3. SINTESI STEREOSELETTIVA DI AZAPIRANOSI E
AZAZUCCHERI PIPERIDINICI
3.1 Introduzione
Nella prima parte della mia tesi sperimentale ho proceduto a verificare la possibilità di utilizzare gli alchil O-imminoglicosidi-2,3-insaturi 3.1 e 3.2 ottenuti dalle reazioni di glicosilazione di alcooli da parte, rispettivamente, degli epossidi 2.2α-Me e 2.2β-Me e delle N-nosil aziridine 2.3α-Me e 2.3β-Me per la sintesi di corrispondenti azapiranosidi (3.3 e 3.4) ed azazuccheri piperidinici (3.5 e 3.6) (Capitolo 2),11 a stereochimica ben definita, come qui sotto riportato in modo generico (Schema 3.1).
N N N N N N Cbz Cbz H H Cbz Cbz Me Me Me Me Me Me OR HO OR NsHN OR NsHN OH OH OR OH OH HO HO OH OH OH OH H2N 3.1 3.3 3.5 3.2 3.4 3.6 N N Me Me O N Ns Cbz Cbz 2.2αααα-Me e 2.2ββββ-Me 2.3αααα-Me e 2.3βββ-Meβ
Schema 3.1. Sintesi di azapiranosi e azazuccheri piperidinici.
Quindi primo scopo della mia attività sperimentale è stato quello di sintetizzare i glicosil donatori a nostra disposizione, gli epossidi 2.2α-Me e 2.2β-Me le N-nosil aziridine 2.3α-Me e 2.3β-Me.
3.2 Sintesi degli epossidi 2.2α-Me e 2.2β-Me e delle N-nosil aziridine 2.3α-Me e
2.3β-Me
La sintesi degli epossidi 2.2α-Me, 2.2β-Me e delle N-nosil aziridine 2.3α-Me e 2.3β-Me, per quanto apparentemente complessa, è risultata essere estremamente efficace e riproducibile e conduce a questi sistemi eterociclici in modo completamente regio- e stereoselettivo. La sintesi prende origine dalla reazione di inserimento del gruppo metilico sull’anello del sistema N-acilpiridinio ed inizialmente conduce attraverso due procedure diverse ai due epossidi 2.2α-Me e 2.2β-Me. La successiva applicazione di un medesimo protocollo sintetico ai due epossidi 2.2α-Me e 2.2β-Me conduce in modo completamente stereoselettivo, alle corrispondenti aziridine di opposta configurazione: solo 2.3α-Me da 2.2β-Me e solo 2.3β-Me da 2.2α-Me.
L’introduzione del gruppo metilico sul C(2) del sale di piridinio [futuro C(5) del sistema immino glicalico] è stata effettuata in modo decisamente semplice e vantaggioso per aggiunta del CH3MgBr (commercialmente disponibile) al sale di acil piridinio 3.7, ottenuto in situ per
reazione della 4-metossi piridina con Cbz-Cl.13 Si ottiene, attraverso l’enol etere 3.8, il metil piridone 3.9 che viene sottoposto ad α-acetossilazione, a riflusso in toluene, con Pb(OAc)4
umido per acido acetico a dare l’α-acetossi derivato 3.10 in cui il gruppo metilico, già presente, ed il gruppo acetossi, ora introdotto, sono in relazione trans. Questo costituisce un aspetto strutturale e stereochimico molto importante per il buon proseguimento della sintesi (Schema 3.2).
N OMe N OMe Cbz Cl N OMe Cbz N O Cbz N O Cbz N O Cbz CbzCl THF -25 °C MeMgBr -45 °C HCl 10% Pb(OAc)4 toluene riflusso HCl/EtOH 50 °C AcO HO 3.8 3.9 3.10 2.38 3.7 Me Me Me Me
Schema 3.2. Sintesi dell’idrossichetone 2.38.
Il risultato completamente stereoselettivo osservato in questa reazione di α-acetossilazione resterebbe giustificato dal fatto che con l’aggiunta dell’agente ossidante al chetone 3.9 si forma un specie intermedia enol-piombo triacetato come 3.11 in cui, caratteristica tipica di questo e di tutti gli altri sistemi immino glicali, il sostituente achilico sul C(2) [futuro C(5)] è disposto esclusivamente in posizione assiale allo scopo di minimizzare la sfavorevole interazione con il gruppo acilico sostituente l’azoto endociclico (gruppo Cbz nel nostro caso). In queste condizioni, mostrate nello Schema 3.3, il gruppo acetato viene trasferito intramolecolarmente in direzione pseudoassiale, attraverso un più favorevole stato di transizione a sedia, a dare l’addotto 3.10 con completa α-stereoselettività.14,15 N CH3 O BnO O Pb O O O CH3 O O CH3 O CH3 3.11 N O AcO Me Cbz 3.10
Schema 3.3. Intermmedio enolpiombo triacetato e trasferimento α stereoselettivo del gruppo acetato.
Il gruppo acetato presente sul C(4) di 3.10 viene quindi idrolizzato in ambiente acido (HCl 10% in EtOH) a dare, dopo circa 24 ore a 40°C, l’α-idrossichetone 2.38 (Schema 3.2).
La riduzione stereoselettiva di 2.38 condotta con Me4NBH(OAc)3 in acetone (HPLC grade)
ed acido acetico glaciale, a temperatura ambiente seguendo la procedura di Evans,16 ha condotto al desiderato diolo trans 3.12 (Schema 3.4).
N O Cbz Me N OH Cbz Me Me4NBH(OAc)3 acetone/AcOH 2.38 3.12 HO HO
Schema 3.4. Riduzione stereo selettiva dell’idrossichetone 2.38 a diolo 3.12.
Anche questo passaggio è risultato essere completamente stereoselettivo e nel diolo 3.12 il gruppo ossidrilico generato sul C(3) dal processo di riduzione viene a trovarsi in relazione trans rispetto al gruppo ossidrilico già presente sul C(4) .
