1-ATPasi da parte di genisteina e resveratrolo è non competitiva.
specialmente uva e mirtilli. Per azione sulla catena respiratoria mitocondriale, sembra inibire gli eventi cellulari associati all'iniziazione, alla promozione e alla progressione del tumore. Attraverso la competizione con UbQ, il resveratrolo riduce l'attività del complesso III e diminuisce i livelli di ROS. Sebbene sia considerato un antiossidante, questa molecola ha dimostrato di indurre l'apoptosi attraverso la via mitocondriale. Sembra che esso interferisca con importanti vie del segnale come PI3K/AKT, JAK/STAT, e la cascata MAPK. Inoltre, riduce l'espressione di HIF-1 α e VEGF indotta dall'acido lisofosfatidico. I cationi N-metilpiridinio e chinolinio inibiscono efficacemente il trasporto elettronico mitocondriale del complesso I. Questo è il caso in particolare dei composti MQ18
(N-metil-2-n-dodecil-3-metilchinolinio) e MP6
(N-metil-4-[2-metil-3-(p-tert-butilfenil)]propilpiridinio), i quali inibiscono selettivamente il trasporto di elettroni, con affinità micromolari, attraverso un'interazione selettiva con uno dei due ubichinoni che legano i siti dell'enzima.
La F0 -F1 -ATP sintasi mitocondriale è il bersaglio principale di una famiglia di macrolidi
citotossici che comprendono la citovaricina e l'ossamicina. Il polichetide naturale apoptolidina, che fa parte di questo gruppo di agenti con attività specifica sui mitocondri, è una delle più selettive molecole citotossiche testate sulle sessanta linee cellulari del NCI, ed è un potente agente pro-apoptotico. Questo studio, insieme ad altri, identifica la F0 -F1 -ATP
sintasi come un bersaglio interessante per lo sviluppo di farmaci antitumorali.
I derivati bis-tetraidrofuranici delle acetogenine Annonaceus, fra cui la rolliniastatina 1 (originariamente isolata da semi di Rollinia membranacea), sono fra i più potenti inibitori del complesso MRC I, inibendo efficacemente la proliferazione delle cellule tumorali; per questo sono stati sviluppati come agenti anti-cancro.
Durante il processo metabolico mitocondriale, a livello dei complessi I o III, possono formarsi specie reattive dell'ossigeno, in particolare l'anione superossido, il perossido d'idrogeno e il radicale ossidrilico. In condizioni fisiologiche, il livello appropriato di tali specie è necessario per la stabilità del bilancio redox e la proliferazione cellulare; un loro accumulo può essere invece tossico per la cellula.
I ROS vengono solitamente eliminati dagli enzimi metabolici, come la superossido-dismutasi (SOD), la catalasi e varie perossidasi. L'inattivazione di questi enzimi o l'aumento della produzione intracellulare di ROS da parte di xenobiotici e da radiazioni ultraviolette o ionizzanti, causano danni che possono portare a morte cellulare. Sebbene gli effetti del
danno cellulare da parte dei radicali liberi dell'ossigeno possano essere associati a disturbi patologici, l'uso di agenti capaci di indurre la produzione di ROS, come il rotenone, potrebbe rappresentare una possibilità interessante per uccidere selettivamente le cellule cancerose. Per esempio, il 2-metossiestradiolo (2-ME), che viene usato nella terapia antitumorale fotodinamica, provoca un accumulo di ROS attraverso l'inibizione dell'attività della SOD, esplicando effetti citotossici. La combinazione di agenti che contemporaneamente inducono un aumento della produzione di ROS e inibiscono la loro eliminazione, potrebbe essere efficace per limitare la crescita delle cellule tumorali. Per esempio, la combinazione di 2-ME con il rotenone aumenta l'accumulo nelle cellule del radicale O2•− e induce l'apoptosi.
Questa classe di mitocani annovera come composto più importante α-TOS (α-tocoferil succinato), oltre a molecole analoghe tipo la N-(idrossifenil)retinamide (fenretide) o 4-HPR.
α-TOS
α-TOS, analogo della vitamina E, provoca la morte selettiva delle cellule cancerose secondo un meccanismo che coinvolge i mitocondri e l'accumulo di ROS. Il bersaglio molecolare di α-TOS non era noto fino a poco tempo fa, quando si è scoperto che questo composto agisce inibendo l'attività dell'enzima SDH (succinato-deidrogenasi) nel complesso II della catena di trasporto elettronico mitocondriale. Il farmaco interagisce con i siti di legame per l'ubiquinone presenti nel complesso II, come confermato con saggi biochimici e analisi computazionale. Modificazioni genetiche delle cellule cancerose a livello della subunità SDHC del complesso II, renderebbero tali cellule resistenti alla morte indotta da α-TOS e dall'accumulo di ROS. Ripristinando, invece, la funzionalità del complesso II, le cellule tornano ad essere sensibili all'azione del composto. Un'analoga resistenza al farmaco si è osservata nelle cellule tumorali quando l'espressione di SDHC è stata ridotta da siRNA. Quindi, è stato ipotizzato che α-TOS agisca sulle cellule cancerose spiazzando il legame
dell'ubiquinone al complesso II dei mitocondri di tali cellule. Come risultato, gli elettroni prodotti dall'azione di SDH si combinano all'ossigeno molecolare per produrre grandi quantità di ROS.
Il meccanismo pro-ossidante dell'altro composto, 4-HPR, non è ancora chiaro, ma probabilmente esso agisce, a concentrazioni micromolari, come inibitore di almeno uno dei complessi della ETC (catena di trasporto elettronica), avendo bisogno, per svolgere la sua azione apoptotica, della respirazione mitocondriale.
