Tecniche sperimentali
3.1 Introduzione
In questo capitolo verranno illustrate tutte le tecniche utilizzate in laboratorio per l’estrazione, la semina e la coltivazione delle cellule di Purkinje e delle cellule mesencefaliche facendo alcuni richiami alle tecniche base di coltivazione cellulare.
È importante sottolineare che l’estrazione e la semina delle cellule sono state effettuate al Dipartimento di Farmacologia dell’Università di Medicina e Chirurgia di Pisa dalla Dott.ssa Francesca Vaglini e dai suoi collaboratori. Le cellule poi sono state coltivate e fotografate nel Laboratorio di Fisiologia Clinica del Prof. Claudio Domenici, dove è stata realizzata la parte sperimentale di questa tesi.
Nella prima parte del capitolo verranno illustrati i tipi di neuroni analizzati, le loro risposte in vitro e le tecniche utilizzate per la dissociazione e la coltivazione di queste cellule.
Successivamente verranno riportate le procedure di immunocitochimica e fluorescenza utilizzate per colorare rispettivamente le cellule mesencefaliche e le cellule di Purkinje ed infine verranno descritte la strumentazione e la metodologia con cui sono state realizzate le fotografie delle cellule tramite microscopio ottico.
3.2 Tipi di neuroni utilizzati
Le cellule utilizzate in questa tesi sono neuroni primari di topo. Le cellule mesencefaliche sono cellule embrionali estratte al tredicesimo giorno di gestazione (E13) da esemplari di topo appartenenti al ceppo SVI, mentre le cellule di Purkinje sono estratte da topi appena nati (P0, post-natal zero).
I neuroni sono stati prelevati dal topo in quanto esso è il principale organismo modello per la sperimentazione nella genetica dei mammiferi ed il surrogato dell’uomo studiato più intensivamente, grazie soprattutto alla consistenza genetica e al modesto costo [59].
Il topo è separato dagli esseri umani soltanto da circa 100 milioni di anni di evoluzione; il suo genoma ha le stesse dimensioni del nostro ed esiste una corrispondenza quasi uno ad uno tra fra geni umani e di topo. Le nostre proteine sono identiche all’80–90% nella loro sequenza amminoacidica e grossi blocchi di somiglianza di sequenza nucleotidica sono evidenti quando si confrontano le sequenze regolatrici di DNA.
Inoltre, i biologi dello sviluppo hanno trovato nuove strade per accedere all’embrione precoce di topo senza ucciderlo e per generare topi su misura con mutazioni in qualunque gene scelto. Le colture nervose dai mutanti neurologici forniscono la preziosa opportunità di analizzare gli effetti di specifici geni sullo sviluppo neurale e sulla funzione.
3.3 Cellule primarie in coltura
I neuroni analizzati in questa tesi, come riportato sopra, appartengono a colture primarie. Le colture primarie si chiamano così perché sono preparate direttamente da un qualsiasi animale. Esse si dicono anche “colture a termine”, per indicare il fatto che le cellule sono in grado di compiere un numero finito di divisioni cellulari in vitro, dopo le quali vanno incontro a degenerazione e morte. Tale fenomeno avviene indipendentemente dalla presenza di metaboliti appropriati per la crescita e si indica come senescenza. Nel ciclo di vita di tali cellule sono state identificate tre fasi (Fig. 3.1):
1. Fase iniziale di adattamento;
2. Fase logaritmica, in cui il numero di cellule cresce in maniera esponenziale (in presenza delle appropriate condizioni);
3. Senescenza e morte.
La senescenza cellulare è un fenomeno controllato a livello genetico e sembra sia dovuto a una riduzione dell’attività dell’enzima teleomerasi, la cui funzione è quella di riparare i teleomeri, brevi sequenze di DNA che si trovano all’estremità del cromosoma. I teleomeri proteggono i cromosomi durante la divisione cellulare e ad ogni duplicazione si accorciano di una certa quantità e, quando la loro lunghezza non è più in grado di proteggere la cellula, essa inizia a riprodursi in modo scorretto generando l’invecchiamento. In genere il numero di “cicli” che una cellula è in grado di effettuare è inversamente proporzionale all’età dell’animale da cui sono prelevati i tessuti; non a caso le cellule di derivazione embrionale sono quelle che possono essere mantenute in coltura in vitro più a lungo.
Le colture primarie hanno il vantaggio di simulare più fedelmente la situazione in vivo, tuttavia esse hanno un limitato potenziale di crescita e un tempo di sopravvivenza piuttosto basso. Un ulteriore svantaggio rispetto alle linee cellulari continue è la difficoltà della procedura di isolamento.
Gli step principali della realizzazione di colture primarie sono i seguenti (Fig 3.2):
1. Isolamento del tessuto 2. Dissociazione
Figura 3.2 Fasi della preparazione di colture primarie
Il tessuto deve essere isolato e mantenuto a 4° per 72 ore.
Dopo questo intervallo di tempo si può passare alla dissociazione che consiste nella separazione delle cellule che costituiscono il tessuto. Esistono diverse tecniche di dissociazione che variano a seconda del tipo di cellula estratto; le tecniche più comuni sono la dissociazione meccanica e la dissociazione enzimatica.
Una volta separate le cellule queste devono essere seminate in apposite piastre contenenti il terreno adatto al tipo di cellula specifico arricchito con fattori di crescita, citochine e metaboliti necessari alla crescita.