La procedura di Evans, inizialmente prevista per gli 1,3-idrossichetoni lineari, non ciclici, è stata qui applicata con successo ad un 1,2-idrossichetone, ciclico. In questa reazione, attraverso l’uso del Me4NBH(OAc)3, si forma, grazie all’ambiente acido, per acido acetico, un intermedio
alcossidiacetossiboroidruro come 3.13 (Schema 3.5) si ha, cioè, una reazione di scambio di un gruppo acetato del reagente con l’ossidrile presente sul C(4) dell’idrossichetone 2.38. Una volta formatosi 3.13, avviene il rilascio intramolecolare di una specie idruro che attacca il carbonio della funzionalità carbonilica protonata dalla faccia α, ovvero dalla stessa parte verso cui si dirige il gruppo ossidrilico già presente. Si forma così, esclusivamente, il diolo trans 3.12, come mostrato nello Schema 3.5.9
Me4NBH(OAc)3 CH3COOH B OCOCH3 OCOCH3 H δ 3.13 3.12 acetone N 2.38 CH3 O BnO O N CH3 O BnO O O H N CH3 O BnO OH OH H 2 3 OH
Schema 3.5. Meccanismo della riduzione stereoselettiva dell’idrossichetone 2.38 a diolo 3.12.
Il trasferimento intramolecolare dell’idruro spiega la stereoselettività della reazione. Se, infatti, fossimo in presenza di un rilascio intermolecolare di uno ione idruro presente nel mezzo di reazione, l’attacco sul carbonio carbonilico potrebbe avvenire teoricamente sia dalla faccia α
che dalla faccia β del sistema. Con la formazione dell’intermedio 3.13 e con il rilascio intramolecolare di una specie idruro (H-), invece, viene garantito un unico attacco, in questo caso dalla faccia α. Determinante anche in questo caso il fatto che l’idrossichetone 2.38, per i motivi sopra già ricordati, si trovi essenzialmente nella corrispondente conformazione con entrambi i sostituenti, l’ossidrile sul C(4) ed il gruppo metilico sul C(5) in relazione trans diassiale tra di loro. Ne segue che una volta formatosi l’intermedio alcossidiacetossiboroidruro 3.13, il trasferimento della specie idruro avviene necessariamente ed esclusivamente dalla faccia α del sistema, portando di conseguenza alla formazione del diolo trans 3.12 quale unico prodotto della reazione.
Perché una reazione di questo tipo abbia successo, è quindi necessaria la presenza di un gruppo ossidrilico sul substrato che deve essere ridotto. A questo riguardo è importante ricordare che il Me4NBH(OAc)3 acquista le proprietà di specie riducente solo quando è complessato con un
gruppo ossidrilico del substrato. Così, nel caso in cui in una molecola siano presenti più gruppi riducibili, il Me4NBH(OAc)3 riduce selettivamente il gruppo carbonilico adiacente al gruppo
ossidrilico cui si lega. Per il buon successo della reazione riveste fondamentale importanza la presenza dell’acido acetico in quanto favorisce la formazione dell’intermedio 3.13 diventando in questo modo l’agente responsabile sia della velocità della reazione che della diastereoselettività. È stato infatti notato sperimentalmente in reazioni di questo tipo, effettuate su altri substrati, che in assenza di acido acetico, la maggior parte del composto di partenza non reagisce o reagisce lentamente. L’aggiunta di sole quantità catalitiche di acido aumenta la velocità della reazione, ma aumenta di poco la selettività. Se la concentrazione dell’acido viene aumentata, anche la diastereoselettività della reazione aumenta.
La trasformazione del diolo trans 3.12 nell’epossido 2.2β-Me è stata effettuata in modo del tutto originale rispetto a quanto fatto precedentemente per la sintesi del corrispondente epossido 2.1β. Il trans diolo 3.12 è stato inizialmente diacetilato (Ac2O/Py) a dare il corrispondente trans
diacetato 3.14. Questo è stato quindi sottoposto a saponifacazione regioselettiva per suo trattamento con soluzione 0.053 N di K2CO3 in una miscela MeOH-H2O 16:1. Dopo 5 h a
–30/-20°C, con costante controllo TLC, si ottiene una miscela di reazione costituita essenzialmente dal desiderato mono acetato 3.15 (57%) accompagnato da minori quantità del diacetato di partenza 3.14 (38%) e del diolo 3.12 (5%) (Schema 3.6). La separazione mediante flash cromatografia permette l’isolamento dell’idrossi acetato 3.15, che prosegue la via sintetica, mentre il diacetato 3.14 ed il diolo 3.12, anch’essi separati e recuperati, vengono opportunamente riciclati.
Temperature di reazione superiori (-10/0°C) e tempi di reazione più brevi (30 minuti) rispetto a quelli impiegati, determinano una maggiore conversione del diacetato di partenza 3.14 (10% di substrato non reagito) con formazione del monoacetato 3.15 (40%), ma al tempo stesso comportano la comparsa di una maggiore quantità del prodotto di completa saponificazione, il diolo 3.12 (50%). N OH Cbz HO N OAc Cbz AcO Ac2O/Py 0 °C N OAc Cbz HO K2CO3/MeOH/H2O OH- 0.053 N - 25 °C N N N Me Me Me O O Me Me O O OH O Me O 3.12 3.14 3.15 OH -Me Me Me OH
-O OBn O OBn O OBn
OH OH
Schema 3.6. Acetilazione del diolo 3.12 e monosapponificazione del diacetato 3.14 con relativo meccanismo
ad elevata regioselattività.
