8
INTRODUZIONE
1 Il fibrinogeno
La coagulazione consiste in una serie di reazioni che hanno come prodotto finale la trasformazione di una proteina solubile, il fibrinogeno (Fg), in fibrina (Fn) a seguito dell’azione della trombina. Il Fg è presente nel plasma a una concentrazione di ca 3 mg/ml. (1,2)
1.1
Sintesi del fibrinogenoGeni codificanti le catene del fibrinogeno e regolazione dell’espressione genica
Il Fg umano è il prodotto di 3 geni che sono localizzati in una regione di 50kb sul cromosoma 4q28 (3-4)in questo ordine FGG (6 esoni 8.5kb),FGA (8 esoni 5.4kb) e FGB (10 esoni 8kb),(5) codificanti
rispettivamente la catena γ,Aα,Bβ. FGB è orientato in una direzione di trascrizione opposta a quella di FGG e FGA. (3)
L’espressione genica è coordinata a livello epatico. L’mRNA dei 3 geni viene prodotto in quantità uguali ed è costitutivamente espresso a livello epatico. (6,7) FGG, ma non FGA e FGB, è inoltre
espresso anche in altri tessuti. La produzione extraepatica è comunque inferiore rispetto a quella epatica. Il Fg di origine extraepatica ha un ruolo indipendente rispetto all’emostasi: per esempio l’importante upregulation del Fg in corso di polmonite da P.carinii suggerisce che il Fg possa contribuire alla patogenesi della fibrosi polmonare. (8) Il mecccanismo di regolazione dell’espressione
extraepatica di FGG non è stato chiarito, mente l’espressione epatica di FGA e FGB è controllata da fattori di trascrizione epato-specifici come HNF-1 (hepatocyte-specific nuclear factor-1).(9)
Il Fg è una proteina di fase acuta, infatti l’espressione epatica è up-regolata da IL-6. (9,10) I FDP
possono stimolare i monociti-macrofagi circolanti a produrre IL-6.(11) Esiste inoltre una regione
glucocorticoidi sensibile a livello dell’estremità 5’ di FGB e FGG, che può mediare l’up regolazione dell’espressione del Fg. (12)
Esistono fattori che inibiscono l’espressione genica del Fg: citochine antiinfiammatorie 4, 10, IL-13(13), paradossalmente anche la citochina proinfiammatoria IL-1β (14), TGF-β. (15)
Sintesi proteica
I sei polipeptidi che vanno a formare il Fg vengono sintetizzati a livello del reticolo endoplasmatico.
(16) Le catene γ e i complessi Aα-γ vanno ad accumularsi formando dei pool intracellulari, mentre le
catene Bβ sono rapidamente incorporate in complessi Bβ-γ, poi in monomeri (Aα,Bβ,γ)1 e infine in
dimeri (Aα, Bβ,γ)2.(pulse- chase experiments). Mentre la sintesi delle catene del Fg, ad eccezione
della catena γ, è epatospecifica, l’assemblamento delle catene per la formazione del dimero non è epatospecifica.(17)
9
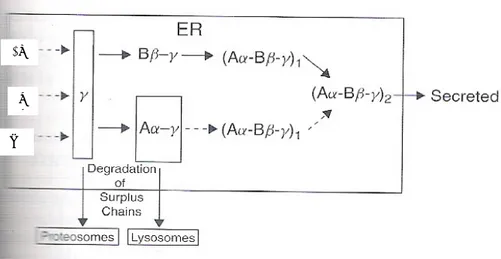
FIG 1.1 Fasi di assemblamento e secrezione della molecola del Fg. Le 3 catene sono sintetizzate
separatamente. Le catene Aα e γ si accumulano come pool intracellulari. I dimeri del Fg vengono inviati all’apparato di Golgi dove avvengono modifiche post-trascrizionali. Le catene in eccesso sono degradate da lisosomi e proteosomi. (da Hemostasis and Thrombosis Basic Principles and Clinical Practice 5th edition) La formazione dei complessi iniziali a due catene (Aα-γ e Bβ-γ) è condizionata dalle metà C terminali delle regioni coiled-coil , dai ponti disolfuro intracatena a livello delle catene Bβ e γ (16)e da alcuni
residui aa nella regione C terminale delle catene γ.(18) I legami disolfuro intracatena sono inoltre
importanti anche per la secrezione della molecola del Fg.(16)