A questo gruppo di mitocani appartiene anche l'anidride arseniosa. Questo composto è stato a lungo usato nella medicina tradizionale cinese per curare un certo numero di malattie. Nel 1970, As2O3 è stata introdotta nella terapia della leucemia promielocitica acuta (APL),
dimostrando di essere molto efficace, con percentuali di completa remissione comprese tra il 65,6% e l'84% osservate nel corso di studi clinici condotti nella regione nord-est della Cina. In vitro, su linee cellulari di APL, esercita una duplice azione, a seconda del dosaggio: ad alte concentrazioni (0.5-0.2 umol/L) induce l'apoptosi, mentre a concentrazioni più basse (0.1-0.5 umol/L) provoca la differenziazione. Numerosi studi hanno dimostrato che As2O3
causa la formazione di superossido e di perossido di idrogeno, portando ad una riduzione del potenziale della membrana mitocondriale e all'apoptosi. Nelle cellule di leucemia, As2O3 è
capace di inibire la respirazione mitocondriale, causando una forte diminuzione nel consumo di ossigeno nelle prime 3h, oltre ad un aumento nella produzione di ROS. Poiché la morte della cellula sopraggiunge 24h dopo la somministrazione del farmaco, l'inibizione della respirazione sembra essere un evento primario, piuttosto che una conseguenza della morte cellulare.
Inoltre, in presenza di rotenone, As2O3 causa una maggiore inibizione, suggerendo che
queste due molecole possono agire sinergicamente. Ciò dimostra che attraverso l'interferenza nella catena di trasporto elettronica mitocondriale, As2O3 può causare una
maggiore perdita di elettroni, e quindi stimolare la produzione di ROS. Uno studio condotto su cellule con respirazione inadeguata, ha messo in evidenza che le cellule rho-0 resistono al trattamento con As2O3, confermando che l'attività sulla catena respiratoria è importante per
Composti attivi sul canale mitocondriale del potassio
L'aumento della permeabilità della membrana mitocondriale ai protoni o al potassio attraverso l'apertura dei canali mitocondriali del potassio, provoca la diminuzione del potenziale della membrana mitocondriale (∆Ψ) (depolarizzazione), il rigonfiamento dei mitocondri, la diminuzione della sintesi di ATP e il rilascio del citocromo c.
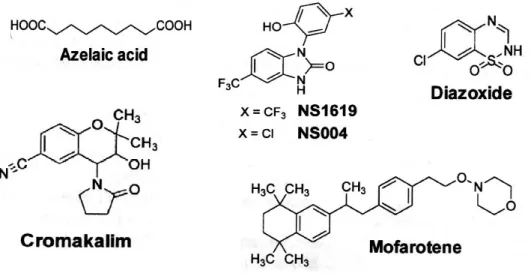
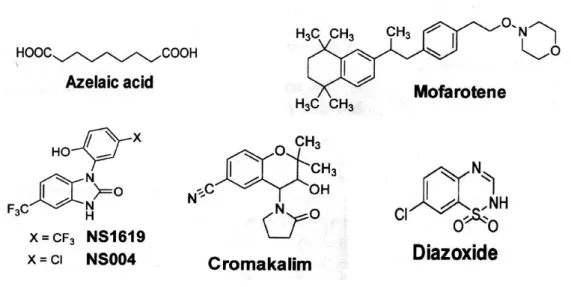
Vari farmaci, come il diazossido, il vasorilasciante cromacalim, ed i suoi analoghi EMD-60480 e EMD-57970, sono stati identificati come molecole capaci di indurre l'apertura del canale del potassio (KCO). Questi derivati sono studiati essenzialmente come agenti vasodilatatori e antipertensivi, ma il cromacalim ha dimostrato di inibire, in maniera dose-dipendente, la crescita del neuroblastoma umano SK-N-MC e dell'astrocitoma umano U-373MG (figura 10).
Figura 10. Farmaci che agiscono sulla catena respiratoria, producendo ROS.
Al contrario, altri KCO attivano la crescita delle cellule tumorali, come ad esempio il minoxidil, che ha dimostrato di stimolare la crescita di cellule tumorali umane della mammella (MCF-7), mentre i bloccanti dei canali del potassio dequalinio e amiodarone inibiscono marcatamente la proliferazione delle stesse cellule. Ciò avviene probabilmente perchè i canali mitocondriali del potassio hanno le stesse proprietà farmacologiche dei canali del potassio della membrana plasmatica. Da qui la necessità di trovare farmaci che interagiscono specificamente con il canale mitocondriale.
La bentotiadiazina diazossido ha dimostrato di agire in maniera 1000 volte più efficace sul trasporto mitocondriale del potassio che sulla membrana plasmatica. Questo composto protegge colture cellulari di neuroni PC12 dalla morte indotta dal rotenone. Esso depolarizza il potenziale della membrana mitocondriale respirazione-dipendente, riduce la velocità di proliferazione e blocca le cellule T di leucemia acuta umana in fase G0/G1.
Sempre nella categoria dei composti KCO, è degno di nota il levosimendano, usato attualmente per il trattamento dell'insufficienza cardiaca acuta e lo scompenso cardiaco. Sembra che la capacità del levosimendano di attivare il flusso di potassio verso la matrice mitocondriale sia responsabile della sua attività anti-ischemica. I KCO possono esercitare effetti farmacologici ben precisi a seconda del tipo di cellula sulla quale agiscono.
Una forte depolarizzazione dei mitocondri, associata all’inibizione di MRC, è stata osservata in cellule di glioma trattate con i derivati attivi sui canali del potassio a larga conduttanza NS1619 (1,3-diidro-1-[2-idrossi-5-(trifluorometil)-fenil]-5-(trifluorometil)-2H-benzimidazol-2-one) e NS004 (5-trifluorometil-1-(5-cloro-2-idrossifenil)-1,3-diidro-2H-benzimidazolo-2-one). Ma questo effetto diretto sui mitocondri non ha prodotto conseguenze sulla sopravvivenza delle cellule di glioma. In maniera analoga, concentrazioni di rotenone che bloccano il flusso elettronico attraverso il complesso I mitocondriale non modificano né la crescita né la diffusione delle cellule di linfoma umano. In altri casi, sembra che ci sia una correlazione tra inibizione di MRC e attività antiproliferativa, come nel caso dell’acido azelaico, un acido bicarbossilico a catena lineare di 9 C di origine naturale, usato per il trattamento del melanoma tipo lentigo maligna, e dell'arotinoide mofarotene (Ro40-8757), che associa l'inibizione della fosforilazione ossidativa con l'inibizione della proliferazione cellulare del linfoma di Burkitt e l'apoptosi.