La ragione della elevata anche se non completa regioselettività osservata nella mono saponificazione del diacetato 3.14, è anche in questo caso riconducubile, a nostro avviso, alla particolare e ricorrente “rigidità” conformazionale di questi sistemi che impone al sostituente sul C(5), il metile nel nostro caso, a disporsi assialmente, al fine di evitare la sfavorevole interazione sterica con il sostituente acilico nell’azoto endociclico. Ne segue che il trans diacetato 3.14 esiste in soluzione essenzialmente nella sua conformazione trans diassiale, come mostrato in Schema 3.6. In queste condizioni, il gruppo carbonilico del gruppo acetossi presente sul C(4) e diretto verso la faccia α appare chiaramente più accessibile al reagente alcalino, in quanto non disturbato dal gruppo metilico adiacente che si dirige verso la opposta faccia β del sistema imminoglicale. Al contrario, lo stesso gruppo metilico sembra invece disturbare, attraverso una sfavorevole interazione di tipo 1,3-sin diassiale, il carbonio carbonilico del gruppo acetossi presente sul C(3) che si dirige verso la medesima faccia β del sistema ciclico (Schema 3.6). Da questi effetti sterici deriva, ragionevolmente, la discreta regioselettività osservata nel processo di mono saponificazione del diacetato 3.14.
Il trans idrossiacetato 3.15 viene quindi mesilato (MsCl/Py) a dare il corrispondente trans mesilossiacetato 3.16 che costituisce il precursore ultimo dell’epossido 2.2β-Me (Schema 3.7).
N Cbz Me N Cbz Me t-BuOK CH3CN OAc MsO N Cbz Me OAc HO MsCl/Py 0 °C 3.15 3.16 2.2ββββ-Me O
Schema 3.7. Sintesi dell’epossido 2.2β-Me a partire dall’idrossiacetato 3.15.
La via sintetica verso l’aziridina 2.3α-Me procede con la formazione del trans azido alcool 3.17. Per questo scopo il trans mesilossiacetato 3.16 viene posto a reagire, in THF, ed in presenza di t-BuOK, con la tetrametilguanidinazide (TMGA) in CH3CN, un classico reagente fornitore di
specie azide, solubile nei solventi organici. Attraverso la formazione dell’epossido 2.2β-Me ed una reazione dello stesso in modo 1,2-regio- ed anti stereoselettivo, con attacco del nucleofilo azide sul carbonio ossiranico allilico C(3), si ottiene il trans azido alcool 3.17 (resa = 60%), quale unico prodotto di reazione. Si può ossevare come nel trans azido alcool 3.17 si abbia, rispetto all’epossido di partenza, completa inversione di configurazione sul C(3) portante l’azido gruppo e questo è il fattore che determinerà, alla fine, l’ottenimento della aziridina 2.3α-Me, avente configurazione opposta rispetto all’epossido 2.2β-Me di provenienza (Schema 3.8).
N Cbz Me N Cbz Me NH2 HO N Cbz Me NHNs HO N Cbz Me NHNs MsO N Cbz Me TMGA THF 0 °C PPh3 supp.pol THF/H2O (20/1) NsCl Py 0 °C MsCl Py 0 °C t-BuOK CH3CN N Cbz Me N3 HO 2.2ββββ-Me 3.17 3.18 3.19 3.20 2.3αααα-Me O N Ns
Dopo purificazione mediante cromatografia flash, la riduzione del trans azido alcool 3.17 al corrispondente trans ammino alcool 3.18 è stata condotta mediante il protocollo che fa uso della PPh3 da noi parzialmente modificata facendo uso della PPh3 supportata su polimero.17 Operando
con questo reagente commercialmente disponibile (Fluka), aggiunto ad una soluzione del trans azido alcool 3.17 in THF-H2O (20:1), si ottiene, dopo 24h di agitazione a temperatura ambiente
seguita da semplice filtrazione, il trans ammino alcohol 3.18, sufficientemente puro da poter essere impiegato direttamente nello stadio successivo. La successiva reazione di mononosilazione regioselettiva (NsCl, 1.1 equiv/Py) trasforma il trans ammino alcool 3.18 nel corrispondente nosil derivato 3.19 che viene mesilato sul residuo gruppo ossidrilico a dare il definitivo trans N-nosil-O-mesilato 3.20, che costituisce il precursore ultimo stabile della aziridina 2.3α-Me. Grazie infatti alla sufficiente acidità del legame N-H del gruppo N-nosilammino e l’appropriata configurazione trans tra lo stesso e l’adiacente gruppo O-mesile quale buon gruppo uscente, il trattamento dell’N-nosil-O-mesilato 3.20 in ambiente alcalino (t-BuOK) determina la formazione della corrispondente aziridina N-nosil sostituita 2.3α-Me (Schema 3.8).
Per la sintesi della diastereoisomerica N-nosil aziridina 2.3β-Me era necessario disporre del corrispondente epossido 2.2α-Me di configurazione opposta, su cui poi applicare la sequenza (azidolisi, riduzione dell’azido gruppo, N-nosilazione e O-mesilazione) sopra descritta a partire dall’epossido 2.2β-Me.
La sintesi dell’epossido 2.2α-Me inizia dal trans idrossi chetone 2.38 che era stato individuato come appropriato sistema su cui effettuare la necessaria inversione di configurazione sul carbonio C(4) portante il gruppo ossidrilico. A questo scopo era stato ritenuto vantaggioso ricorrere ad una epimerizzazione termica condotta sul corrispondente mesil derivato. Così, il trans idrossichetone 2.38 è inizialmente trasformato (MsCl/Py) nel corrispondente mesilossichetone 3.21 e questo sottoposto ad epimerizzazione a riflusso in MeCN in presenza di AcONa (2-3 equiv). Dopo 72 h viene raggiunto l’equilibrio che corrisponde ad una miscela 4:6 (1H NMR) del mesilato trans di partenza 3.21 e dell’epimero cis 3.22 desiderato. I due mesilati epimeri non sono separabili attraverso le comuni tecniche cromatografiche, per cui la sintesi prosegue utilizzando la miscela grezza di reazione. La riduzione (NaBH4/CeCl3)18 della miscela
di 3.21 e 3.22 è completamente stereoselettiva su entrambi gli epimeri e conduce ad una corrispondente miscela dei due idrossimesilati cis 3.24 e trans 3.23, rispettivamente da 3.21 e 3.22 (Schema 3.9).