Eterogeneità della molecola del fibrinogeno
L’eterogeneità della molecola del Fg dipende da modificazioni a vario livello durante e dopo la biosintesi: splicing alternativo dell’mRNA, solfatazione di aa, fosforilazione,vario grado di glicosilazione, proteolisi, polimorfismi genetici . Questi meccanismi sono alla base di oltre un milione di forme diverse di Fg .Questa eterogeneità può cambiare le proprietà del coagulo di Fn e influenzare il processo di guarigione delle ferite.(19)
1.2
Distribuzione e metabolismo del fibrinogeno nell’organismoDa 1,7 a 5,0 g/die di Fg sono sintetizzati dal fegato. Tale produzione può essere incrementata di ben 20 volte. Circa il 75% del Fg sintetizzato si trova nel plasma, mente il restante 25% è distribuito nello spazio interstiziale e nella linfa. L’emivita della molecola è di 3-5 giorni. La modalità di catabolismo del Fg rimane ad oggi sconosciuta (trombina e plasmina contribuiscono a ridurre i valori del Fg solo per il 2-3%).(20,21)
Il Fg è inoltre presente anche negli α granuli delle PLT. Il Fg all’interno delle PLT non presenta la catena γ’. (22)La maggior parte degli studi svolti dimostra che il Fg nei megacariociti e nelle PLT
origina dall’endocitosi recettore mediata del Fg plasmatico, che viene poi immagazzinato negli α granuli. (23) Il recettore interessato è l’integrina α
IIbβ3, coinvolta anche nell’aggregazione delle PLT.
Infatti nei soggetti con tromboastenia di Glanzmann gli α granuli non contengono Fg. (24)
Meno della metà dei livelli di Fg plasmatico sono determinati geneticamente, infatti i livelli di Fg aumentano con l’età, gravidanza, menopausa, fumo, ipertensione, diabete mellito tipo 2, infezioni, obesità, mentre calano a seguito di un consumo moderato di alcol. L’associazione di elevati livelli di
Bβ
γ γ Aα
10
Fg e CVD ha fatto ipotizzare che il Fg possa essere un fattore di rischio per CVD, tuttavia un’altra interpretazione è possibile: dal momento che il Fg è una proteina di fase acuta i livelli elevati di Fg potrebbero riflettere l’infiammazione cronica a livello della placca aterosclerotica vascolare.(25)
1.3
Struttura della molecola del fibrinogenoIl fibrinogeno è una glicoproteina ricca di acido sialico, di peso molecolare ca 340 kD, lunga 45 nm, formata da 2 subunità identiche, ognuna costituita a sua volta da tre coppie distinte di catene (Aα,Bβ e γ), unite da legami disolfuro.(26) La molecola di Fg contiene 4 catene saccaridiche: una per
ciascuna catena Bβ e una per ciascuna catena γ. La catena α è invece priva di oligosaccaridi.
Le sequenze amminoacidiche delle 3 catene sono omologhe , si ritiene infatti che originino tutte da un comune precursore ancestrale.(27) Tuttavia ci sono importanti differenze strutturali e funzionali
tra le 3 catene.
Caratteristiche delle 3 catene del Fg:
Catena α Catena β Catena γ
Peso molecolare (28) 67 kDa 54 kDa 47 kDa
Aminoacidi (29) 610 461 411
Fp FpA (16 aa) FpB (14 aa) Nessuno
Carboidrati (30,31) Nessuno 2 oligosaccaridi 2 oligosaccaridi
TAB 1A
Esistono varianti strutturali meno comuni:
- la catena γ’ in cui gli amminoacidi dal 408 al 411 sono sostituiti con una catena di 20 aa che termina con leucina in posizione 427.(32) La catena γ’ costituisce circa l’8% delle totale delle
catene γ del Fg. Il 15% delle molecole di Fg sono eterodimeri γA/γ’ (17), mentre <1% sono omodimeri γ’/γ’.
