56
2.1.
C
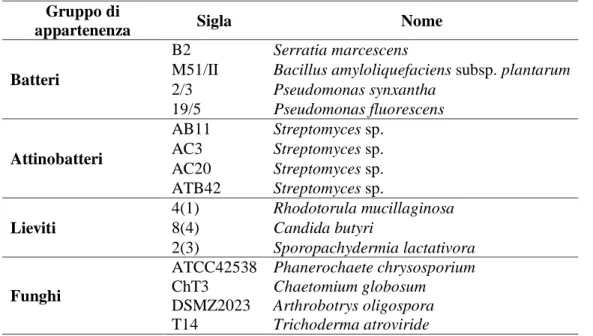
EPPI MICROBICI UTILIZZATINell’ambito della presente tesi, sono stati utilizzati 15 diversi ceppi microbici che includevano varie specie batteriche, tra le quali alcuni attinobatteri, alcuni lieviti e funghi filamentosi, come elencati in Tabella 2.
Tabella 1: Ceppi microbici utilizzati, divisi per gruppi tassonomici Gruppo di
appartenenza Sigla Nome
Batteri
B2 Serratia marcescens
M51/II Bacillus amyloliquefaciens subsp. plantarum
2/3 Pseudomonas synxantha 19/5 Pseudomonas fluorescens Attinobatteri AB11 Streptomyces sp. AC3 Streptomyces sp. AC20 Streptomyces sp. ATB42 Streptomyces sp. Lieviti 4(1) Rhodotorula mucillaginosa 8(4) Candida butyri 2(3) Sporopachydermia lactativora Funghi
ATCC42538 Phanerochaete chrysosporium
ChT3 Chaetomium globosum
DSMZ2023 Arthrobotrys oligospora
T14 Trichoderma atroviride
Tali microrganismi, presenti nella collezione del DBPA (Dipartimento di Biologia delle Piante Agrarie) dell'Università di Pisa, sono stati scongelati e mantenuti su mezzi specifici riportati nel paragrafo 2.3.1.
57
2.2.
L
A PROVA DI COMPOSTAGGIO PRESSOL
’
AZIENDAPresso la Cooperativa Olivicola di Arnasco, in provincia di Savona, a febbraio 2011 è stata allestita la prova di compostaggio delle sanse vergini d'oliva, di varietà Pignola e Taggiasca. Tale prova è stata condotta all’aperto, in un luogo soleggiato, ad un’altezza di circa 250 m sul livello del mare.
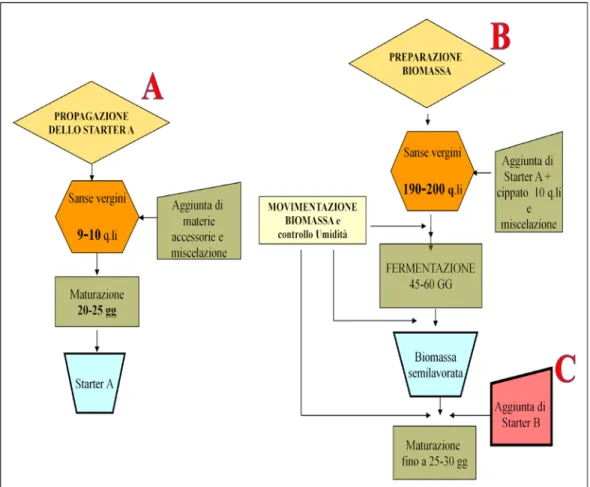
È possibile scomporre l’allestimento di questa sperimentazione in tre fasi principali, come schematizzato in Figura 13.
Figura 1: Schema della prova di compostaggio allestita ad Arnasco
A. Propagazione dello “starter A”
Un volume di 1 L di ciascun ceppo dei microrganismi starter, riportati in Tabella 2 ad eccezione di Streptomyces sp. ATB42, Sporopachydermia
58
una densità di 2,3 e 1,5∙109 UFC/mL per i batteri e gli attinobatteri rispettivamente, e fino alla densità di 6,1 e 3,4∙108 UFC/mL per i lieviti e i funghi filamentosi rispettivamente, ed è stato poi adsorbito in una miscela di sansa esausta e spezzato di favino, in ragione di 16 kg di sansa e 4 kg (20% p/p) di favino (Echeverria et al., 2011b).