N O Cbz N O Cbz MsCl/Py 0 °C N O Cbz N O Cbz AcONa MeCN riflusso N OH Cbz MsO N OH Cbz NaBH4 MeOH -45 °C
HO MsO MsO MsO
MsO N Cbz t-BuOK CH3CN 2.38 3.21 3.21 3.22 3.24 3.23 2.2αααα-Me Me Me Me Me Me Me Me O
Schema 3.9. Sintesi dell’epossido 2.2α-Me a partire dall’intemedio idrossichetone 2.38.
Il motivo della completa stereoselettività osservata nella riduzione di entrambi i mesilossichetoni diastereoisomeri 3.21 e 3.22, è al solito riconducibile alla “rigidità, di fatto, di entrambi i composti, che esistono in soluzione esclusivamente nella corrispondente conformazione 3.21’ e 3.22’ con il gruppo metilico in posizione assiale. L’esclusivo attacco assiale della specie idruro, come normalmente osservato con il NaBH4, conduce selettivamente ai
due prodotti di riduzione, come riportato nello Schema 3.10.
N OH Cbz MsO NaBH4 MeOH -45 °C N O Cbz N O Cbz MsO MsO N OH Cbz MsO 3.21 3.22 3.24 3.23 Me Me Me Me N OBn O Me OMs O N OBn O Me N OBn O Me N OBn O Me H-BH3 -Cl3Ce O MsO H-BH3 -Cl3Ce OMsHO MsO HO 3.24' 3.21' 3.22' 3.23'
I due idrossimesilati cis 3.24 e trans 3.23 sono separati mediante flash cromatografia e mentre l’idrossimesilato cis 3.24 può essere teoricamente riciclato dopo ossidazione al mesil chetone 3.21, l’idrossimesilato trans 3.23 costituisce il precursore ultimo stabile dell’epossido 2.2α-Me (Schema 3.11). N Cbz Me t-BuOK 2.2αααα-Me O N Cbz Me OH MsO 3.23
Schema 3.11. Sintesi dell’epossido 2.2α-Me dal precursore idrossimesilato 3.23.
Come precedentemente anticipato, una volta ottenuto l’epossido 2.2α-Me abbiamo proceduto verso l’aziridina 2.3β-Me (Schema 3.12).
N Cbz Me N Cbz Me HO N3 N Cbz Me HO NH2 N Cbz Me HO NHNs TMGA CH3CN 0 °C PPh3 supp. pol. THF/H2O (20/1) NsCl Py 0 °C N NHNs Cbz MsO N Cbz Me 2.2αααα-Me 3.24 3.26 3.27 3.28ββββ 2.3ββββ-Me Ms/Cl Py 0 °C t-BuOK Me N Ns N Cbz Me HO N3 3.25 O
Schema 3.12. Sintesi dell’aziridina 2.3β-Me a partire dall’epossido 2.2α-Me .
Così, l’azidolisi di 2.2α-Me con TMGA conduce all’azido alcool trans 3.24, accompagnato da un altro prodotto di azidolisi (30%) che è risultato essere il corrispondente azido alcool diastereoisomero cis 3.25. Questa miscela non viene separata, ma direttamente utilizzata nella successiva reazione di riduzione con PPh3 supportata su polimero. Si ottiene, dopo filtrazione, un
grezzo di reazione costituito essenzialmente dal trans ammino alcool 3.26 che, per successive reazioni di N-nosilazione e di O-mesilazione, conduce prima all’N-nosil derivato 3.27 e quindi al trans N-nosil-O-mesilato 3.28β, che costituisce l’ultimo precursore stabile della N-nosil aziridina 2.3β-Me.
3.3 Sintesi di azapiranosidi ed azazuccheri piperidinici
Come primo approccio per verificare la possibilità di trasformare i glicosidi-2,3-insaturi di tipo 3.1 e 3.2 in corrispondenti azapiranosidi ed azazuccheri piperidinici 3.3 e 3.6, come tali o parzialmente protetti, sono stati presi inizialmente in esame l’i-propil N-Cbz-immino-α-O-glicoside 3.29α e l’i-propil 4-N-(nosilammino)-β-O-N-Cbz-immino-α-O-glicoside 3.30β ottenuti per glicosilazione dell’isopropanolo da parte, rispettivamente, dell’epossido 2.2α-Me, derivato del D,L -imminoallale e della N-nosil aziridina 2.3β-Me, derivata del D,L-imminogalattale, più facilmente accessibili dal punto di vista sintetico (Schema 3.13). La scelta dell’alcool, in questo caso l’i-PrOH, è stata dettata da puri motivi di praticità, selettività e rapidità della reazione e purezza del glicoside in ciascun caso ottenuto e non da considerazioni di altro tipo legate alla natura dei composti che andavamo preparando. Infatti, come si può ricavare dallo Schema generico di trasformazione (Schema 3.1), il gruppo alcossi presente sul C(1) dell’alchil O-imminoglicoside impiegato, con relativa configurazione α o β, mentre rimane presente nel corrispondente azapiranoside ed anzi può risultare importante nella costruzione dello stesso attraverso il suo effetto dirigente di tipo sterico e/o elettronico, viene irrimediabilmente perso nella successiva fase di trasformazione nel corrispondente azazucchero piperidinico. Ragionevolmente, l’eventuale successo ottenuto nella trasformazione a “veri” azazuccheri di questi due imminoglicosidi presi a modello, poteva poi essere trasferito anche ai corrispondenti glicosidi ottenuti dall’epossido 2.2β-Me ed N-nosil aziridina 2.3α-Me, diastereoisomeri.