Composti che alterano il potenziale di membrana
Non c’è dubbio che i mitocondri giochino un ruolo centrale nella morte cellulare programmata, infatti alcune modificazioni mitocondriali sono state descritte come tappe cruciali dell’apoptosi: diminuzione del potenziale della membrana mitocondriale (∆Ψ), blocco del trasporto elettronico e della fosforilazione ossidativa, sintesi delle specie attive dell’ossigeno e rilascio di fattori pro-apoptotici, come citocromo c, Smac/Diablo, AIF, ecc.,
che inducono l’attivazione delle caspasi. Poiché i mitocondri svolgono un ruolo chiave nell’induzione dell’apoptosi, è di grande interesse la possibilità di sfruttare la loro funzione pro-apoptotica per ridurre la crescita e la sopravvivenza delle cellule tumorali. Sono stati considerati essenzialmente due approcci farmacologici correlati all’apoptosi: l’inibizione della famiglia delle proteine Bcl-2 e la formazione dei pori di transizione di permeabilità. L’inibizione della permeabilità della membrana mitocondriale (MMP), che blocca il rilascio del citocromo c, contribuisce alle funzioni anti-apoptotiche della proteina Bcl-2 situata nella membrana mitocondriale esterna.
Sono state messe a punto strategie diverse per annullare la proprietà anti-apoptotica delle proteine della famiglia Bcl-2. L’inibizione dell'espressione di Bcl-2 o dei suoi affini Bcl-XL
è stata ottenuta mediante un anticorpo a singola catena o con oligonucleotidi antisenso a singolo filamento, i quali possono ibridare con il mRNA bersaglio e inibire il suo trasferimento all’interno della proteina Bcl-2. Infatti, l’associazione di Genasense (Oblimersen sodium, gp3139), un oligonucleotide antisenso 18-merfosforotiato attivo su mRNA-Bcl-2, con farmaci citotossici convenzionali, ha dato risultati incoraggianti, anche se la monoterapia con l’oligonucleotide antisenso è fortemente compromessa.
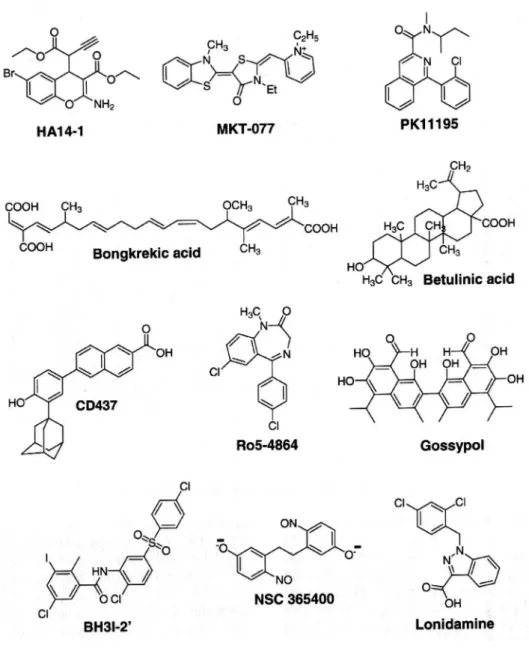
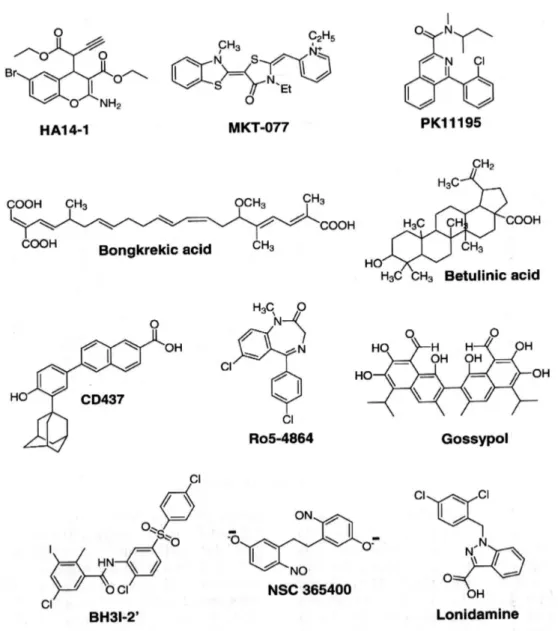
Una strategia alternativa è lo sviluppo di piccole molecole capaci di mimare il dominio di dimerizzazione BH3, identificato in praticamente tutte le proteine correlate a Bcl-2. L’ interazione di tali composti con Bcl-2 induce apoptosi caspasi-dipendente delle cellule cancerose. È il caso del derivato del cromene, permeabile alle cellule, HA14-1 (etil 2-amino-6-bromo-4-(1-ciano-2-etossi-2-ossoetil)4H-cromene-3-carbossilato), il quale si lega alla tasca di superficie del Bcl-2 e stimola l’apoptosi. Altre molecole di natura non peptidica, antagoniste di Bcl-2 e Bcl-XL sono state sviluppate recentemente, come il derivato della diazocina diossido NSC365400 e il composto tiazolidinico BH31-2, che si è mostrato un inibitore del peptide BH3, che si lega a Bcl-XL (figura 11).
Figura 11. Farmaci che agiscono su Bcl-2 o sui pori di transizione della membrana.
Il composto più recente di questo gruppo di molecole è il gossipolo, farmaco polifenolico di derivazione naturale che, oltre ad essere un contraccettivo maschile, possiede in vivo proprietà antitumorali. Questo composto rappresenta il prototipo della piccola molecola che interagisce con BH3, capace di inibire Bcl-2, Bcl-XL e Mcl-1; infatti, sembra che sia in grado di agire direttamente sulle molecole Bcl-2 presenti nella membrana mitocondriale esterna. Il gossipolo è capace di bloccare l'eterodimerizzazione di Bcl-XL con Bax o Bad, come di
promuovere l'attivazione della caspasi-3 ed il rilascio del citocromo c in cellule che sovraesprimono Bcl-2 e Bcl-XL. Poiché tale composto mostra una marcata capacità di distruggere vari tipi di cellule cancerose, la sua attività antitumorale è stata testata nel corso
di sperimentazioni cliniche su pazienti con tumore della prostata refrattario alla terapia ormonale e su soggetti con tumore mammario in fase avanzata. Attualmente, numerosi derivati del gossipolo vengono studiati come potenziali agenti antitumorali; tra questi si può citare l’enantiomero (-) del gossipolo, che ha dimostrato di esercitare un'azione selettiva nei confronti delle proteine della famiglia Bcl-2, superando così la resistenza all’apoptosi.