Il giorno 01 febbraio 2011 è stata preparata, su un telo di nylon, una matrice pre-inoculo costituita da 10 q.li di sansa vergine seccata all’aria, a cui sono stati aggiunti i microrganismi starter preparati precedentemente, 1‰ di estratto di lievito e una quantità d’acqua tale da portare l’umidità del cumulo al 60%. Il cumulo ottenuto è stato quindi lasciato riposare per circa 4 settimane, in modo da consentire ai microrganismi starter di colonizzare la matrice.
Dato che la temperatura giornaliera in quel periodo era di circa 5°C, non è stato necessario rivoltare la massa per eliminare gli eccessi di temperatura.
Figura 2: Fotografie di alcune fasi della preparazione della matrice pre-inoculo: ceppi microbici costituenti lo “starter A” (A) e aggiunta dell'estratto di lievito alla sansa (B)
B Preparazione e compostaggio della biomassa
Il cumulo, dell’altezza di 1 m e lunghezza 8 m con sezione triangolare, è stato allestito in data 24 febbraio 2011 (tempo 0 gg), con 200 q.li di sansa vergine, lo “starter A” e 10 q.li di cippato, ed è stato rivoltato subito per una maggiore omogeneità.
L’intero processo di compostaggio e finissaggio, compresa la maturazione (dopo raggiungimento della temperatura ambientale), è durato 7 mesi. Durante la
59
maturazione è stata controllata l’umidità della biomassa ed è stata reintegrata con acqua ogni volta che si è mostrata inferiore al 50%. Inoltre, per mezzo di due termometri inseriti nel cumulo, è stato possibile controllare la temperatura della biomassa, che è stata rivoltata meccanicamente ogni volta che ha superato i 50-55°C.
Figura 3: fotografia del cumulo in allestimento (A) e particolare del termometro inserito nel cumulo (B)
C Propagazione e inoculo dello “starter B”
Lo “starter B” era costituito dal microrganismo Trichoderma atroviride (T14) ed è stato preparato utilizzando 10 sacchetti di plastica sterile, ognuno dei quali conteneva 200 g di crusca e 200 mL di acqua. Tali sacchetti sono stati sterilizzati mediante due cicli di autoclave successivi, entrambi a 121°C per 20' e sono stati lasciati raffreddare. Dopo aver fatto sviluppare Trichoderma atroviride T14 su piastre di malto per 7 giorni, queste ultime sono state tagliate, sotto cappa sterile, a cubetti di circa 5 mm3 ciascuno e inserite nei sacchetti contenenti la crusca sterilizzata, nella quantità di 1 piastra a sacchetto. Tali sacchetti, una volta chiusi con cotone cardato, che consente al fungo di respirare, sono stati agitati e lasciati a temperatura ambiente per circa 10 giorni, in modo da favorire la colonizzazione del substrato da parte del fungo.
Il 06 giugno 2011, è stata preparata una matrice pre-inoculo prelevando circa 40 kg di biomassa semilavorata e inoculandola con 4 kg di “starter B”. Tale matrice è stata mescolata ed è stato formato uno strato di circa 5-10 cm, che è stato fatto riposare per circa 20 giorni. Durante tale periodo, per evitare la
60
disidratazione della matrice e la conseguente inibizione di crescita del fungo, la stessa è stata inumidita con acqua.
Il giorno 02 luglio 2011 sono stati prelevati circa 7,50 q.li di biomassa semilavorata e sono stati inoculati con la matrice pre-inoculo. Anche in questo caso la miscela ottenuta è stata lasciata a riposo per circa 20 giorni, in modo da favorire lo sviluppo del fungo.
Figura 4: Fotografia di Trichoderma atroviride inoculato su crusca (A) e fotografia della matrice pre-inoculo dello “starter B” (B)
Infine, il 19 luglio 2011 (tempo 145 gg dall’inizio del processo) è stato effettuato l’inoculo dello “starter B” nell’intero cumulo, circa 150 q.li, ed è stato eseguito un rivoltamento della biomassa in maturazione. Tale biomassa è stata quindi lasciata riposare per circa 2 mesi, ed è stata rivoltata ogni volta che la temperatura del cumulo superava i 40°C.
2.2.1. Campionamenti
Nell'arco della sperimentazione, per valutare l'evoluzione microbica durante il processo di compostaggio della sansa, sono stati prelevati diversi campioni a tempo 0 gg, 35 gg, 100 gg, 145 gg e 200 gg. Inoltre è stato prelevato un campione dello “starter B”.