Così, utilizzando una procedura precedentemente messa a punto nel nostro laboratorio, per trattamento di una soluzione del trans idrossimesilato 3.23 in i-PrOH con t-BuOK, secondo condizioni di protocollo A, si ottiene l’i-propil N-Cbz-immino-α-O-glicoside 3.29α19 che viene purificato mediante cromatografia flash. Allo stesso modo il trattamento di una soluzione del trans N-nosil-O-mesilato 3.28 in i-PrOH con K2CO3 conduce all’i-propil
4-N-(nosilammino)-β-O-glicoside 3.30β,11 anch’esso poi purificato mediante cromatografia flash (Schema 3.13). Come già discusso nelle pagine precedenti, in entrambi i casi si assiste ad un completo trasferimento
della configurazione del sistema eterociclico (epossido o aziridina) al carbonio anomerico C(1) dell’O-glicoside sintetizzato, nel concetto di glicosilazione completamente regio- e stereoselettiva, direttamente substrato-dipendente (vedi anche Capitolo 2, Paragrafo 2.3, 2.5)
N Cbz N Cbz N Cbz t-BuOK 3.29αααα OH MsO HO O-i-Pr 3.23 2.2αα−αα−−−Me Me O i-PrOH N Cbz N Cbz N Cbz t-BuOK 3.30ββββ NHNs MsO NsHN O-i-Pr 3.28ββββ 2.3ββββ−−−Me− Me N i-PrOH Ns Me Me Me Me
Schema 3.13.Sintesi dell’i-propil-α-O-glicoside 3.29α e dell’i-propil-β.O-glicoside 3.30β in condizioni di
protocollo A.
Per continuare la sintesi di un corrispondente azazucchero, si poneva a questo punto il problema di funzionalizzare il doppio legame presente in 3.29α in modo possibilmente completamente regio- e stereoselettivo, al fine di evitare la formazione indesiderata di diastereoisomeri e/o regioisomeri. Varie possibilità erano chiaramente a disposizione, tra cui, da ricordare, la diidrossilazione diretta, la diidrossilazione indiretta attraverso idrolisi di un intermedio epossido, la ammino idrossilazione, la idroborazione-ossidazione, la ossimercuriazione-demercurazione. Tra queste abbiamo scelto, come primo approccio, e come anche rappresentato nello Schema 3.1, la diidrossilazione diretta attraverso il classico protocollo OsO4 (catalitico)/N-metilmorfolina-N-ossido (NMO) che, introducendo due gruppi ossidrilici in
modo completamente sin-stereoselettivo, non avrebbe comportato, quantomeno, alcun problema di regioselettività. Inoltre, considerato il meccanismo della reazione di diidrossilazione (reazione in un unico stadio) e tenuto conto della struttura del i-propil O-immino-glicoside 3.29α ed in particolare della configurazione relativa (α) di entrambi i due sostituenti allilici, il gruppo
O-i-propile sul C(1) ed il gruppo ossidrilico sul C(4) c’era ragionevolmente da attendersi un risultato ampiamente, se non completamente stereoselettivo. Evidentemente, erano plausibili due possibilità: una β-stereoselettività nel caso in cui i gruppi sostituenti sopra ricordati, tutti diretti verso la faccia α, avessero esercitato solo un effetto sterico, dirigendo, di conseguenza, l’attacco elettrofilo verso la opposta faccia β del sistema imminoglicosidico insaturo. In questo caso l’effetto sterico dei due sostituenti allilici avrebbe potuto essere indebolito dalla opposta disposizione, se influente, del gruppo metilico sul C(5). Alternativamente, nel caso in cui i due gruppi allilici, ed in particolare il gruppo ossidrilico sul C(4), avessero esercitato un effetto dirigente collegato alla loro capacità di dare luogo a legami ad idrogeno con la specie elettrofila, avremmo assistito ad un attacco elettrofilo altamente o completamente α stereoselettivo. In questo secondo caso, infatti, l’attacco elettrofilo sarebbe ragionevolmente avvenuto verso la stessa faccia (α) verso cui si dirigono i due gruppi allilici.
N Cbz N Cbz 3.29αααα 3.31 3.31b HO HO O-i-Pr O-i-Pr N Cbz HO O-i-Pr Me Me OH OH OH OH β-diidrossilazione
(effetto sterico dei sostituenti)
α-diidrossilazione (effetto di coordinazione dei sostituenti)
Me
Schema 3.14. Reazione di diidrossilazione dell’i-propil α-O-imminoglicoside 3.29α e possibili prodotti
azapiranosidici.
La reazione di diidrossilazione (OsO4/NMO) dell’i-propil α-O-imminoglicoside 3.29α è
risultata completamente β-stereoselettiva e conduce al solo i-propil α-O-N-Cbz-imminomannopiranoside 3.31 ad indicare come l’effetto sterico costituisca l’esclusivo fattore di controllo della reazione stessa.
N Cbz N Cbz 3.29αααα 3.31 HO HO O-i-Pr Me O-i-Pr OH OH OsO4 NMO Me
Schema 3.15. Sintesi dell’ i-propil α-O-N-Cbz-imminomannopiranoside 3.31.
L’esatta determinazione strutturale dell’imminopiranoside 3.31 rivestiva una particolare importanza, non solo per sé stessa, ma anche in quanto avrebbe dimostrato a posteriori la struttura del glicoside insaturo 3.29α di provenienza e, quindi, di conseguenza avrebbe sostenuto quello che costituiva l’aspetto più importante di tutta questa fase della ricerca, ovvero l’esattezza del processo di glicosilazione direttamente substrato-dipendente. La struttura del glicoside insaturo 3.29α, al pari di quella di tutti gli altri glicosidi a suo tempo sintetizzati (Schema 2.12), per quanto sufficientemente certa sulla base di analogie con il sistema 6-benzilossi sostituito studiato in precedenza nel nostro laboratorio,7 mancava di una dimostrazione inequivocabile e definitiva.