Le cellule che in vivo fanno parte di tessuti solidi crescono in vitro aderendo alla superficie delle piastre di coltura. L'adesione alle piastre è una condizione necessaria per la crescita in vitro e si tratta di un fenomeno attivo che, per avvenire, richiede l'interazione di recettori di membrana, le integrine, con le proteine adesive, quali la fibronectina, adsorbite sulla superficie della piastra di coltura.
Le cellule in coltura crescono in apposite fiasche, piastre o provette di plastica contenenti un terreno appropriato, all’interno di un apparecchio denominato incubatore (Fig. 3.3) in cui l’atmosfera è mantenuta a concentrazioni fisse di CO2, a un’umidità del 90% e ad una temperatura di 37°C [60, 61, 62].
Figura 3.3 Incubatore
3.4 Neuroni primari in coltura
Le colture di cellule primarie neuronali derivano spesso da animali embrionali. L’utilizzo di cellule embrionali presenta infatti numerosi vantaggi:
• i neuroni sono meno suscettibili al danno durante la dissociazione quando i loro soma sono ancora piccoli e quando non hanno ancora formato grandi alberi assonici e dendritici e non sono altamente innervati;
• i neuroni sono meno dipendenti dalle cellule target per il supporto trofico al primo stage del loro sviluppo;
• tra i neuroni primari, quelli embrionali sono quelli che possono essere mantenuti in coltura in vitro più a lungo
Quando le cellule dal cervello embrionico vengono dissociate e messe in coltura, i neuroni che hanno completato la divisione in situ estendono i processi e diventano elettricamente attivi; ma se il tessuto è rimosso durante la
neurogenesi, è raro osservare cellule che si dividano in coltura e acquisiscano successivamente il fenotipo neurale.
I neuroni nelle colture primarie non si dividono ma, in condizioni favorevoli, possono essere mantenuti in coltura per diverse settimane, durante le quali sviluppano assoni e dendriti, stabiliscono sinapsi, formando una densa rete, ed esprimono i recettori e i canali ionici caratteristici delle corrispondenti cellule in situ.
Inoltre, i neuroni primari mantengono le loro identità individuali, probabilmente perché, generalmente, quando vengono messi in coltura, sono post-mitotici e hanno compiuto la loro differenziazione. Durante i primi giorni di coltura, prima che la rete diventi troppo complicata, singoli neuroni possono essere osservati nella loro interezza e questo permette la diretta osservazione della crescita degli assoni e del loro modo di diramarsi [63, 64]. Al fine di poter studiare in vitro la crescita dell’albero dendritico e dell’assone di un singolo neurone e la formazione di interconnessioni tra neuroni diversi, è molto importante quindi scegliere, a seconda del particolare tipo neuronale a cui si è interessati, a quale fase dello sviluppo estrarre il tessuto.
Per quanto riguarda i neuroni mesencefalici è stato visto che lo sviluppo del mesencefalo inizia allo stadio E10 e termina allo stadio E16. Solamente dallo stadio E13 è possibile però estrarre il tessuto senza danneggiarlo mentre nei giorni successivi l’estrazione diventa più semplice ma la vitalità delle cellule decresce. Lo stadio a cui è opportuno dissociare il mesencefalo per preparare una coltura di neuroni primari è quindi l’E13.
Il cervelletto completa il suo sviluppo molto più tardivamente rispetto al mesencefalo; solamente dopo la nascita infatti si formano tutti e tre gli starti da cui è composto ed esso comincia ad acquisire quella forma foliata che lo caratterizza. Per questo motivo le cellule di Purkinje non vengono estratte da embrioni me da animali post-natali [65, 66].
3.5 Colture di cellule mesencefaliche
3.5.1 Preparazione delle colture
La preparazione delle colture mesencefaliche è una procedura molto delicata e relativamente complessa, che ha richiesto differenti passaggi [67].
I cervelli prelevati dall’embrione sono stati messi in una soluzione bilanciata osmoticamente al pH fisiologico. Una soluzione di sale bilanciata (phosphate buffered saline: PBS) consiste in una semplice miscela di sali quali Na, K, Mg, Ca, Cl, PO4 e HCO3 alle concentrazioni approssimativamente del fluido extracellulare, insieme con glucosio.
Il mesencefalo è stato sezionato sotto controllo microscopico e piazzato in un mezzo nutriente avente la seguente composizione:
• 50% MEM • 50% F12
• 10% di Nu-serum
• 4,5 mg/ml di glucosio al 30% • 2 mM di glutamina.
Dopo la dissezione, il tessuto ottenuto è stato disperso in una sospensione di singole cellule. La dissociazione è stata realizzata sia con trattamento enzimatico che meccanico (triturazione e filtrazione). Il tessuto è stato meccanicamente disperso, usando delle pipette Pasteur, centrifugato a 100 g per 3 minuti e risospeso nel mezzo di coltura.
Le cellule sono state contate con un emocitometro e piastrate, a diverse densità, in piastre multiwell da 6 pozzetti, ognuno dal diametro di 2,4 cm.
La superficie di ogni pozzetto era stata preventivamente ricoperta con 15 µg/ml di poli-D-lisina in soluzione acquosa per 1 ora a 37°C, e sciacquata tre volte con acqua sterile.
Le cellule sono state mantenute a 37°C in atmosfera contenente il 5% di CO2. Per prevenire la crescita delle cellule gliali e ottenere delle colture neuronali virtualmente pure, dopo 72 ore dalla piastratura è stata aggiunta citosina arabinoside o Ara-C alla concentrazione di 10 µM.