Attivazione del poro di transizione della permeabilità
Le alterazioni nella permeabilità della membrana mitocondriale svolgono un ruolo chiave nella via apoptotica.
I mitocondri usano pompe-canale e le vie ossidative per mantenere un potenziale di membrana costante di circa – 180 mV, attraverso il loro doppio strato lipidico. Un valore simile non viene raggiunta in alcun altro organulo; il potenziale che si registra nei mitocondri è doppio rispetto a quello delle cellule eccitabili della membrana plasmatica, e circa sei volte maggiore di quello delle cellule non eccitabili. Le caratteristiche peculiari della membrana mitocondriale la distinguono dagli altri organelli intracellulari, offrendo un’opportunità unica di bersagliare selettivamente i mitocondri.
Un improvviso crollo del potenziale di membrana (∆Ψ), evidenziato valutando la perdita di fluorescenza di fluorocromi cationici lipofili, come la rodamina123, è spesso associato alla formazione di un megaporo, il poro di transizione della permeabilità, nella membrana interna.
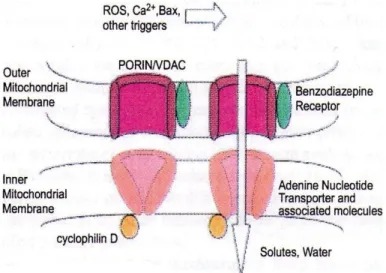
La composizione e la struttura del poro PTP deriva dall’associazione di diverse proteine, ovvero l’adenina nucleotide traslocasi (ANT), situata nella membrana mitocondriale interna, il canale ionico voltaggio-dipendente (VDAC), localizzato nella membrana esterna, il recettore periferico delle benzodiazepine (PBR) e la peptidil-prolil isomerasi ciclofilina D (Cyp-D) (figura 12).
Figura 12. Poro di transizione della permeabilità mitocondriale.
Questo complesso proteico crea un canale che collega la matrice mitocondriale al citosol. L'apertura di questo canale permette il libero passaggio di molecole di dimensioni fino a 1,5 kDa, e la dissipazione del gradiente protonico che danneggia le funzioni della catena respiratoria. L’ingresso di soluti e acqua provoca il rigonfiamento della matrice e la rottura della membrana esterna, determinando il rilascio di proteine, come il citocromo c, che attivano le caspasi.
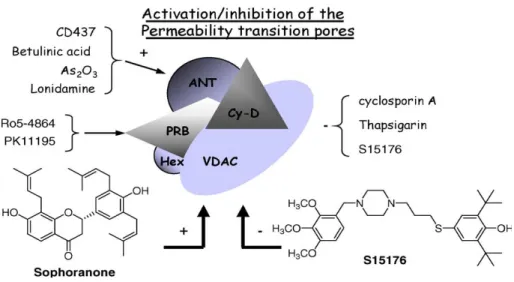
Un certo numero di farmaci chemioterapici sperimentali agiscono direttamente su MMP inibendo l'apertura del poro, legandosi ad uno dei componenti della proteina PTP. Per esempio, la ciclosporina A, un potente immunosoppressore, si lega con elevata affinità alla ciclofilina D, determinando l’inibizione dei fattori che aprono il PTP. Questa inibizione non dipende dall’azione immunosoppressiva della ciclosporina A, dal momento che l’analogo N-metil-Val-4-ciclosporina è capace di bloccare il PTP ma non è in grado di inibire la calcineurina né bloccare i geni delle citochine. Il legame della ciclosporina A al PTP impedisce la morte cellulare dovuta a necrosi da stress ossidativo, la tossicità del trasportatore del Ca2+ e l’ischemia.
L’apertura del PTP può essere inibita non solo attraverso il legame diretto di un farmaco, ma anche impedendo l’accumulo di Ca2+ con composti quali la tapsigarina, un inibitore della Ca2+ -APTasi del reticolo endoplasmatico, che determina apoptosi in varie linee cellulari. La tapsigarina e la ciclosporina A inibiscono entrambe l’apertura del PTP (figura 13).
Figura 13. Schema del poro di transizione mitocondriale (PTP) e farmaci che interagiscono con proteine
costituenti il PTP.
La situazione contraria, invece, è stata osservata recentemente con il complesso dinucleare oro(I)-carbene e con il composto vegetale soforanone, un flavonoide isoprenoide-sostituito isolato dalle radici di Sophora subprostrata, ed estratto dal tradizionale medicinale cinese Shan Dou Gen. Il soforanone stimola la produzione di ROS all’esterno dei mitocondri, induce l’apertura del PTP ed il rilascio del citocromo c, provocando l’apoptosi delle cellule di leucemia U937. Il bersaglio specifico del soforanone non è ancora stato individuato, comunque la sua attività selettiva sui mitocondri giustifica gli studi rivolti alla scoperta di una sua potenziale attività antitumorale.
Una molecola particolarmente interessante, attiva sui mitocondri, è l' N-metilpiridinio o F16 (figura 14), che inibisce selettivamente la proliferazione delle cellule dell’epitelio mammario che sovraesprimono il protoncogene erb-2/neu e altri oncogeni, come c-myc e v-Ha-ras.
Figura 14.