Infatti nei sistemi insaturi quali il glicoside 3.29α, gli esperimenti NOE, che avrebbero potuto essere di valido aiuto nella assegnazione strutturale, per quanto non fossero mai in contraddizione con le strutture assegnate, non erano, d’altro canto, mai così evidenti come sarebbe stato opportuno fossero e come ci saremmo potuti aspettare da molecole così semplici. D’altro canto, anche tutte le consuete considerazioni strutturali basate sulle costanti di accoppiamento dei protoni del sistema ciclico, in molecole come 3.29α non erano in alcun modo possibili. Ora, con la sintesi dell’imminopiranoside 3.31, avevamo la possibilità di eseguire uno studio approfondito della sua struttura il cui risultato avrebbe confermato o, alternativamente, smentito quanto ammesso fino a questo momento riguardo agli O-glicosidi-2,3-insaturi come 3.29α, sia dal punto di vista strutturale che meccanicistico relativamente alla loro formazione.
Prima di entrare nel particolare della indagine strutturale condotta sull’imminopiranoside 3.31, è opportuno portare all’attenzione un aspetto molto importante di questi sistemi imminoglicalici N-Cbz sostituiti derivati da piperidine e piperidine-2,3-insature. Sulla base di studi conformazionali condotti allo scopo, nel sistema piperidino-2,3-insaturo come quello presente nel i-propil α-O-imminoglicoside 3.29α ed in tutti i glicosidi ad esso strutturalmente riconducibili, non vi è di fatto alcun equilibrio conformazionale, essendo presente in soluzione solo il conformero in cui il gruppo alchilico (il metile, nel caso in esame) sul C(5) adiacente
all’azoto imminico è necessariamente disposto in modo assiale. Questa disposizione è infatti la sola in grado di minimizzare la sfavorevole interazione sterica tra il gruppo alchilico stesso ed il gruppo benzilossicarbonilico del sistema uretanico adiacente (allylic strain, A1,3).7,11
N Cbz 3.29αααα 3.31 HO O-i-Pr Me OH OH OsO4 NMO unico conformero N O OBn OH Me O-i-Pr
Schema 3.16 . Razionalizzazione della reazione di osmilazione dell’ i-propil-α-O-glicoside-2,3-insaturo 3.29α.
Nel caso specifico relativo all’i-propil-α-O-glicoside-2,3-insaturo 3.29α, quanto detto sopra implica che anche il gruppo isopropossi presente sul C(1) sia pseudoequatoriale e che il gruppo – OH sul C(4) sia pseudoassiale. Tutta questa rigidità conformazionale tipica di questi sistemi imminoglicalici-2,3-insaturi, decade del tutto con la scomparsa del doppio legame a seguito della sua funzionalizzazione (nel presente caso attraverso diidrossilazione) con formazione di composti cicloesanici a sedia che esistono normalmente come un equilibrio tra i due possibili conformeri ed in cui il prevalere di un conformero sull’altro resta determinato dalla natura assiale od equatoriale dei sostituenti, secondo le consuete regole che classicamente governano questo tipo di equilibri. Così, tornando al nostro sistema saturo e completamente funzionalizzato costituito dall’ imminopiranoside 3.31, esso potrà esistere come un equilibrio dei suoi due possibili conformeri 3.31’ e 3.31”, come qui sotto mostrato.
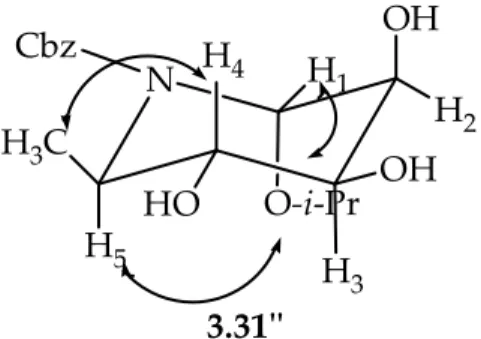
N Me OH OH O-i-Pr HO Cbz N H1 O-i-Pr H2 OH OH H3 OH H4 CH3 H5 N i-Pr-O H1OH H2 H3 OH H4 HO H5 H3C 3.31 3.31' 3.31'' Cbz Cbz
Tra questi, il conformero 3.31’’ portante il più importante gruppo metilico in posizione equatoriale, dovrebbe essere favorito all’equilibrio e quindi essere presente in largo eccesso rispetto al conformero 3.31’ in cui lo stesso gruppo verrebbe a trovarsi in una più sfavorevole posizione assiale. Questa preferenza conformazionale resta dimostrata dal fatto che la costante di accoppiamento H(5)-H(4) è elevata (J4,5= 9.5 Hz) a dimostrazione della loro disposizione
1,2-trans diassiale. L’esistenza poi di chiari ed inequivocabili NOEs tra H(5) ed il –CH- ed i due metili del gruppo –O-i-Pr indicano chiaramente come anche quest’ultimo gruppo sia disposto assialmente, confermando di fatto quanto più ci stava a cuore, ovvero che il processo di glicosilazione dell’epossido 2.2α-Me era proceduto in modo completamente α-stereoselettivo come sempre ammesso. La presenza, poi, di un segnale relativo al protone H(4) in forma di doppio-doppietto con elevate costanti di accoppiamento [H(4), J = 9.5, 8.5 Hz] indica come anche il protone H(3) sia assiale e che, quindi conseguentemente, il processo di diidrossilazione sia avvenuto sulla faccia β del sistema insaturo. Tutto ciò è poi confermato dalla costante di accoppiamento del protone anomerico H(1) che è piccola (J1,2 = 2.6 Hz), in quanto corrisponde
ad un accoppiamento 1,2-trans-diequatoriale. Riassumendo, la presenza di NOE tra H(5) ed il gruppo isopropilico conferma la configurazione relativa al processo di glicosilazione dell’epossido 2.2α-Me, mentre la J3,4= 8.5 Hz conferma la configurazione del processo di
diidrossilazione del glicoside insaturo 3.29α ed entrambe le osservazioni confermano la struttura del prodotto saturo, il glicoside 3.31. Altri effetti NOE consistenti con la struttura data sono riportati nella Figura 3.1.
N O-i-Pr H1 OH H2 H3 OH H4 HO H5 H3C 3.31'' Cbz
Figura 3.1. Effetti NOE riscontrati nell’i-propil-α O-N-Cbz-imminomannopiranoside 3.31.