L’Ara-C, usato generalmente come farmaco antitumorale, è un antimetabolita, ovvero una sostanza la cui struttura chimica è simile ad altri metaboliti normali presenti nell’organismo. Questa analogia strutturale comporta o il blocco di un sistema enzimatico di cui il metabolita fisiologico funziona da substrato, o la sintesi di un prodotto inattivo o con diversa funzione. Si tratta più precisamente di un antipirimidinico: inibisce a vari livelli la sintesi delle pirimidine (citosina e timidina) costituenti del DNA.
3.5.2 Risposta dei neuroni mesencefalici in coltura
Nella fase iniziale del lavoro sulle cellule mesencefaliche è stata effettuata un’analisi di densità per determinare quale fosse la densità ottimale di semina cioè la minima possibile che consentisse alle cellule di sopravvivere. Minore è la densità più facile risulta l’analisi della morfologia e della topologia della rete. Nel precedente lavoro di tesi la densità di semina utilizzata era di 500000 cellule per cm2. Questa densità è piuttosto elevata per cui dopo qualche giorno è difficile visualizzare le interconnessioni tra i vari nodi. Si è cercato quindi di abbassare la densità di semina. Dato che era stato osservato che le cellule a densità minore muoiono, si è ipotizzato che la morte fosse causata dal fatto che il farmaco Ara-C in parte uccide anche le cellule neurali non gliali. Le colture quindi sono state realizzate senza l’aggiunta di tale farmaco.
Le densità analizzate sono state: • 50000 cellule/cm2 ; • 100000 cellule/cm2 ; • 150000 cellule/cm2 ; • 250000 cellule/cm2 ; • 500000 cellule/cm2 .
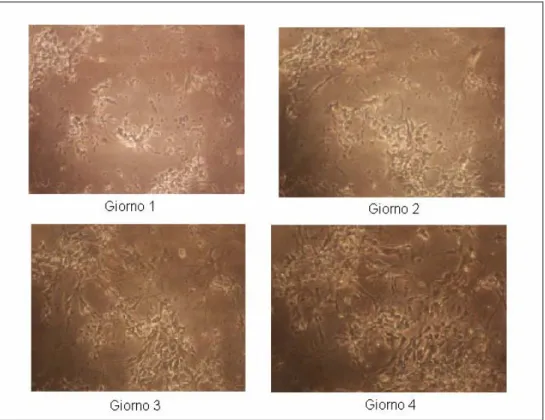
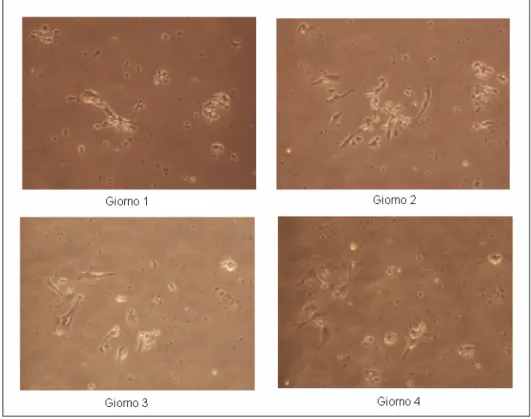
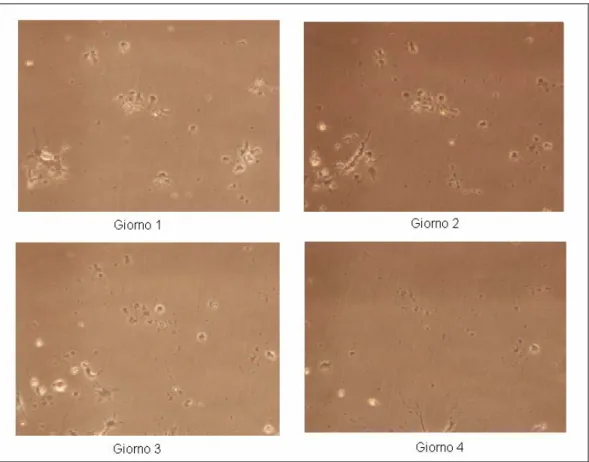
Da questa analisi si è dedotto che la minima densità di semina che consente la crescita cellulare è di 150000 cellule/cm2 per cui le colture successive sono state realizzate tutte a questa densità. A densità superiore infatti le cellule crescono ma sono molto vicine tra loro ed è difficile vedere bene le interconnessioni, mentre a densità inferiori le cellule dopo poco muoiono. Dalle immagini riportate delle reti (Fig. 3.4-3.8) si osserva come già nei primi quattro giorni il rate di crescita sia diverso per diverse densità di coltura.
Figura 3.4 Neuroni mesencefalici di topo seminati con densità di 500000 cellule/cm2
Figura 3.6 Neuroni mesencefalici di topo seminati con densità di 150000 cellule/cm2
Figura 3.8 Neuroni mesencefalici di topo seminati con densità di 50000 cellule/cm2
Una volta stabilità la densità ottimale, le colture successive sono state seminate a questa densità. È stato osservato che le cellule si mantengono vive in coltura per circa 20 giorni, ma dopo 7 giorni la situazione rimane stabile e non cambia la struttura della rete.