Questo composto lipofilo, che presenta una struttura piuttosto semplice, scoperto attraverso lo screening di una libreria di composti, si accumula all’interno dei mitocondri, principalmente nella matrice, causando, mediante la dissipazione del gradiente protonico che si forma attraverso la membrana mitocondriale interna (diminuzione di ∆Ψ), danneggiamento mitocondriale, apertura del poro di transizione della permeabilità, rilascio di citocromo c, blocco del ciclo cellulare, fino alla morte della cellula. Il danneggiamento delle funzioni mitocondriali da parte di F16 potrebbe essere all’origine della sua capacità di indurre apoptosi o necrosi a seconda della struttura genetica della cellula tumorale bersaglio. L'interesse nella ricerca di molecole analoghe che inducano tossicità mitocondriale, è comunque condizionato dall'osservazione che la struttura del composto F16 somiglia al 1-metil-4-fenilpiridinio (MPP+), una nota tossina che causa parkinsonismo (metabolicamente prodotta per ossidazione della neurotossina nigrostriatale MPTP).
L'acido betulinico, un triterpene lupano-simile, la lonidamina, l’anidride arseniosa (una delle cure più efficaci per la leucemia promielocitica acuta) e l'acido 6[3-adamantil-4-idrossifenil]-2-naftalen carbossilico (CD437) inducono la MMP attraverso un’azione diretta su ANT (figura 11).
Figura 11. Farmaci che agiscono su Bcl-2 o sui pori di transizione della membrana.
Comunque, anche se queste molecole hanno un effetto simile sulla MMP, esse inducono l’apoptosi secondo meccanismi diversi. L’inibizione della fosforilazione ossidativa indotta dall’inibitore della F0-F1-ATPasi oligomicina rende più sensibili le cellule alla morte cellulare indotta da lonidamina, mentre sia l’inibizione della fosforilazione ossidativa che la glicolisi sono necessarie per sensibilizzare le stesse cellule alla morte provocata da anidride arseniosa.
Si è cercato quindi di comprendere l’esatto bersaglio dell’anidride arseniosa.
Utilizzando un anticorpo policlonale VDAC, capace di inibire selettivamente il rilascio di citocromo c mediato da VDAC e indotto da Bax e Bak, e liposomi contenenti VDAC, si è arrivati a definire che il VDAC è il bersaglio principale dell’anidride arseniosa. In effetti, il
meccanismo d’azione dell'As2O3 è pleiotropico, dal momento che implica dissipazione del ∆Ψ, conseguente rilascio del citocromo c e inibizione della respirazione mitocondriale attraverso l’aumento della sintesi di ROS, riuscendo a promuovere l’apoptosi in cellule di leucemia primaria. Questo composto è anche in grado di causare la degradazione della proteina di fusione PML-RARα in cellule affette da leucemia promielocitica acuta.
La situazione risulta più chiara nel caso della lonidamina (LND), che induce l’apoptosi attraverso un’azione diretta sul poro di transizione della permeabilità mitocondriale.
LND, che deriva dall'acido carbossilico 3-indazolico, mostra proprietà sia antispermatogeniche che antineoplastiche. Essa ha vari effetti sulle cellule, sebbene la sua attività principale sia quella di regolare l'energia nelle cellule cancerose. La lonidamina ha dimostrato di bloccare la glicolisi attraverso l'inibizione della esochinasi II, che si trova spesso in quantità elevata nelle cellule cancerose. L'attività di questo composto si basa sulla deplezione dell'ATP, sull'inibizione del consumo di ossigeno nelle cellule di tumore ascitico di Ehrlich e della produzione di lattato in condizioni aerobie e anaerobie. Fenomeni apoptotici di scarsa rilevanza si sono osservati in corso di terapia con la sola lonidamina o con radioterapia, mentre un significativo aumento dell'apoptosi si osserva trattando cellule di melanoma maligno resistente alla radioterapia con l'associazione dei due agenti terapeutici. LND aumenta anche la citotossicità di diversi farmaci antitumorali, tra cui doxorubicina, cisplatino, carmustina (BCNU) e 4-idroperossiciclofosfamide (4-HC). Numerosi studi clinici con lonidamina da sola e in associazione con altri agenti chemioterapici sono stati condotti su pazienti con tumore del polmone non a piccole cellule, del seno, ovarico e glioblastoma.
Un composto più recente è l'oxamato, che inibisce la lattato deidrogenasi (LDH), un enzima NADH-dipendente responsabile della trasformazione del piruvato a lattato.
In diverse linee cellulari epiteliali di tumore mammario nel topo, l'attività dell'LDH-A è maggiore, se paragonata a quella nelle cellule sane. Alcuni studi hanno dimostrato, attraverso la misurazione del lattato prodotto, che la velocità di glicolisi in cellule HeLa S3 trattate con oxamato viene ridotta; inoltre, anche la crescita cellulare e l'assorbimento di glucosio vengono inibiti da questa molecola. Un altro studio sostiene che l'enzima LDH sia bersaglio del composto utilizzando l'α- chetobutirrato come sostituto del piruvato: l'addizione di α- chetobutirrato blocca l'inibizione della crescita e l'assorbimento del glucosio in presenza di oxamato nelle cellule HeLa. Tuttavia, l'α- chetobutirrato, da solo, inibisce la crescita cellulare. La sintesi di LDH-A umano ricombinante mostra che l'oxamato inibisce in modo competitivo l'enzima, e ciò viene confermato anche in vitro. L'inibizione della crescita è stata osservata sia in vitro che in vivo trattando cellule di adenocarcinoma mammario MDA-MB-231 con oxamato. La perdita di ATP nelle cellule HeLa per azione di questo composto, sembra anche contribuire all'inibizione della crescita e ad aumentare la citotossicità mediata da doxorubicina. Inoltre, l'oxamato ha dimostrato di inibire l'attività dell'aspartato aminotrasferasi (AAT) umana ricombinante, che lavora in associazione con la malato deidrogenasi, un enzima TCA che ossida il malato ad ossaloacetato, per facilitare il trasporto dei due metaboliti fra i mitocondri e il citosol.