L’imminopiranoside 3.31 è stato quindi sottoposto al protocollo da noi individuato per la sua trasformazione in azazucchero piperidinico. Questo protocollo consiste essenzialmente nella deprotezione del gruppo uretanico mediante idrogenolisi catalitica (H2-Pd/C), una procedura che
nel composto in esame determina anche l’allontanamento del gruppo isopropossi, come sarà poi descritto nello Schema 3.18.20 Poiché un protocollo di questo tipo avrebbe condotto ad un composto finale particolarmente polare e quindi possibilmente difficile da estrarre e purificare, abbiamo pensato di applicare la procedura sopra descritta non al glicoside 3.31, bensì al suo tri-acetil derivato 3.32. Così il trattamento dell’imminopiranoside 3.31 con Ac2O/Py trasforma lo
stesso nel tri-acetil derivato 3.32 che viene quindi sottoposto a idrogenolisi catalitica. Si ottiene un grezzo di reazione che dopo purificazione mediante TLC preparativa fornisce l’azazucchero 3.37, sotto forma di triacetil derivato che non è stato ulteriormente elaborato (Schema 3.18).
N Me OAc OAc O-i-Pr AcO 3.32 H2 Pd/C N Me OAc OAc O-i-Pr AcO O OBn O OH 3.33 + PhCH3 -CO2 H N O-i-Pr AcO Me OAc OAc 3.34 - i-Pr-O -N H + Me OAc OAc AcO -H+ N Me OAc OAc AcO 3.35 3.36 H2 Pd/C H N OAc OAc Me AcO 3.37 3.31 Ac2O Py
Schema 3.18. Sintesi dell’azazucchero piperidinico 3.37.
In questo ultimo passaggio, come già anticipato, assieme alla deprotezione del gruppo amminico si ha la perdita del gruppo isopropossi tipico dei sistemi glicosidici. Questo inevitabile evento è conseguenza della instabilità dell’emiaminale 3.34 che si ottiene al momento della idrogenolisi del gruppo uretanico con formazione del gruppo amminico endociclico libero. La spontanea eliminazione del gruppo alcossi con contemporanea formazione dell’immina ciclica 3.36, poi ridotta nell’ambiente di reazione, conduce al sistema azazucchero piperidinico 3.37 privo del sostituente sul C(2) (Schema 3.18). Comunque, anche se non completamente portata a
termine, la procedura per la trasformazione del glicoside insaturo 3.31 nell’azazucchero 3.37 era stata individuata e realizzata.
Sono quindi passata a verificare la possibilità di effettuare la medesima trasformazione in azazucchero sull’i-propil 4-ammino-β-O-glicoside 3.30β. Il protocollo da impiegare non poteva essere dello stesso tipo utilizzato in precedenza per l’O-glicoside 3.29α, in quanto ora avrebbe dovuto prevedere ad un certo punto anche la deprotezione del gruppo N-nosilammino e questa doveva necessariamente essere effettuata prima dell’allontanamento del gruppo protettivo N-Cbz. E’ stato così pensato di deproteggere subito il gruppo N-nosilammino. In questo modo, il glicoside 3.30β è stato trattato con PhSH/K2CO3 a dare un grezzo di reazione costituito dal
corrispondente O-glicoside-4-ammino sostituito 3.38 e dal tiofenolo in eccesso che viene direttamente sottoposto come tale ad acetilazione (Ac2O/Py). La successiva purificazione
mediante flash cromatografia di questo secondo grezzo di reazione permette di recuperare puro l’N-acetil derivato 3.39. Questo nuovo glicoside è stato quindi sottoposto a reazione di diidrossilazione con protocollo OsO4/NMO. Anche nel presente caso viene ottenuto in modo
completamente stereoselettivo un solo prodotto che ad appropriata analisi strutturale, risulta essere l’i-propil 4-desossi-4-acetilammino-β-O-N-Cbz-imminogulopiranoside 3.40. Anche in questa circostanza, la selettività faciale della reazione di diidrossilazione risente dell’effetto sterico dei due sostituenti allilici, il gruppo –NHAc ed il gruppo –O-i-Pr, che fiancheggiano il doppio legame. In quanto diretti entrambi verso la faccia β del sistema ciclico insaturo, i due sostituenti allilici dirigono l’attacco elettrofilo dell’OsO4 dalla opposta faccia α in modo completo
N N N N O-i-Pr Me NsHN H2N Me O-i-Pr Me O-i-Pr AcHN O-i-Pr Me AcHN OH OH Cbz Cbz Cbz Cbz PhSH K2CO3 Ac2O Py OsO4 NMO 3.30ββββ 3.38 3.39 3.40
Schema 3.19. Sintesi dell’i–propil β-O-N-Cbz-imminomannopiranoside 3.40.
La struttura di 3.40 è stata confermata dalle seguenti considerazioni e misure. Per quanto detto a proposito del piranoside 3.37, anche 3.40 esiste come un equilibrio tra i due conformeri a sedia 3.40’ e 3.40”. La presenza di NOE tra il protone H(5) ed il protone anomerico H(1) dà evidenza, al tempo stesso, della configurazione β sul carbonio anomerico e della prevalente presenza, all’equilibrio, del conformero 3.40 con il gruppo metilico equatoriale (Schema 3.20). L’osservazione, poi, della struttura di doppietto del segnale relativo al protone anomerico H(1) con valore elevato della relativa costante di accoppiamento H(1)-H(2) (J1,2= 11.3 Hz)permette di
dimostrare come il processo di diidrossilazione sia avvenuto con completa selettività β-faciale. I dati raccolti sono consistenti solo con la struttura 3.40, correttamente assegnata all’unico prodotto della reazione di diidrossilazione di 3.39.