- la variante AαE (<2%) con un aa in più in posizione 236 all’estremità C-terminale. (33)
Il dimero del Fg è tenuto insieme da 5 ponti disolfuro simmetrici che si formano a livello dell’estremità N-terminale delle 3 catene (un legame tra le due catene Aα 28,due tra le 2 catene γ8- γ9, e infine due tra Aα 36 e Bβ 65). Solo i due legami Aα-Bβ e i 2 legami tra le catene γ sono sufficienti e necessari per garantire la formazione del dimero, mentre i due legami tra le catene Aα non sono sufficienti a mantenere la struttura dimerica. (17,34) Altri ponti disolfuro non simmetrici
intercatene sono detti anelli disulfurici. (35)
Le due catene γ sono disposte in maniera antiparallela, così come i ponti disolfuro tra le 2 catene.(26)
11
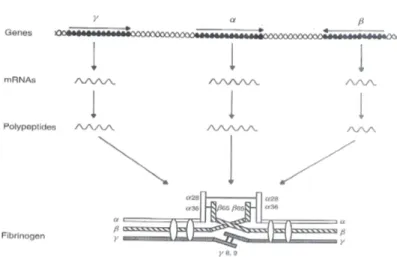
FIG 1.2 Rappresentazione schematica della biosintesi epatica del Fg. Le frecce poste al di sopra dei singoli geni
indicano la direzione di trascrizione. L’m-RNA è processato e trasportato dal nucleo al citoplasma dove avviene la sintesi dei polipeptidi, che vengono poi assemblati a formare la molecola del Fg. (da Hemostasis and Thrombosis Basic Principles and Clinical Practice 5th edition)
1.3.1
Struttura della molecola alla microscopia elettronicaIl Fg è stata la prima macromolecola studiata al microscopio elettronico nel 1959 da Hall e Slayter.
(37)
È composta da tre strutture nodulari collegate tra loro mediante rod like strands lineari e presenta una regione centrale E e due regioni terminali D, due regioni αC e due regioni BβN.
Le regioni αC sono le prime porzioni della molecola del Fg ad essere clivate dalla plasmina nel processo di fibrinolisi portando alla formazione del frammento X. Queste regioni costituiscono la cosiddetta protuberanza Aα, dal momento che emergono dalla regione D. (38)
Anche le regioni BβN (o regioni βN nella Fn perché FpB è stato rimosso) vengono rimosse dalla molecola di Fg mediante l’azione della plasmina e sono localizzate alle estremità N terminali delle catene β.
Sulla catena Aα sono presenti 2 siti suscettibili di proteolisi da parte della plasmina. Il clivaggio plasmina mediato di parte della catena Aα (che corrisponde alla regione αC) dà luogo alla formazione del cosiddetto frammento X. L’altro sito d’azione della plasmina è comune a tutte e 3 le catene del Fg ed è localizzato nella regione coiled-coil e comporta la formazione di un frammento Y e un frammento D. Infine il frammento Y verrà tagliato in 2 frammenti , rispettivamente D e E. (39,40)
12
1.3.2
Analisi cristallografica della molecola del fibrinogenoPiù recentemente, nel 2001, l’analisi cristallografica del fibrinogeno del pollo (e del Fg bovino modificato) ha fornito maggiori informazioni sulla struttura della molecola.(41,42) Il fibrinogeno del
pollo presenta un’elevata omologia con il Fg umano, però manca di una porzione flessibile all’estremità C-terminale della catena α che impedisce la cristallizzazione del Fg umano.
Le 3 catene del Fg formano una struttura a tripla elica detta coiled coil lineare, che collega una struttura nodulare centrale detta regione E (formata dalle estremità N terminali di tutte e sei le catene del Fg legate da ponti disulfurici), con due strutture nodulari distali dette regioni D (formate dalle estremità C terminali delle sole catene Bβ e γ).
La regione E comprende 4 domini strutturali distinti: γN domain (estremità N terminali delle due catene γ), funnel-shaped domain (estremità N terminali delle catene Aα e Bβ) e 2 coiled-coil E domains (formati dalle estremità C terminali di tutte e 3 le catene del Fg).
Ciascuna regione D comprende 7 domini strutturali: ciascuna delle estremità C terminali delle catene β e γ forma 3 domini (NH2 terminal A-domain , central B domain e COOH-terminal Polimerization-domain) per un totale di 6 domini complessivi, c’è inoltre un coiled-coil D domain (estremità N terminali di tutte e 3 le catene del Fg).(43,44)
È stata inoltre studiata la struttura della regione αC che comprende una parte compatta (αC domain) e una parte flessibile (αC connector).(45)
Le due regioni BβN formano 2 domini che contengono importanti siti funzionali che vengono esposti dopo il clivaggio del FpB .(46)
13
FIG 1.4
A. Composizione polipeptidica del Fg. La catena Aα in blu, la Bβ in verde e la γ in rosso. FpA e FpB sono in magenta;i ponti disulfidici sono rappresentati con linee nere; la freccia singola indica i punti di clivaggio delle regioni αC e BβN, mente le triple frecce indicano i punti di clivaggio proteolitico tra le regioni D e E. B. Struttura cristallografica del Fg. La struttura nodulare centrale è formata dalle porzioni N-terminali di
tutte e 6 le catene, è connessa alle strutture nodulari β e γ distali (formate dalle porzioni C-terminali delle catene rispettivamente β e γ) da due strutture a tripla elica coiled-coil, alla cui formazione contribuiscono tutte e 3 le catene.