Un altro farmaco selettivo verso i mitocondri da menzionare, è l’imexone (figura 14), un imminopirrolidone contenente aziridina, il quale mostra la capacità di inibire selettivamente la crescita del mieloma multiplo. Questo derivato cianoaziridinico, nelle cellule con mieloma, provoca stress ossidativo, alterazioni mitocondriali e apoptosi, attraverso il legame covalente con composti sulfidrilici biologicamente importanti. Le cellule di mieloma RPMI8226/I resitenti all’imexone presentano importanti alterazioni morfologiche dei mitocondri ed una maggiore espressione delle proteine mitocondriali con funzione anti-apoptotica, come la Bcl-2. La potente azione pro-apoptotica dell’imexone associata alla sua capacità di interagire selettivamente con i mitocondri, rende questa molecola un composto interessante per lo sviluppo di nuovi aziridino-imminopirrolidoni ad azione antitumorale. Per quanto riguarda il composto dicloroacetato (DCA), sembra che uno dei meccanismi d'azione sia l'inibizione della chinasi piruvato deidrogenasi (PDK), che a sua volta provoca l'attivazione della piruvato deidrogenasi (PDH). PDH è responsabile della trasformazione del piruvato ad acetil-CoA, che può, in questo modo, entrare nel ciclo degli acidi tricarbossilici. La fosforilazione di PDH ad opera dell'enzima PDK causa l'inibizione
dell'attività della piruvato deidrogenasi. Alcuni studi hanno dimostrato che il dicloroacetato è in grado di ridurre l'iperpolarizzazione del potenziale della membrana mitocondriale nelle cellule di glioblastoma, di tumore al polmone non a piccole cellule e al seno, mentre lo stesso non avviene nelle cellule sane. L'analisi dei parametri metabolici nel corso del trattamento con DCA di cellule di tumore polmonare ha dimostrato che il composto è capace di ridurre sia la glicolisi che l'ossidazione degli acidi grassi, mentre aumenta l'ossidazione del glucosio. In più, il dicloroacetato è in grado di indurre l'apoptosi, una forte produzione di H2O2 e di attivare il canale del potassio Kv1.5 nelle cellule A549. Il trattamento di cellule di carcinoma della testa e del collo con DCA ha indotto una riduzione dose-dipendente della forma fosforilata dell'enzima PDHα nelle cellule primarie UM-22A. Dato che la membrana mitocondriale presenta un potenziale negativo, altri agenti sono caratterizzati da un gruppo carico positivamente, che si serve delle forze elettrostatiche per raggiungere il suo bersaglio.
La rodamina 123 e composti analoghi, contenenti gruppi cationici all’interno di una struttura altrimenti non polare, hanno la capacità di attraversare la membrana lipidica mitocondriale sfruttando il gradiente di potenziale negativo di questo organello come forza motrice elettrostatica. In accordo con l’equazione di Nernst, esso può comportare un aumento dell’accumulo di 100-500 volte. La rodamina 123 e i suoi analoghi sono stati usati per valutare l’accumulo di questo gruppo di coloranti fluorescenti nei mitocondri, e come risultato del successo e della riproducibilità della loro selettiva incorporazione all’interno dei mitocondri, i coloranti contenenti rodamina sono stati messi a punto per saggi mitocondriali correntemente utilizzati.
Rodamina 123
composti ad essa legati, nei mitocondri. Il farmaco antitumorale cisplatino è stato selettivamente introdotto nei mitocondri di cellule cancerose utilizzando questo metodo, ed approcci dello stesso genere sono stati sperimentati con altre piccole molecole. Il risultato più importante degli studi sulla rodamina è stato la scoperta dell’utilità dell’effetto
chaperone, il quale è stato ulteriormente sfruttato per lo sviluppo di sali lipofili di
trifenilfosfonio (TPP).
Questo ultimo gruppo di composti comprende la maggior parte degli agenti non-peptidici mitocondri-specifici sintetizzati fino ad oggi (figura 15).
Figura 15. Composti cationici lipofili.
Dal momento che l’assorbimento e il profilo di selettività dei derivati TPP alchilici sono simili a quelli degli analoghi strutturalmente più complessi della rodamina, sono stati sintetizzati vari cationi TPP lipofili legati a molecole antiossidanti, con interessanti risultati. Altri cationi lipofili, che mostrano selettività verso i mitocondri, potrebbero costituire un’opportunità importante per lo sviluppo di veri e propri farmaci (figura 16).
Figura 16. Altri composti cationici lipofili.
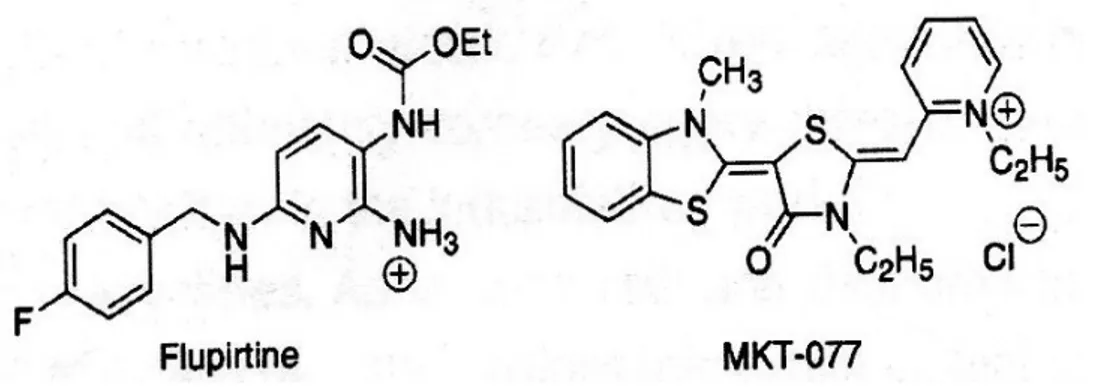
La flupirtina è un analgesico non-oppioide localizzato all’interno dei mitocondri che può proteggere dal danno cellulare indotto da N-metil-D-aspartato e dal danno ischemico, oltre a
prevenire l’aumento indotto da glutammato, dei livelli di Ca2+ , causando apoptosi.
Il composto MKT-077 si accumula nei mitocondri grazie alla sua funzione amminica in forma cationica ed al suo scaffold relativamente non-polare. Tuttavia, invece di agire in modo protettivo, MKT-077 mostra una tossicità selettiva verso i mitocondri di cellule cancerose; tale selettività è legata al potenziale della membrana mitocondriale, che nelle cellule tumorali risulta aumentato rispetto al valore che si registra nelle cellule sane.