N Me OH OH O-i-Pr AcHN Cbz N O-i-Pr H1 OH H2 H3 OH H4 AcHN CH3 H5 N H1 O-i-Pr H2 OH OH H3 NHAc H4 H5 H3C 3.40 3.40' 3.40'' Cbz Cbz
L’imminoallopiranoside 3.40 è stato quindi sottoposto a reazione idrogenolisi (H2/Pd-C)
allo scopo di rimuovere il gruppo protettivo –Cbz. Il grezzo di reazione ottenuto viene immediatamente acetilato a dare un nuovo grezzo che, per quanto alquanto sporco, dà evidenza, all’esame 1H NMR, della presenza del tetracetil derivato 3.42: 1H NMR δ 5.01 (dd, 1H, J = 11.8, 3.5 Hz), 4.10-4.33 (m, 1H), 3.35-3.68 (m, 3H), 2.08 (s, 3H), 2.07 (s, 3H), 1.96 (s, 3H), 1.88 (s, 3H). Sfortunatamente, il tentativo di separazione/purificazione effettuato su tale grezzo mediante TLC preparativa non ha avuto successo, in quanto è stato recuperato un prodotto solo non ancora puro, in quanto molto simile al grezzo (Schema 3.21).
N Me OH OH O-i-Pr AcHN Cbz 3.40 N N H Ac OAc OAc OH OH Me AcHN Me AcHN Ac2O H2 Pd/C 3.41 3.42
Schema 3.21. Trasformazione dell’i-propil β-O-N-Cbz-imminoallopiranoside 3.40 nell’azazucchero piperidinico 3.42.
Riteniamo sia necessario ripetere la procedura di trasformazione di 3.42 nel corrispondente azazucchero piperidinico utilizzando una strategia leggermente diversa da quella sopra impiegata e che preveda l’effettuazione della reazione di idrogenolisi possibilmente su substrati non acetilati.20 Non avendo avuto più tempo a disposizione, non ho potuto dare seguito a queste nostre intenzioni.
Considerata la semplicità e l’efficacia dimostrativa delle strutture degli O-glicosidi-2,3-insaturi ottenuti nelle reazioni di glicosilazione degli alcooli da parte degli epossidi 2.2α-Me e 2.2β-Me e delle aziridine 2.3α-Me e 2.3β-Me, attraverso la reazione di diidrossilazione, abbiamo voluto ripetere una corrispondente dimostrazione anche sul i-propil 4-ammino-α-O-imminoglicoside 3.43α ottenuto quale unico prodotto nella reazione di glicosilazione dell’i-PrOH da parte della N-nosil aziridina 2.3α-Me seguendo la procedura classica K2CO3 (3 equiv) in
i-PrOH (protocollo A) (Schema 3.22).11 Il glicoside 3.43α, preso come appropriato campione della reazione di glicosilazione completamente α-stereoselettiva, cui l’aziridina 2.3α-Me dà luogo nelle sue reazioni con gli alcooli (vedi paragrafo 2.4), riveste, come anche il precedente glicoside 3.29α per quanto riguarda gli epossidi, un ruolo centrale nel concetto di glicosilazione
direttamente substrato-dipendente con cui erano sempre state descritte le reazioni di questi sistemi eterociclici derivati da imminoglicali con gli alcooli. Infatti, come descritto nel Capitolo 2, paragrafo 2.3 di questa tesi, nelle loro reazioni con i nucleofili i derivati imminoglicalici mostrano prevalentemente una selettività β (selettività prodotto-dipendente) come conseguenza del preferenziale attacco pseudoassiale da parte del nucleofilo.8 Avere con l’aziridina 2.3α-Me, come già visto in precedenza con l’epossido 2.2α-Me, un processo di addizione nucleofila completamente α-stereoselettiva costituisce un risultato decisamente importante, in quanto in contrasto con il consueto comportamento di questi sistemi, che merita quindi di essere confermato in modo non equivoco. Lo studio precedentemente condotto sull’imminopiranoside 3.29α aveva di fatto confermato il processo di O-glicosilazione substrato-dipendente nel sistema ossiranico costituito dall’epossido 2.2α-Me: ora era opportuno procedere allo stesso modo anche con il sistema aziridinico costituito dalla aziridina 2.3α-Me.
N Me OH OH O-i-Pr NsHN Cbz N H1 O-i-Pr H2 OH OH H3 NHNs H4 CH3 H5 N O-i-Pr H1 OH H2 H3 OH H4 NsHN H5 H3C 3.44 3.44' 3.44'' N Cbz NsHN O-i-Pr Me N Cbz N Me Ns K2CO3 i-PrOH OsO4 NMO 2.3αααα-Me 3.43αααα Cbz Cbz N Cbz Me 3.28αααα NHNs MsO
Schema 3.22. Sintesi ed equilibrio conformazionale del prodotto 3.44.
Così l’i-propil 4-ammino-α-O-glicoside 3.43α è stato trattato come di consueto con protocollo OsO4/NMO. Viene ottenuto un unico prodotto, risultato di una reazione
completamente selettiva, la cui struttura è risultata essere quella mostrata in 3.44. La dimostrazione strutturale di 3.44 deriva dalle seguenti considerazioni e misure. Innanzi tutto l’equilibrio conformazionale all’interno di 3.44 è ragionevolmente spostato verso il conformero
3.44’, con il gruppo metilico ed –NHAc equatoriali, come confermato dalla presenza di NOE tra H(5) ed il gruppo -O-i-Pr. La presenza di questo NOE associato alla completa assenza di alcun effetto NOE tra H(1) e H(5) confermano la configurazione necessariamente α del O-glicoside e quindi la correttezza dell’assegnazione a suo tempo data al carbonio anomerico del glicoside 3.43α di provenienza e quindi in definitiva al processo di glicosilazione della aziridina 2.3α-Me.11 La natura di doppietto del protone anomerico H(1) con costante di accoppiamento H(1)-H(2) piccola (J1,2 = 2.7 Hz) indica poi come il processo di diidrossilazione sia avvenuto con
completa selettività faciale sulla faccia β del sistema insaturo, ovvero, correttamente, dalla parte opposta a quella verso cui si dirigono i due sostituenti allilici, un comportamento, questo, già osservato anche nella diidrossilazione dei sistemi insaturi esaminati in precedenza.