3.6 Colture di cellule di Purkinje
3.6.1 Preparazione delle colture
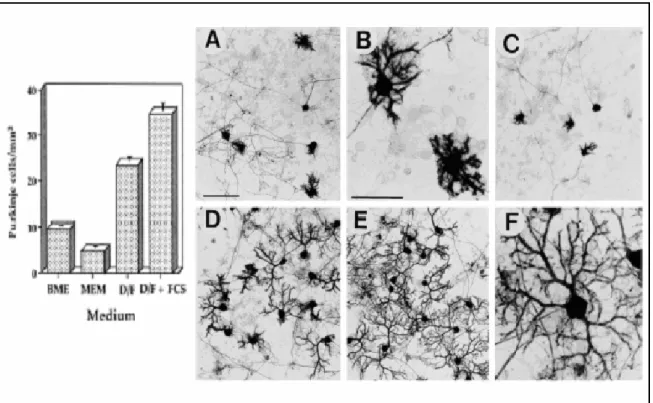
Le cellule di Purkinje hanno un rate di sopravvivenza piuttosto basso per cui è difficile che realizzando colture dissociate esse riescano a non morire. Furuya e collaboratori hanno dimostrato che il rate di sopravvivenza può essere aumentato realizzando delle colture monostrato miste cioè non solo contenenti le cellule di Purkinje ma anche altre cellule del cervelletto quali le cellule granulari e gli interneuroni [68]. È stato dimostrato inoltre che il mezzo di coltura influenza molto la crescita delle cellule di Purkinje. Esperimenti in cui sono utilizzati diversi mezzi di coltura hanno rivelato che il massimo rate di sopravvivenza si ottiene utilizzando come mezzo di coltura il DMEM/F-12 arricchito con l’1% di FCS (Fetal Calft Serum). Tale mezzo, oltre ad aumentare la sopravvivenza, ha anche l’importante vantaggio di promuovere la differenziazione dendritica. Le cellule coltivate in questo mezzo infatti, sviluppano un albero dendritico molto più ramificato rispetto a quello sviluppato da cellule coltivate in altri mezzi. In Fig. 3.9 sono riportate delle foto di cellule di Purkinje mantenute in diversi mezzi di coltura.
Figura 3.9 Istogramma della densità di cellule di Purkinje coltivate in mezzi di coltura
diversi (sinistra) e immagini di cellule di Purkinje in tali mezzi (destra)
La procedura utilizzata in questo studio è stata ottimizzata in un lavoro successivo realizzato da Tabata e collaboratori [69]. La preparazione delle colture di Purkinje utilizzate in questa tesi è stata effettuata seguendo il protocollo utilizzato in questo studio.
I cervelletti sono prelevati da topi P0 (Post-natal zero, cioè il giorno di nascita) in PBSw/CaMg contenente gentamicina (10 μg/ml). Per cambiare soluzione i cervelletti, contenuti all’interno di una Falcon, sono stati quindi cenrifugatti alla temperatura di 4°C alla velocità di 1000 rpm per 1-3 minuti ed è stato aspirato il surnatante. La dissociazione del tessuto è stata realizzata sia per via enzimatica che per via meccanica. La dissociazione enzimatica è stata ottenuta aggiungendo prima tripsina (0,1% w/v in PBS) mantenuta a 33° per 13-15 minuti e, dopo 2 lavaggi in PBS, con DNAsi (5 U/ml in PBS). Mantenendo i cervelletti all’interno di questa soluzione, la dissociazione meccanica è stata realizzata tramite triturazione utilizzando una Pasteur fino a
che non si sono ottenuti aggregati invisibili. Sono stati aggiunti quindi 5 ml di PBS alla sospensione di cellule ed è stata effettuata una centrifugazione alla temperatura di 4°C a alla velocità di 1200 rpm per 5 minuti. Dopo la rimozione del surnatante, le cellule sono state contate e la densità è stata portata a 5 × 106 cellule/ml utilizzando il mezzo di coltura.
Il mezzo di coltura ha la seguente composizione: • 100 ml di DMEM/F-12 in rapporto 1:1 • 100 µl di Putrescina
• 30 nM di NaSeO3 • 1,4 mM di L-glutamina • 10 µg/ml di gentamicina.
Il pH è stato aggiustato al valore di 7.2 con NaOH. Dopo 3 ore di incubazione, è stato messo in ogni pozzetto 1ml del terreno preparato prima con aggiunta di:
• 20 µg/ml di transferrina • 40 nM di progesterone • 20 µg/ml di insulina
• 0,5 ng/ml di tri-iodotironina.
Aggiungendo queste sostanze si ottiene un terreno contenente approssimativamente l’1% di FCS.
Una volta pilastrate, le cellule sono state mantenute all’interno dell’incubatore e una volta ogni 3 giorni è stato sostituito metà del vecchio terreno con terreno nuovo arricchito con:
• 100 µg/ml di BSA (Bovine Serum Albumin) • Ara-C.
La BSA costituisce un nutriente per le cellule e quindi ne favorisce la crescita mentre l’ Ara-C, come è già stato detto, inibisce la crescita delle cellule gliali.
3.6.2 Risposta delle cellule di Purkinje in coltura
Le cellule di Purkinje hanno mostrato di riuscire a sopravvivere in coltura dai 20 ai 25 giorni sviluppando un albero dendritico altamente ramificato.
Le colture seguite in questo lavoro di tesi sono state due: la prima è stata seguita dall’undicesimo al ventitreesimo giorno di coltura, la seconda si è iniziata a seguire prima per analizzare anche le prime fasi della crescita. Le cellule di questa coltura tuttavia sono sopravvissute solo fino al diciassettesimo giorno, probabilmente perché la manipolazione durante i primi giorni di crescita le ha in parte danneggiate.
Nelle Fig. 3.10 e 3.11 è illustrata l’evoluzione di due cellule di Purkinje, ognuna appartenente ad una coltura.