Durante l’ultimo rivoltamento della biomassa in compostaggio, una parte dell’argilla presente sul suolo è stata mescolata al compost. Pertanto durante il campionamento successivo (a 200 gg) è stato prelevato, e successivamente analizzato, anche un campione di argilla presente nel cumulo, in modo da poter
61
verificare la presenza nel compost dei microrganismi presenti nell'argilla ed eventualmente escluderli dall’analisi del compost.
Ciascun campionamento è stato eseguito prelevando il campione da zone diverse della matrice in analisi, sia in aree più superficiali che più interne del cumulo. Le aliquote prelevate sono state miscelate in sacchetti sterili, in modo da avere un campione il più omogeneo possibile.
Tale campione è stato utilizzato nell'arco delle 48 ore per l'analisi microbiologica, mentre un'aliquota è stata conservata a -20°C e utilizzata successivamente per le analisi molecolari.
Figura 5: Fotografie di un campionamento della matrice in compostaggio
62
2.3.
A
NALISI MICROBIOLOGICHE COLTURA-DIPENDENTI
2.3.1. Mezzi di coltura
Al fine del mantenimento dei microrganismi starter sono stati preparati diversi mezzi di coltura specifici per i vari microrganismi, in particolare il “Nutrient agar” per la crescita dei batteri, il “Waksman agar” per gli attinobatteri, il “Glucose yeast extract agar” per i lieviti e i funghi filamentosi e infine il “Malt extract agar” e il “Potato dextrose agar, PDA” per Trichoderma spp.
Nell’ambito dell’analisi microbiologica dei campioni di sansa in compostaggio, tali mezzi sono stati addizionati con antibiotici in modo da aumentare la selettività dei mezzi stessi. Per la ricerca di Trichoderma spp. è stato utilizzato il “Trichoderma-selective medium”.
2.3.1.1. Nutrient agar
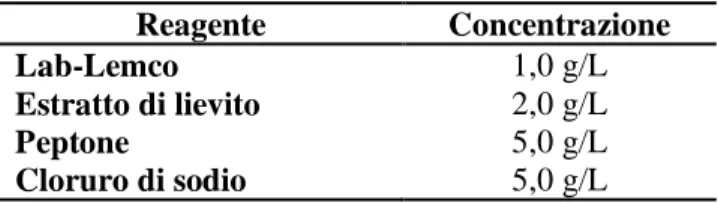
Il mezzo è stato preparato utilizzando 13 g/L di “nutrient broth” della ditta Oxoid, la cui composizione è riportata in Tabella 3. A tale mezzo sono stati aggiunti 20 g/L di “agar bacteriological” della Oxoid e acqua deionizzata per arrivare a volume; infine il pH è stato aggiustato a 7.0. Il mezzo è stato sterilizzato in autoclave per 15 minuti alla temperatura di 121°C e alla pressione di 1atm.
Tabella 2: Composizione “Nutrient broth” (Oxoid) Reagente Concentrazione
Lab-Lemco 1,0 g/L
Estratto di lievito 2,0 g/L
Peptone 5,0 g/L
Cloruro di sodio 5,0 g/L
Successivamente, sono stati aggiunti 250000 UI/L di nistatina e 100 mg/L di cycloheximide che impediscono l'eventuale crescita di eumiceti nel mezzo di coltura.
Tale substrato è stato utilizzato anche per verificare la presenza di Bacillus
63
e avente una doppia resistenza antibiotica. Pertanto nel “Nutrient agar” sono stati aggiunti i due antibiotici battericidi: 100 mg/L di streptomicina e 75 mg/L di rifampicina.
2.3.1.2. Waksman agar
Il “Waksman agar” è stato preparato miscelando diversi reagenti, e la sua composizione è descritta in Tabella 4. Dopo aver aggiustato il pH a 7.0, si è proceduto a sterilizzare tale soluzione in autoclave per 20 minuti alla temperatura di 121°C e alla pressione di 1atm.
Anche in questo caso per impedire l'eventuale crescita di lieviti e funghi sono stati aggiunti 250000 UI/L di nistatina e 100 mg/L di cycloheximide.
Tabella 3: Composizione “Waksman agar”
Reagente Concentrazione
Glucosio 10,0 g/L
Cloruro di sodio 5,0 g/L
Bacteriological Peptone (Oxoid) 5,0 g/L Estratto di carne (Difco) 3,0 g/L Agar bacteriological (Oxoid) 20,0 g/L Acqua deionizzata
2.3.1.3. Glucose yeast extract agar
Per preparare questo mezzo di coltura sono stati utilizzati 37 g/L di “Glucose yeast extrat agar” della Oxoid, la cui composizione è descritta in Tabella 5. A tale estratto è stata aggiunta una quantità di acqua deionizzata tale da arrivare al volume desiderato, ed è stato aggiustato il pH a 7.6. Successivamente il mezzo di coltura è stato sterilizzato in autoclave per 10 minuti alla temperatura di 115°C e alla pressione di 1atm.