Per quanto i cationi lipofili proteggano dal danno mitocondriale, la loro dipendenza dal potenziale di membrana mitocondriale costituisce tuttavia la limitazione più importante. Infatti, con l'aumento del numero di cationi lipofili all’interno dell’organello, il gradiente di potenziale diminuisce fino ad un valore in cui si ha la rapida fuoriuscita dell’inibitore dal mitocondrio, con perdita di attività, finché non viene ripristinata l’entrata. Quindi, a meno che l’inibizione sia irreversibile, non si può verificare un'attività costante del catione lipofilo. Questo inconveniente può costituire un problema, legato alla velocità con cui le specie cationiche entrano ed escono dai mitocondri.
Il decalinio è un composto lipofilico con due cariche positive e una catena laterale alifatica di 10 atomi di carbonio. Clinicamente, questa molecola è stata usata come antibiotico topico per mezzo secolo. Come altri cationi lipofilici, si ritrova principalmente nei mitocondri delle cellule tumorali. Il trattamento prolungato con questo composto altera drasticamente la morfologia dei mitocondri, causando il cambio globulare e la distribuzione perinucleare. Esso inibisce, inoltre, la capacità della calmodulina, una proteina che lega il calcio, di attivare la fosfodiesterasi, per cui impedisce la proliferazione cellulare. Il decalinio bersaglia selettivamente le cellule cancerose, danneggia la proliferazione, la migrazione e l'invasione delle cellule di carcinoma, e prolunga la sopravvivenza dei topi in cui erano state impiantate cellule di tumore della vescica e del colon.
DERIVATI A STRUTTURA PROTEICA, PEPTIDICA E PEPTIDOMIMETICA
Proteine
Affinché filamenti polipeptidici vengano correttamente riconosciuti e introdotti nei mitocondri, i precursori delle proteine che sono sintetizzati nel citosol spesso necessitano di una specifica sequenza aminoacidica che viene riconosciuta attraverso una via di ingresso. Mentre i precursori proteici sono capaci di piegarsi ed aggregarsi, le proteine chaperone del citosol si mantengono in una forma adatta all’ingresso nella membrana. La proteina modificata viene quindi legata da traslocasi delle membrane esterna ed interna (TOM e TIM), che la trasportano attraverso il doppio strato lipidico.
L’introduzione e il riconoscimento della proteina sono generalmente diretti da una sequenza segnale N-terminale o, meno frequentemente, C-terminale, costituita da circa 20-30 residui aminoacidici, i quali vengono tagliati da peptidasi mitocondriali (MPP) sia durante l’ingresso che all'interno della matrice mitocondriale. Il confronto di presequenze conosciute rivela che esse non hanno una struttura primaria comune . In questi casi, però, può essere presente una comune struttura secondaria, oppure residui basici (arginina), idrofobici (alanina, leucina) e polari (serina). Proteine come il citocromo c e le superossido dismutasi, vengono introdotte senza che subiscano grandi modifiche, dal momento che esse contengono gli elementi necessari al riconoscimento nella sequenza primaria. Le regioni N-terminali sono strutturate in modo da ripiegarsi in eliche anfifiliche. Si pensa che questa anfifilicità e le cariche positive dovute ai residui basici, siano i fattori principali che rendono possibile l’ingresso delle proteine, mediato da interazioni elettrostatiche tra le cariche positive presenti sull’elica, e le cariche negative dei recettori della membrana esterna. Di conseguenza, in un evento elettroforetico, la presequenza proteica viene trasportata attraverso la membrana interna da un potenziale di membrana molto ampio (solitamente 150-180 mV).
Manganese superossido dismutasi (MnSOD)
Le superossido dismutasi (SOD) catalizzano la dismutazione del superossido in ossigeno e perossido d’idrogeno; per questo motivo costituiscono una parte importante della difesa antiossidante nella maggior parte delle cellule esposte all’ossigeno. Si conoscono varie famiglie di SOD, e l’attività di ogni famiglia dipende da un cofattore metallico “redox-attivo” come manganese, ferro, rame o nickel. Un singolo cofattore metallico dovrebbe catalizzare l’ossidazione di un singolo elettrone e la riduzione di due anioni superossido separati, per dare, rispettivamente, ossigeno e perossido d’idrogeno.
Queste reazioni si autolimitano, e non richiedono fonti esterne di equivalenti redox. Il meccanismo generale della degradazione catalitica dell’anione superossido è:
La famiglia delle manganese superossido dismutasi (MnSOD) è costituita da dimeri o tetrametri di subunità di circa 21 Kda, contiene importanti omologie di sequenza e ripiegamenti proteici ben conservati attraverso i vari philum. Le forme dimeriche di MnSOD sono tipiche dei batteri, mentre negli eucarioti è più diffusa la forma tetramerica. Nel sito attivo dell’enzima, un singolo atomo di manganese catalizza la reazione di disproporzione del superossido, ed è coordinato, in una geometria trigonale bipiramidale, a tre residui di istidina, uno di aspartato e una molecola di acqua (figura 17).
Nel caso di MnSOD eucariotico (SOD2), il polipeptide viene inizialmente codificato da un gene nucleare come precursore polipeptidico, contenente la sequenza necessaria per il mitocondrio alla sua estremità amminica. Il meccanismo attraverso il quale SOD acquisisce il cofattore manganese è ancora sconosciuto; comunque, la forma di SOD contenente manganese si trova generalmente nella matrice mitocondriale. Forme mutanti, enzimaticamente inattive, di SOD2 di Saccharomyces cerevisiae, mancanti della sequenza adatta ai mitocondri (chiamate SOD2P) si accumulano più velocemente nel citosol che nei mitocondri. L'inattività di queste forme è stata attribuita alla carenza di manganese, e recenti studi hanno ipotizzato che SOD2P devono entrare nel mitocondrio per acquisire effettivamente il loro cofattore. È stato quindi proposto che, non appena il precursore di SOD2 entra nei mitocondri, il manganese viene inserito all’interno del polipeptide, ed il complesso, così ottenuto, viene assemblato nella struttura quaternaria dell’enzima.