Figura 3.10Evoluzione di una cellula di Purkinje appartenente alla seconda coltura
Figura 3.11 Evoluzione di una cellula di Purkinje appartenente alla seconda coltura
Dalle foto si vede come le cellule accrescano le loro dimensioni e sviluppino un numero sempre maggiore di dendriti.
La sopravvivenza e la dendrogenesi delle cellule sono favorite sia dal mezzo di coltura utilizzato, che è stato dimostrato essere il mezzo ottimale per la
coltivazione di tali cellule, sia dalla presenza di altre cellule del cervelletto, in particolare le cellule granulari, che forniscono loro i nutrienti necessari per la crescita.
Si osserva poi che dopo i primi giorni di coltura le cellule granulari e le cellule gliali diminuiscono in grande quantità rendendo più visibili le cellule di Purkinje. L’apoptosi delle cellule gliali è dovuta al farmaco Ara-C che viene somministrato alle cellule ogni tre giorni insieme al terreno di coltura mentre la morte delle cellule granulari potrebbe riflettere il naturale processo apoptotico che avviene in vivo.
3.7 Colorazione delle cellule
Una volta che le cellule hanno raggiunto una configurazione stabile, sono state colorate. La colorazione permette di vedere meglio al microscopio la struttura dendritica di una singola cellula e le interconnessioni tra i neuroni che formano una rete. Essa tuttavia prevede la fissazione delle cellule per cui non si concilia con la volontà di realizzare uno studio in vivo ed è per questo che le cellule sono state colorate solamente l’ultimo giorno di coltura.
La tecnica utilizzata per la colorazione delle cellule di Purkinje è stata quella della immunocitochimica con rilevamento dell’anticorpo tramite digestione enzimatica, mentre le reti di neuroni mesencefalici sono state colorate tramite fluorescenza. Nel primo caso la colorazione ha avuto lo scopo di riconoscere le cellule di Purkinje dalle altre cellule presenti all’interno della coltura. Come è già spiegato infatti, sono state trattate colture arricchite in Purkinje e non colture di cellule di Purkinje dissociate, per cui sono presenti nella piastra di coltura tutte le cellule che costituiscono il cervelletto. Questo rende meno semplice la loro identificazione dato che spesso si confondono con alte cellule
di morfologia simile. L’utilizzo di un anticorpo specifico per le Purkinje, ovvero la Calbindina, favorisce la loro identificazione ed inoltre permette di mettere in risalto la struttura morfologica della cellula.
La colorazione in fluorescenza invece è state realizzata utilizzando il complesso Rodamina-Falloidina che, legandosi ai filamenti di actina del citoscheletro cellulare, ha consentito di mettere in risalto i nodi che costituiscono le reti e le loro intersconnessioni. Le immagini ottenute fotografando le reti colorate, sono immagini a due colori (le cellule in rosso e lo sfondo in nero) in cui l’organizzazione dei neuroni all’interno della rete è bene evidente. A queste immagini è stato possibile applicare tecniche che permettono di valutare l’organizzazione di un sistema, quali l’analisi frattale e l’analisi di clustering, che verranno illustrate nel prossimo capitolo.
3.7.1 Immunocitochimica
3.7.1.1 La tecnica dell’immunocitochimica [70]
L’immmunocitochimica comprende una serie di tecniche in cui un anticorpo è utilizzato per legare in modo specifico un antigene cellulare ad un colorante ben visibile al microscopio. In particolare l’immunocitochimica viene utilizzata per la localizzazione di antigeni in cellule in coltura e si differenzia dall’immunoistochimica applicata invece ai tessuti.
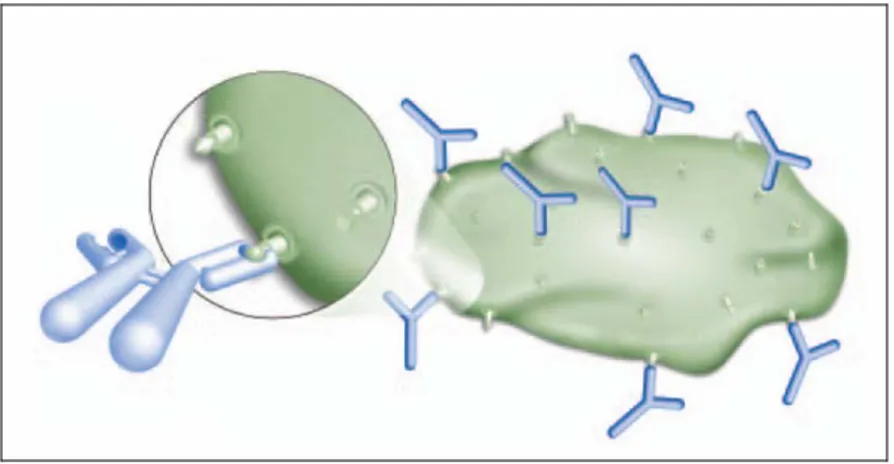
Le tecniche immunochimiche si basano sull’estrema specificità, a livello molecolare, di ciascun anticorpo per il suo antigene (Fig.3.12), perfino in presenza di livelli elevati di molecole contaminanti.
Figura 3.12 Legame di anticorpi con i loro specifici antigeni
Esistono moltissimi tipi di antigene, di anticorpi, di affinità antigene-anticorpo e di metodi di rilevamento. Le condizioni ottimali per l’immunocitochimica, che dipendono da queste variabili, devono essere determinate a seconda del caso specifico.