Tabella 4: Composizione “Glucose yeast extract agar” (Oxoid) Reagente Concentrazione
Yeast extract 5,0 g/L
Glucose 20,0 g/L
64
Una volta che il mezzo è raffreddato sono stati aggiunti 100 mg/L di oxytetracyclina; questo antibiotico ha lo scopo d'impedire l'eventuale crescita batterica, poiché la presenza di colonie batteriche nel mezzo di coltura potrebbe inibire lo sviluppo degli eumiceti.
2.3.1.4. Malt extract agar
Per preparare questo mezzo di coltura sono stati utilizzati 50 g/L di “Malt extrat agar” della ditta Difco, la cui composizione è descritta in Tabella 6. A tale estratto è stata aggiunta una quantità di acqua deionizzata tale da arrivare al volume desiderato, ed è stato aggiustato il pH a 7.0. Successivamente il mezzo è stato sterilizzato in autoclave per 10 minuti alla temperatura di 115°C e alla pressione di 1atm.
Tabella 5: Composizione “Malt extract agar” (Difco) Reagente Concentrazione
Malt extract 30,0 g/L
Mycological peptone 5,0 g/L Agar bacteriological 15,0 g/L
2.3.1.5. Potato dextrose agar (PDA)
Questo mezzo è stato ottenuto aggiungendo acqua deionizzata al “Potato dextrose agar” della Oxoid, la cui composizione è riportata in Tabella 7. Il mezzo è stato autoclavato alla temperatura di 121°C per 15 minuti e alla pressione di 1 atm.
Tabella 6: Composizione “Potato dextrose agar” (Oxoid) Reagente Concentrazione Estratto di patata 4,0 g/L
Destrosio 20,0 g/L
65
2.3.1.6. Trichoderma-selective medium
Il “Trichoderma-selective medium” è stato ottenuto miscelando i reagenti elencati in Tabella 8. Una volta aggiustato il pH a 7.6, tale mezzo è stato sterilizzato in autoclave per 15 minuti alla temperatura di 121°C e alla pressione di 1atm.
Successivamente sono stati aggiunti 100 mg/L di chloramphenicolo e 100 mg/L di streptomicina, che hanno un'azione battericida.
Tabella 7: Composizione “Trichoderma-selctive medium”
Reagente Concentrazione
Solfato di magnesio 7H2O 0,2 g/L
Mono-idrogeno fosfato di potassio 0,9 g/L
Nitrato di ammonio 1,0 g/L
Cloruro di potassio 0,15 g/L
Rose Bengal 0,15 g/L
Glucosio 3,0 g/L
Agar bacteriological (Oxoid) 20,0 g/L Acqua deionizzata
2.3.2. Analisi microbiologica
Una quantità di 10 g di campione è stata diluita in 90 mL di soluzione fisiologica NaCl 9∙1000, contenente 2 gocce di Tween 80. Quest'ultimo composto è un agente emulsionante che favorisce il rilascio dei microrganismi dalle particelle di terreno.
La sospensione ottenuta, che corrisponde alla diluizione 10-1 del campione, è stata mantenuta in agitazione per 15 minuti. Da tale sospensione è stata prelevata un'aliquota di 1 mL ed è stata trasferita in una provetta contenente 9 mL di soluzione fisiologica NaCl 9∙1000; successivamente questa diluizione è stata agitata su vortex in modo da assicurare un’omogenea distribuzione dei microrganismi presenti, e mediante trasferimento sequenziale di 1 mL, sono state ottenute le successive diluizioni del campione fino alla diluizione 10-8.
Ciascuna diluizione del campione è stata inoculata su piastre Petri contenenti i diversi terreni di coltura, in quantità di 100 µL per piastra, ed è stata distribuita uniformemente sulla superficie del terreno mediante semina per
66
spatolamento. Ai fini di ottenere un risultato statisticamente valido, sono state allestite 3 piastre per ciascuna diluizione.
Prima di inoculare le piastre contenenti il terreno selettivo per Bacillus
amyloliquefaciens subsp. plantarum le diverse sospensioni sono state mantenute a
80°C per 10 minuti, in modo da eliminare la maggior parte dei batteri non termo-tolleranti.