MnSOD gioca un ruolo essenziale nella protezione dallo stress ossidativo, e la trasformazione di questo peptide tetramerico nell’enzima attivo contenente manganese, è cruciale per la sopravvivenza. Per esempio, nei topi neonati, la perdita di MnSOD è letale, mentre nei moscerini della frutta adulti, la sua modificazione provoca una drastica riduzione della durata della vita. Al contrario, per il suo ruolo di modulatore negativo dell’apoptosi, l’inibizione di MnSOD nelle cellule cancerose è considerata un bersaglio interessante per future terapie antitumorali.
Il gene MnSOD è indotto dal fattore di necrosi tumorale alfa (TNFα), e garantisce la protezione contro l’apoptosi TNF indotta. Per queste ragioni, anche modeste quantità di questo enzima sono considerate cruciali per la resistenza delle cellule tumorali a stimoli infiammatori, radiazioni ionizzanti e farmaci antitumorali comunemente utilizzati.
Peptidi e peptidomimetici
Sono stati sintetizzati e testati vari peptidi contenenti antiossidanti, in grado di permeare le cellule e di bersagliare i mitocondri; tra questi, i peptidi di tipo SS, che portano legata una 2’,6’-dimetiltirosina (Dmt), ed i peptidomimetici XJB, che rilasciano il gruppo 4-amino-TEMPO (4-AT), un radicale nitrossido stabile.
i mitocondri, che contengono, come caratteristica strutturale comune, l'alternanza di residui aromatici e basici (figura 18).
Figura 18.Struttura di peptidi SS attivi e di controllo.
Prima della scoperta delle loro capacità antiossidanti, erano già stati ampiamente studiati per la loro elevata affinità e selettività per il recettore oppioide µ, e sono risultati sorprendentemente potenti e a lunga durata d’azione.
Le capacità antiossidanti di SS-02 e SS-31 dipendono probabilmente dai loro residui dimetiltirosinici (Dmt) e, più specificamente, i tetrapeptidi SS-31 e SS-02 si sono dimostrati ugualmente efficaci nello smaltimento di H2O2 e nell’inibizione dell’ossidazione di acido
linoleico in vitro. Questo risultato indica che la specifica posizione del residuo di Dmt nella sequenza del peptide antiossidante è ininfluente. I residui basici sono responsabili della localizzazione nella membrana mitocondriale interna, mentre le funzioni Dmt fenoliche di SS-02 e SS-31 sembrano responsabili della riduzione chimica delle specie reattive dell’ossigeno e dei legami perossido. Il tetrapeptide SS-20, nel quale Dmt è sostituito con un residuo di fenilalanina, è stato impiegato come controllo e, in accordo con quanto precedentemente ipotizzato, fu dimostrata la sua incapacità di smaltire ROS.
indurre l’apoptosi nelle cellule trattate. L’apoptosi causata da t-BHP viene stimolata dalla transizione di permeabilità mitocondriale (MPT), che provoca l’aumento della permeabilità delle membrane mitocondriali a piccole molecole di peso molecolare inferiore a 1500 Da. In una serie di studi in vitro, i peptidi SS-02 e SS-31 hanno dimostrato di essere efficaci agenti antiossidanti, riuscendo inoltre ad inibire l’induzione di MTP ed a migliorare parzialmente l’apoptosi delle cellule trattate con t-BHP.
I peptidi XJB ed i derivati peptidomimetici si basano sulla sequenza aminoacidica degli antibiotici attivi sulla membrana, della classe della Gramicidina (GS) (figura 19). Le loro proprietà antiossidanti dipendono dalla presenza del radicale libero stabile, 4-amino-TEMPO (4-AT). Tra i vantaggi di 4-amino-TEMPO c’è la capacità di sfruttare la risonanza di spin elettronico (ESR) per misurare la distribuzione del livello di spin e individuare localmente fenomeni di stress ossidativo nell’ambiente cellulare.
Figura 19. Strutture dell'antibiotico ciclodecapeptidico e dell'XJB-5-131.
Oltre al derivato XJB-131, sono stati studiati altri composti contenenti due differenti emisegmenti GS, Leu-D-Phe-Pro-Val-Orn e D-Phe-Pro-Val-Orn-Leu, insieme al gruppo
Figura 20. Strutture degli analoghi dell'XJB-5-131.
Per incubazione di cellule embrionali di topo sia con XJB-5-125 che con XJB-7-75 si ha diminuzione, in maniera dose-dipendente, dell’eliminazione di fosfatidilserina (PS) indotta da actinomicina D (ActD), raggiungendo una concentrazione di quasi 1000 volte inferiore rispetto a quanto avviene con i composti che non contengono il residuo 4-AT. I derivati emi-GS-TEMPO inibiscono quasi completamente la produzione di superossido in cellule embrionali di topo trattate con ActD. Il frammento peptidico isostero XJB-5-208 non mostra effetti anti-apoptotici, e, in accordo all’ipotesi funzionale, frammenti di controllo con sequenze leggermente alterate come XJB-5-197 e XJB-5-194, non dimostrano capacità protettive. Gli studi successivi di approfondimento hanno utilizzato il peptide isostero XJB-5-131, dal momento che la sostituzione di un legame ammidico con un alchene conduce ad un’estesa biodisponibilità, probabilmente dovuta all’aumentata resistenza contro l’azione della proteasi.
I livelli di assorbimento di XJB-5-125, XJB-5-131 e XJB-5-208, come pure di 4-AT non legato, sono stati definiti, dopo la loro incubazione con cellule embrionali di topo, mediante spettroscopia ESR e ESI-MS. XJB-5-125, XJB-5-131 e XJB-5-208 sono stati velocemente
rinvenuti nei mitocondri, mentre non è trovata traccia di 4-AT. Le caratteristiche di biodistribuzione della molecola non-anti-apoptotica XJB-5-208 hanno suggerito che il semplice accumulo di nitrossidi nei mitocondri non è sufficiente per prevenire l’apoptosi indotta da ActD. Da ciò è stato dedotto che sia il gruppo nitrossido che la sequenza invertita sembrano essenziali per le proprietà antiapoptotiche dei derivati emi-GS-TEMPO.