Nonostante la possibilità di adattare la procedura al caso specifico, si possono elencare una serie di fasi comuni a tutte le procedure di immunocitochimica:
1. Fissazione: le cellule devono essere immerse in un fluido fissante che ha lo scopo di preservare e stabilizzare il campione in modo che mantenga inalterata la propria struttura, proteggere il campione dai danni osmotici ed impedire la solubilizzazione dell’antigene mantenendolo nella sua posizione originaria
2. Permeabilizzazione: nel caso in cui l’anticorpo sia intracellulare, si devono utilizzare dei detergenti che rendono permeabile la membrana cellulare
3. Bloccaggio dei siti aspecifici: per prevenire il legame dell’anticorpo secondario con siti aspecifici, la coltura viene incubata con il siero pre-immune dell’animale in cui è stato prodotto l’anticorpo
4. Incubazione con anticorpo primario: le cellule vengono fatte incubare con l’anticorpo primario, prodotto in una specie differente da quella da cui proviene il campione, che si lega agli specifici antigeni. È molto importante determinare sperimentalmente la concentrazione di anticorpo e il tempo di incubazione ottimali
5. Incubazione con anticorpo secondario: la rilevazione dell’anticorpo primario legato all’antigene si effettua utilizzando un secondo anticorpo che riconosca l’anticorpo primario. Più molecole di anticorpo secondario legano diversi epitopi dell’anticorpo primario per cui si ha un’amplificazione del segnale
6. Lavaggi: dopo ogni incubazione i campioni devono essere lavati per portare via l’eccesso di soluzione
Il rilevamento dell’anticorpo può essere sia enzima-mediato sia tramite fluorescenza. Nel primo caso si utilizza un enzima che catalizza la trasformazione di uno specifico substrato in un prodotto colorato che precipita e che è ben visibile al microscopio ottico. Nel secondo caso si utilizza invece un fluorocromo legato all’anticorpo secondario e la cui emissione può essere visualizzata utilizzando un microscopio a fluorescenza.
In entrambi i casi si può ottenere un’amplificazione del segnale utilizzando anticorpi secondari poli-coniugati, interazioni Avidina-Biotina a altri sistemi disponibili in commercio.
3.7.1.2 La tecnica applicata alle cellule di Purkinje
L’anticorpo primario utilizzato per la colorazione delle cellule di Purkinje è la Calbindina D28K, un anticorpo policlonale intracellulare prodotto nel coniglio
e diretto contro la calbindina, una proteina vitamina D-dipendente che si lega al calcio e che, all’interno del cervelletto, è espressa esclusivamente dalle cellule di Purkinje [66].
Il sistema di rivelazione dell’anticorpo secondario utilizzato è invece quello per via enzimatica, in particolare sfruttando la reazione tra il complesso Avidina-Perossidasi biotinilata e Diaminobenzoidina (DAB). L’Avidina ha quattro siti di legame per la biotina per cui può legarsi sia alla Perossidasi biotinilata sia all’anticorpo secondario biotinilato. Si forma così un array tridimensionale che consente un’amplificazione del segnale. Viene aggiunta quindi la Diaminobenzidina che subisce una reazione chimica, catalizzata dalla Perossidasi, trasformandosi in un precipitato di colore marrone visibile al microscopio [71].
Il protocollo di immunocitochimica seguito per la colorazione delle cellule di Purkinje è il seguente:
1. Fissazione delle cellule: dopo aver decantato il mezzo di coltura sono stati effettuati 3 lavaggi in PBS ed è stata aggiunta Paraformaldeide al 4% in PBS per 1 ora a T ambiente. È importante che la parformaldeide sia portata ad un PH neutro (circa 7,2) affinché non danneggi le cellule. Questo è stato fatto aggiungendo NaOH sotto controllo di un PHmetro 2. Permeabilizzazione della membrana: dopo aver aspirato la
paraformaleide sono stati realizzati 3 lavaggi da 8 minuti con una soluzione di Tryton X 100 allo 0,05% in PBS
3. Bloccaggio dei siti aspecifici: Incubazione per 30 minuti con normal goat serum in soluzione al 10% in PBS + Tryton X 100
4. Incubazione con anticorpo primario (Calbindina D28K anti-topo fatto in coniglio): dopo aver aspirato il normal goat serum senza lavare è stata
aggiunta una soluzione di anticorpo primario diluita in PBS + Tryton X 100. Sono state sperimentate tre diverse diluizioni in diversi pozzetti: 1:250, 1:500 e 1:1000. Le cellule sono state incubate nella soluzione di anticorpo in frigo a 4° C over-night
5. Incubazione con anticorpo secondario (Ig G biotinilato anti-coniglio fatto in capra): il giorno seguente è stato aspirato l’eccesso di anticorpo e, dopo 3 lavaggi da 8 minuti in PBS + Tryton X 100, è stata aggiunta al soluzione di anticorpo secondario in PBS + Tryton X 100 + 0,08% di normal goat serum, con diluizione 1:200. Le cellule sono state incubate con l’anticorpo secondario per 1 ora a T ambiente.
6. Sistema di rivelazione: dopo aver aspirato la soluzione di anticorpo secondario in eccesso sono stati effettuati 3 lavaggi da 8 minuti in PBS + Tryton X 100 ed è stata aggiunto il sistema di rivelazione costituito da Avidina e Perossidasi biotinilata entrambe al 2% in PBS
7. Aggiunta del substrato per l’enzima: dopo aver aspirato la soluzione in eccesso sono stati effettuati 3 lavaggi da 8 minuti in PBS ed è stata aggiunta la Diaminobenzidina all’1%. La Diaminobenzidina è stata poi inattivata con la candeggina dato che è cancerogena.