Le piastre sono state incubate in termostato alla temperatura di 28°C in presenza di ossigeno per un periodo di tempo variabile dipendente dalla velocità di crescita dei microrganismi ricercati. In particolare, le piastre contenenti “Nutrient agar” sono state mantenute in incubazione per 2 giorni, quelle con “Waksman agar” per 5 giorni e le piastre per gli eumiceti per 4 giorni.
Le colonie batteriche e le colonie fungine che si sono formate, sono state contate nelle diluizioni in cui il numero di colonie sviluppate era statisticamente valido ed era possibile distinguere una colonia dall'altra. Il risultato è stato espresso in UFC/mL (Unità Formanti Colonia per mL), ricavato mediante la seguente equazione:
𝑈𝑈𝑈𝑈𝑈𝑈
𝑚𝑚𝑚𝑚 = 𝑛𝑛° 𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑐𝑛𝑛𝑐𝑐𝑐𝑐 𝑝𝑝𝑐𝑐𝑝𝑝 𝑝𝑝𝑐𝑐𝑝𝑝𝑝𝑝𝑝𝑝𝑝𝑝𝑝𝑝 × 10𝑛𝑛 × 10
dove n rappresenta la diluizione seminata sulle piastre su cui è stata effettuata la conta.
La ricerca di Trichoderma spp. sul mezzo selettivo per questo fungo è stata effettuata solo sullo “starter B” e sui campioni prelevati successivamente all'inoculo dello stesso.
2.3.3. Isolamento di attinobatteri
Al fine di isolare ceppi di attinobatteri, sviluppati sulle piastre di “Waksman agar”, in coltura pura, alcune colonie scelte in modo casuale all’interno di ciascuna morfologia sono state prelevate, strisciate sul medesimo mezzo e incubate in termostato a 28°C per 5 giorni, ripetendo tale procedimento per 4 volte.
Da ciascuna delle colture purificate è stato prelevato un inoculo con un’ansa sterile e stemperato in tubi contenenti 4 mL di mezzo non agarizzato. I tubi sono
67
stati incubati in aerobiosi a 28°C per 48 ore. Dopo il periodo di incubazione, 800 µL di ciascuna sospensione sono stati inseriti in microtubi contenenti 500 µL di gliecerolo al 60% e congelati a -80°C.
68
2.4.
A
NALISI MOLECOLARI COLTURA-INDIPENDENTI
La tecnica molecolare utilizzata per l’analisi dei campioni di sansa d’oliva in compostaggio è stata la PCR-DGGE. Mediante tale tecnica è possibile monitorare l’evoluzione dei microrganismi che si sviluppano nella matrice durante il processo e valutare la presenza dei microrganismi inoculati.
2.4.1. Estrazione del DNA da coltura pura
Per estrarre il DNA dalle colture pure dei microrganismi inoculati nelle sanse in compostaggio è stato utilizzato il kit “MasterPureTM Yeast DNA Purification” dell'Epicentre.
Per ogni ceppo sono state preparate delle colture liquide: 4 mL di mezzo liquido sono stati inoculati con una colonia prelevata da coltura pura, e sono stati mantenuti in agitazione per un giorno alla temperatura di 28°C.
È stato prelevato un volume di 1 mL dalle colture ed è stato centrifugato a 10000 rpm per 10 minuti a temperatura ambiente. Successivamente è stato eliminato il surnatante ed il pellet è stato risospeso aggiungendo 300 µL di Yeast Cell Lysis Solution e agitando sul vortex. Tale sospensione è stata incubata prima a 65°C per 15 minuti e poi a 4°C per 5 minuti. In seguito sono stati aggiunti 150 µL di MPC Protein Precipitation Reagent e la sospensione è stata agitata su vortex per 10 secondi; è stata fatta quindi una centrifuga a 10000 rpm per 10 minuti a temperatura ambiente. Il surnatante è stato prelevato e trasferito in una nuova eppendorf, ad esso è stato aggiunto un ugual volume di isopropanolo (Sigma) ed è stato incubato a temperatura ambiente per 5 minuti. Successivamente il campione è stato centrifugato a 10000 rpm per 10 minuti e il surnatante è stato eliminato. Il pellet ottenuto è stato lavato con 1 mL di etanolo al 70% ed è stato nuovamente centrifugato a 10000 rpm per 5 minuti. Dopo aver eliminato l'etanolo, il pellet è stato lasciato asciugare all'aria ed è stato risospeso con 50 µL di acqua DEPC (EuroClone). Il DNA estratto è stato mantenuto in frigo a 4°C.