La reazione si è avvenuta molto rapidamente ed in pochi secondi si è formato un precipitato marrone nel pozzetto.
Delle tre diluizioni di anticorpo primario quella più idonea è risultata essere quella 1:250 in quanto negli altri due casi l’anticorpo risulta essere troppo diluito e la colorazione che si ottiene non è sufficiente.
Si riportano alcune delle fotografie delle cellule di Purkinje colorate utilizzando l’anticorpo primario con diluizione 1:250. Le fotografie sono state realizzate al microscopio ottico sia con contrasto di fase che senza ad ingrandimenti di 10 e 20 X (Fig. 3.13).
Figura 3.13 Cellule di Purkinje colorate con Calbindina e fotografate al microscopio ottico con (A e C) e senza (B e D) contrasto di fase
3.7.2 Fluorescenza
3.7.2.1 La colorazione in fluorescenza [72]
La colorazione in fluorescenza si basa sull’utilizzo di fluorocromi che vengono fatti legare a specifici traccianti che hanno come target componenti della cellula.
Utilizzando un microscopio a fluorescenza si può rendere visibile l’emissione del fluorocromo inviando un fascio luminoso in direzione del campione (Fig 3.14).
Figura 3.14 Spettro di emissione e di assorbimento tipico di un fluorocromo
La luce proveniente dalla lampada e diretta verso il campione attraversa prima il collimatore, poi il filtro che assorbe il calore e infine il diaframma. La luce di eccitazione colpisce quindi il campione che emette fluorescenza. La luce fluorescente viene infine diretta all’oculare dopo essere stata filtrata e collimata (Fig 3.15).
Figura 3.15 Percorso della luce all’interno di un microscopio a fluorescenza
3.7.2.2 La tecnica applicata alle cellule mesencefaliche
Il flouorocromo utilizzato per la colorazione delle cellule mesencefaliche è la rodamina. Essa è venduta in un preparato insieme alla falloidina che costituisce il tracciante per il legame alla cellula [73].
La falloidina è una tossina che si estrae dalla cappella del fungo Amanita phalloides ed ha la capacità di legarsi ai filamenti di actina che costituiscono il citoscheletro ed in particolare alle interfacce tra le diverse subunità prevenendo quindi la loro dissociazione e depolimerizzazione.
Figura 3.16 Formula molecolare della falloidina
La falloidina di per sé non è fluorescente per cui deve essere legata alla rodamina un fluorocromo molto utilizzato in biologia grazie al suo basso costo e alla sua facilità di utilizzo. La formula chimica della rodamina è la seguente: C28H31N2O3Cl [74].
La rodamina emette una colorazione rosso-arancione nel campo dell’infrarosso (Fig. 3.17).
Figura 3.17 Emissione ed assorbimento della rodamina
Il protocollo utilizzato per la colorazione delle cellule mesencefaliche è il seguente:
1. Fissazione eseguita utilizzata la stessa procedura utilizzata per le cellule di Purkinje
2. Lavaggi in PBS (3 lavaggi di 8 minuti)
3. Incubazione con il complesso rodamina-falloidina per 1 ora 4. Lavaggi in PBS (3 lavaggi di 8 minuti)
La struttura delle reti di neuroni mesencefalici è stata messa bene in evidenza dalla colorazione. Si riportano le foto di una stessa rete realizzate a 10 e a 20X (Fig 3.18).
Figura 3.18 Rete di neuroni mesencefalici colorati in fluorescenza e fotografati al
microscopio ad ingrandimenti di 10 (A) e 20X (B)
3.8 Tecnica di realizzazione delle fotografie alle cellule
L’obiettivo di questa tesi è lo studio della dinamica di formazione delle reti e di crescita delle cellule di Purkinje.
Per poter effettuare uno studio dinamico è necessario innanzi tutto che le cellule siano mantenute vive e non vengano fissate, inoltre è necessario fotografare sempre la stessa area della piastra in cui le cellule sono seminate
in modo tale da seguire l’evoluzione di una specifica rete o di una specifica cellula.
Le foto sono state realizzate mediante una telecamera collegata ad un microscopio ottico e per ritrovare lo stesso punto della piastra, sono state realizzate delle griglie utilizzando il programma CorelDraw che sono state attaccate sul fondo della piastra.
3.8.1 La strumentazione
Per la realizzazione delle fotografie è stato utilizzato un microscopio ottico a cui è stata collegata una telecamera.
In Fig. 3.19 si vede come è costituita la strumentazione.
La telecamera utilizzata è una telecamera FireWire cioè permette la trasmissione di dati: la telecamera viene connessa alla porta FireWire del computer e il segnale video viene trasmesso tramite il cavo al PC. In questo modo è possibile visualizzare sul monitor ciò che l’obiettivo sta catturando [75]. Sul computer è stato installato un software realizzato appositamente per la telecamera e che consente di comunicare con essa tramite un’interfaccia grafica che viene aperta quando si vogliono realizzare le fotografie.
I vantaggi di questa tecnica sono:
• Le cellule sono più facilmente visibili e più facilmente rintracciabili rispetto a quando vengono viste nell’obiettivo del microscopio. Questo è molto utile nel presente lavoro in cui si vogliono andare a fotografare sempre la stessa rete o la stessa cellula
• Tramite l’interfaccia grafica è possibile regolare alcune caratteristiche dell’immagine quali il contrasto, la luminosità, il guadagno, etc., è possibile zoomare alcune aree dell’immagine e realizzare dei video
• L’immagine risulta essere ingrandita ulteriormente rispetto all’ingrandimento dato dal microscopio grazie al sistema ottico di cui è dotata la telecamera
• Le immagini possono essere direttamente salvate sul computer.
Il sistema utilizzato è quindi un sistema meccatronico il cui schema è riportato in Fig. 3.20.
Figura 3.20 Schema tipico di un sistema meccatronico
I sensori sono costituiti dai CCD (Charge Coupled Device): essi captano le immagini attraverso una matrice di punti (pixel) ognuno dei quali è in grado di riprodurre una gamma di gradazioni di intensità luminose dal nero al bianco e contiene l’informazione relativa ad un colore [76].
In Fig.3.21 è schematizzata la struttura di un CCD.
L’alimentazione della telecamera è esterna e avviene tramite collegamento alla corrente elettrica mentre meccanica, elettronica e controllo sono tutti integrati al suo interno.
L’interfaccia (Fig. 3.22) infine è quella che viene visualizzata sul monitor e consente la visualizzazione, la manipolazione e il salvataggio delle immagini.
Figura 3.22 Interfaccia grafica della telecamera FireWire
Nella finestra più grande del pannello si vede ciò che la telecamera sta catturando, mentre nella colonna a destra vengono visualizzate le immagini salvate.
3.8.2 Le fotografie
Utilizzando la strumentazione descritta nel paragrafo precedente è stato possibile fotografare in maniera piuttosto semplice ad efficiente le cellule.
Per quanto riguarda le cellule di Purkinje la frequenza con cui sono state realizzate le foto è stata di 24 ore, mentre le cellule mesencefaliche sono state fotografate ogni 24 ore i primi due giorni e gli ultimi giorni mentre nei giorni centrali (terzo, quarto e quinto giorno) la frequenza è stata aumentata a 4 ore dato che è stato visto che in questi giorni la formazione di nuove connessioni e il riarrangiamento della rete è molto rapido.
Le immagini ottenute sono digitali, cioè costituite da pixel, per cui possono essere elaborate ed analizzate con le tecniche che verranno descritte nel prossimo capitolo. Le immagini sono state salvate nel formato bitmap (BMP) in quanto l’immagine non viene compressa e non si ha perdita di dati.
Le fotografie sono state realizzate ad ingrandimenti di 10X e 20X regolando l’obiettivo del microscopio (Fig.3.23). Esse risultano poi ulteriormente ingrandite grazie al sistema ottico della telecamera.
Figura 3.23 Diversi ingrandimenti di un microscopio ottico
3.8.3 Preparazione delle griglie
Per poter fotografare sempre le stessa area della piastra, ovvero le stesse cellule a intervalli di tempo differenti, sono state preparate, usando il programma CorelDraw, delle apposite griglie micrometriche.
3.8.3.1 CorelDraw
CorelDraw è uno dei più rinomati programmi di grafica vettoriale, inserito in una suite di grafica professionale che comprende, oltre a CorelDraw, altri programmi come CorelPhotoPaint, per la grafica pittorica, CorelCapture, per la cattura delle immagini a video, Coreltrace, per il tracciamento delle immagini bitmap e tanti altri.
CorelDraw deve molto della sua popolarità alla sua estrema semplicità di utilizzo, alla totale personalizzazione dell'interfaccia, ma soprattutto alla sua flessibilità ed intuitività che rende la realizzazione di qualsiasi progetto una vera passeggiata.
Consente di creare e modificare le immagini vettoriali.
La grafica vettoriale è la grafica orientata agli oggetti. Ogni oggetto è composto da singoli elementi, ognuno dei quali è un elemento a sé stante, indipendente da tutti gli altri e con le sue proprietà (colore di riempimento, colore di contorno, spessore del contorno ecc..).
Ogni elemento trova le proprie posizione e forma grazie ad una serie di coordinate e impostazioni matematiche.
Un’immagine vettoriale si può spostare e modificare, si può ingrandire o rimpicciolire a piacimento, mantenendo inalterate chiarezza e definizione. Si pensi ad un disegno rappresentante una casa: in questo caso la porta, la finestra, i vetri e tutto quello che concorre a formare l'immagine saranno oggetti divisi l’uno dall'altro, ognuno con le proprie caratteristiche ed ognuno di essi si potrà ridimensionare, ricolorare, cancellare senza intaccare gli altri elementi che compongono l’immagine. Inoltre le immagini vettoriali richiedono poca memoria per essere eseguite.
3.8.3.2 Realizzazione delle griglie
Prendendo ispirazione dalla camera di Burker, si è scelto di disegnare una serie di quadrati, ciascuno dei quali è stato suddiviso, attraverso due linee orizzontali e due verticali, in nove quadratini minori, di lato 1 mm, e quindi in nove aree.
Ciascun quadrato principale è inoltre stato numerato, così da poterlo identificare in maniera univoca (Fig. 3.24).
Figura 3.24 Grigia realizzata per una piastra multiwell a 12 pozzetti
Tali griglie sono state stampate su lucido e poi incollate sotto la piastra di coltura, contenente le cellule aderite alla superficie inferiore.
L’osservazione al microscopio delle piastre ha permesso di identificare le zone ottimali per l’acquisizione delle immagini.
Si sono scelte quelle regioni la cui area corrispondeva all’area racchiusa da uno dei quadratini minori di uno dei quadrati principali.
Ricordando la posizione del quadratino scelto all’interno del quadrato principale (alto a destra, alto a sinistra, ecc..) e il numero di questo ultimo, ogni volta è stato possibile ritrovare la stessa area e fotografarla.