69
2.4.2. Estrazione del DNA dalle sanse in compostaggio
Le aliquote dei campioni conservate in congelatore a -20°C sono state utilizzate per estrarre il DNA presente. Il DNA di ciascun campione è stato estratto in 3 replicati.
Per l'estrazione del DNA dalle matrici in esame è stato utilizzato un kit commerciale: il “PowerSoil™ DNA Isolation Kit Sample” della MO-BIO
Laboratories.
Una quantità di 250 mg di campione è stata prelevata e posta in appositi tubi (PowerBead Tubes) contenenti un buffer che favorisce la dispersione delle particelle di terreno, la dissoluzione degli acidi umici e protegge gli acidi nucleici dalla degradazione. Dopo aver agitato sul vortex questa sospensione, sono stati aggiunti 60 µL di soluzione C1, che contiene l’SDS e altri agenti che favoriscono la lisi cellulare; i tubi sono stati posti su un supporto per il vortex e agitati orizzontalmente per 15 minuti alla velocità massima.
Successivamente, la sospensione è stata centrifugata a 10000 rpm per 30 secondi a temperatura ambiente ed è stato recuperato il surnatante, a cui sono stati aggiunti 250 µL di soluzione C2. Tale reagente favorisce la precipitazione dei materiali organici e non organici ad eccezione del DNA. La miscela ottenuta è stata quindi agitata su vortex per 5 secondi, incubata per 5 minuti a 4°C e nuovamente centrifugata a temperatura ambiente per 1 minuto a 10000 rpm. Il surnatante è stato recuperato e sono stati ripetuti gli stessi passaggi, aggiungendo, al posto della soluzione C2, 200 µL di soluzione C3.
Al fine di eliminare ogni traccia di eventuali residui contaminanti, al surnatante ottenuto sono stati aggiunti 1,2 mL di soluzione C4, caratterizzata da un'alta concentrazione salina. Infatti, in presenza di alte concentrazioni di sali, il DNA, a differenza degli altri materiali organici e non organici, si lega alla silice. Pertanto 675 µL della miscela ottenuta sono stati caricati su un tubo contenente un filtro in silice (Spin Filter) e centrifugati a 10000 rpm per 1 minuto a temperatura ambiente; il liquido è stato scaricato e l'operazione è stata ripetuta per 2 volte, in modo da centrifugare tutto il surnatante rimanente.
A questo punto il DNA estratto era legato alla membrana del filtro. Sono stati aggiunti 500 µL di soluzione C5, a base di etanolo, che servivano a
70
rimuovere i residui di sale, di acidi umici e di altri contaminanti. La soluzione é stata centrifugata a temperatura ambiente per 30 secondi a 10000 rpm, ed il liquido é stato rimosso. Una seconda centrifuga da 1 minuto a 10000 rpm a temperatura ambiente ha rimosso l'etanolo.
Il filtro contenente il DNA é stato trasferito in un nuovo tubo dove sono stati aggiunti 100 µL di soluzione C6; tale reagente è un buffer di eluizione sterile contenente Tris 10 mM, che favorisce il rilascio del DNA dalla membrana in silicio. È stata fatta quindi una centrifuga a 10000 rpm per 30 secondi a temperatura ambiente della miscela ed il surnatante ottenuto è stato conservato in congelatore a -20° C.
2.4.3. Amplificazione della regione V3-V5 dell’rDNA 16S batterico
La regione batterica V3-V5 dell’rDNA 16S è una regione iper-variabile
presente all'interno di porzioni genomiche altamente conservate, che permette di differenziare i microrganismi a livello di taxa.
Tale regione è stata amplificata utilizzando lo strumento iCycleriQ Multicolor Real-Time PCR Detection System della BIORAD. I primers impiegati nell'amplificazione sono il 341F e il 907R, la cui sequenza è mostrata in Tabella 9. In rosso è riportata la sequenza della coda GC legata all'estremità 5' del primer forward, che impedisce la completa denaturazione dell'amplicone durante la DGGE successiva. Il frammento ottenuto in seguito all'amplificazione ha una lunghezza di 560 bp.
Tabella 8: Sequenza dei primers che amplificano per la regione V3-V5 dell’rDNA 16S
Primer Sequenza
341F-GC CGCCCGCCGCGCCCCGCGCCCGTCCCGCCGCCCCCGC
CCGCCTACGGGAGGCAGCAG
907R CCGTCAATTCCTTTRAGTTT
La reazione di amplificazione è stata condotta in un volume finale di 50 µL, contenente, 10 µL di buffer Phusion HF 5X che include MgCl2 7,5 mM
(Finnzymes), 1,5 µL di DMSO 100% (Finnzymes), una concentrazione 10 µM di ciascun primer, 0,2 mM di ciascun dNTP (EuroClone), 1 U di Taq Phusion DNA polymerase (Finnzymes) e 1 µL di DNA campione diluito 100 volte.
71
Il programma di amplificazione che è stato utilizzato è mostrato in Figura 18.
Figura 6: Programma di amplificazione della regione V3-V5 dell’rDNA 16S
La corretta amplificazione del frammento è stata poi verificata mediante elettroforesi su gel d'agarosio.
2.4.3.1. Elettroforesi su gel d'agarosio
L'elettroforesi è stata condotta su gel di agarosio all' 1,5%, contenente 0.5 µg/mL di bromuro di etidio; la corsa ha avuto luogo in tampone Tris-Borato-EDTA (TBE) 1X, costituito da Tris 0,89 M, Tris-Borato-EDTA Na2·2H2O 0,20 M e da acido
borico 0,89 M.
Ad ogni corsa sono stati caricati 5 µL di ciascun amplificato, a cui sono stati aggiunti 2,5 µL di tampone di caricamento 3X (15% di Ficoll, 66 mM di EDTA, 19,8 mM di Tris-HCL, 0,102% di SDS e 0,09% di blu di bromofenolo) e 5 µL di
marker 100 pb DNA Ladder, composto da 70 µL di acqua DEPC, 20 µL di
tampone di caricamento 6X e 10 µL di DNA Ladder.
2.4.4. Amplificazione della regione D1-D2 dell’rDNA 26S
eucariotico
Negli eucarioti l’rDNA 26S è un gene che codifica per una sub-unità ribosomiale, e quindi è altamente conservato in tutte le specie viventi. La regione D1-D2 dell’rDNA 26S è invece una regione variabile, che permette di studiare e
distinguere microrganismi tassonomicamente diversi.
Denaturazione •30" a 98°C Amplificazione •10" a 98°C •10" a 52°C •18" a 72°C Estensione finale •10' a 72°C 35 c icl i
72
Tale regione è stata amplificata utilizzando lo strumento iCycleriQ Multicolor Real-Time PCR Detection System della BIORAD. I primers impiegati nell'amplificazione sono NL1-GC e LS2, la cui sequenza è mostrata in Tabella 10. Anche in questo caso, in rosso è riportata la sequenza della coda GC legata all'estremità 5' del primer forward. Il frammento ottenuto in seguito all'amplificazione ha una lunghezza di 250 pb.
Tabella 9: Sequenza dei primers che amplificano per la regione D1-D2 dell’rDNA 26S
Primer Sequenza
NL1-GC GCGGGCCGCGCGACCGCCGGGACGCGCGAGCCGGCG
GCGGGCCATATCAATAAGCGGAGGAAAAG
LS2 ATTCCCAAACAACTCGACTC
La reazione di amplificazione è stata condotta in un volume finale di 50 µL, contenente, 10 µL di buffer Phusion HF 5X che include MgCl2 7,5 mM
(Finnzymes), 1,5 µL di DMSO 100% (Finnzymes), una concentrazione 10 µM di ciascun primer, 0,2 mM di ciascun dNTP (EuroClone), 1 U di Taq Phusion DNA polymerase (Finnzymes) e 2 µL di DNA campione diluito 10 volte.
Il programma di amplificazione utilizzato è mostrato in Figura 19.
Figura 7: Programma di amplificazione della regione D1-D2 dell’rDNA 26S
La corretta amplificazione del frammento è stata verificata mediante elettroforesi su gel d'agarosio, come descritto nel paragrafo 2.4.3.1.
Denaturazione •30" a 98°C Amplificazione •10" a 98°C •10" a 48°C •15" a 72°C Estensione finale •10' a 72°C 35 c icl i
73
2.4.5. Elettroforesi su gel di poliacrilammide con gradiente di
denaturanti chimici (DGGE)
Gli amplificati batterici ed eucariotici sono stati analizzati mediante elettroforesi su gel di poliacrilammide con gradiente di denaturanti chimici, parallelo al campo elettrico, utilizzando lo strumento DCodeTM Universal Mutation Detection System della BIORAD.
È stato utilizzato un gel di poliacrilammide all'8% p/v, utilizzando 20 mL di soluzione di acrilammide e bisacrilammide in rapporto 37,5:1, a cui sono stati aggiunti 2 mL di Tris-Acetato-EDTA (TAE) 50X e le soluzioni denaturanti, preparate considerando come 0% la soluzione senza denaturanti, e come 100% una soluzione contenente urea 7 M (42% g/v) e formammide al 40% v/v. Infine è stata aggiunta acqua deionizzata per arrivare al volume di 100 mL. Tali soluzioni sono state degassate mediante una pompa ad acqua per 30 minuti, in beute caudate; dopo la degassazione sono state distribuite in tubi da 15 mL e conservate in congelatore a -20°C.
Il gel di poliacrilammide utilizzato ha le dimensioni di 16 cm2 e lo spessore di 1 mm; il volume necessario per la sua preparazione è quindi di 25,6 mL ed è stato realizzato utilizzando 15 mL di ciascuna soluzione denaturante. La reazione di polimerizzazione è stata fatta partire aggiungendo ad ogni soluzione 80 µL di ammonio persolfato al 10% e 29 µL di TEMED.
Il gel è stato quindi caricato con 20 µL di amplificato a cui sono stati aggiunti 20 µL di tampone di caricamento 2X, costituito da glicerolo al 70%, xylene cyanol allo 0,05% e blu di bromofenolo allo 0,05%. Il marcatore è stato realizzato con 20 µL di tampone di caricamento 2X, a cui sono stati aggiunti, per i procarioti 4 µL degli amplificati di ciascun ceppo in coltura pura e per gli eucarioti 3 µL degli amplificati di ciascun ceppo in coltura pura (vedi Tabella 1).
La corsa è avvenuta in tampone Tris-Acetato-EDTA (TAE) 50X, costituito da Tris 2 M, EDTA Na2·2H2O 0,10 M e da acido acetico glaciale 1 M, ed è stata
condotta a 90 V per 16 ore, alla temperatura di 60°C, utilizzando un gradiente 30%-65% per i procarioti e un gradiente 20%-75% per gli eucarioti.
Dopo la corsa, i gel sono stati colorati per 30 minuti in 500 mL di tampone TAE 1X contente 50 µL di SYBR Green gold (Molecular Probes) e i profili
74
ottenuti sono stati acquisiti mediante il programma “LISCAP per ImageMaster VDS System”.
2.4.6. Analisi dei profili DGGE
I profili ottenuti mediante la tecnica PCR-DGGE sono stati analizzati mediante il programma ImageMaster 1D Elite v3.00 (Pharmacia Biotech). Le
lanes sono state normalizzate mediante sottrazione del background, e le immagini
del gel sono state allineate per dare una curva densitometrica. I frammenti sono stati assegnati e abbinati automaticamente e poi controllati manualmente. La posizione dei frammenti è stata convertita in valori Rf compresi tra 0 e 1 e la similarità dei profili è stata calcolata utilizzando il coefficiente Dice per il profilo DGGE di ciascun campione. I coefficienti di similarità calcolati sono stati utilizzati per costruire un dendrogramma, usando il metodo di clustering UPGMA (Unweighted Pair Group Method Using Arithmetic Average).
I dati relativi ai frammenti DGGE, ovvero numero dei frammenti e intensità dei picchi ottenuti per ciascun campione, sono stati utilizzati per stimare quattro diversi indici, trattando ciascun frammento come un’unità tassonomica operazionale (OTU). La Richness (S) esprime il numero di OTUs presenti in un campione ed è stata determinata dal numero di frammenti rilevati in quel campione. L’indice si Shannon-Weaver di diversità generale (H) e l’indice di
Simpson di dominanza (D) sono stati calcolati dalle seguenti equazioni: 𝐻𝐻 =
− ∑(𝑃𝑃𝑐𝑐 ∙ ln 𝑃𝑃𝑐𝑐) e 𝐷𝐷 = ∑ 𝑃𝑃𝑐𝑐2 , rispettivamente, dove l’importanza relativa di
ciascuna OTU, 𝑃𝑃𝑐𝑐 = 𝑛𝑛𝑐𝑐/𝑁𝑁; ni è l’intensità di ciascun picco e N è la somma di tutte
le intensità dei picchi di un campione. La Evenness (E) permette di individuare le