C. Sono mostrate i domini αC collegati alla molecola da una parte flessibile (αC connectors) e le regioni BβN. Si evidenziano inoltre il funnel-shaped domain al centro che comprende il FpA.(magenta). Si rilevano i singoli domini delle regioni D: A,B,P . Con gli asterischi sono indicati gli holes a livello del dominio P sulla catena γ e β. In grigio sono indicati i punti di clivaggio tra i frammenti D e E.
(da L Medved, Weisel JW, on behalf of fibrinogen and FXIII subcommittee of the scientific standardization committee of the international society on thrombosis and haemostasis. Recommendations for nomenclature on fibrinogen and fibrin J Thromb Haemost 2009;7:355-9)
14
1.4
Formazione del coagulo1.4.1
Azione della trombina sul fibrinogenoIl fibrinopeptide A (FpA FpA sito di clivaggio Arg16-Gly17) e il fibrinopeptide B (FpB FpB sito di clivaggio Arg 14-Gly15) sono presenti all’estremità N-terminale delle catene Aα e Bβ e vengono clivati dalla trombina. La trombina si lega al Fg attraverso un sito di riconoscimento detto exosite 1.(47) I Fp costituiscono meno del 2% della molecola del Fg, ma il loro clivaggio espone dei siti di
legame detti knobs “A” e “B” nella regione E, che sono complementari a siti detti holes “a” e “b” esposti costantemente a livello delle regioni D.(48,49)
EA Porzione N terminale della catena α compresa tra i residui 17-20
Porzione della catena β compresa tra i residui 15-42 (50)
Da Porzione della catena γ tra residui 337-379(51)
EB Porzione sulla catena β che inizia con β15-18
Db Localizzato nella porzione C terminale della catena β (51)
TAB 1B
Una volta avvenuto il clivaggio dei Fp dalla molecola del Fg (Aα,Bβ,γ)2 si ottengono monomeri
solubili di Fn (α,β,γ).(52) Di regola FpA è rimosso più rapidamente di FpB con la formazione di desA
fibrina (α,Bβ,γ)2. Poi quando anche FpB è stato rimosso ho desAB fibrina (α,β,γ)2. (53,54)La specificità
della trombina nel clivaggio dei Fp è legata maggiormente alla struttura primaria, piuttosto che a quella terziaria del Fg. (55)
1.4.2 Fibrinopeptidi
Il rilascio del solo FpA è sufficiente per iniziare il processo di assemblamento della fibrina , ma la stabilità delle protofibrille e la formazione di fibre spesse di fibrina richiedono il clivaggio anche del FpB.(56)
Sono state identificate ben 3 forme diverse di FpA: 70% FpA classico non modificato, 10% des-Ala FpA (forma che manca di una parte all’estremità NH2 terminale), 20% P-FpA (forma con aa Ser3
fosforilato). L’FpA rilasciato in grandi quantità viene poi degradato lentamente e rimosso dal circolo. Dopo il rilascio del FpB un enzima carbossipeptidasi B simile rimuove un’arginina a livello dell’estremità C terminale del FpB portando alla formazione del des-Arg FpB. L’FpB, a differenza del FpA, viene prodotto solo in modeste quantità e viene degradato molto rapidamente. (57)
1.4.3 Assemblamento delle molecole di fibrina
Inizialmente, grazie all’interazione knobs-holes (end-to-middle domain D:E il domain E centrale di ciascun monomero può interagire con 4 domains D di altri monomeri di fibrina), si formano dimeri e oligomeri di fibrina. Con il procedere dell’attività catalitica della trombina si formano strutture più complesse dette protofibrille, costituite da una doppia fila di polimeri di fibrina e caratterizzate dalla presenza di interazioni knobs-holes (una terza molecola di fibrina fa da ponte tra le due file di polimeri di fibrina offrendo gli A e B knobs), ma anche di interazioni non covalenti end-to-end tra le regioni D di protofibrille adiacenti.(58-60)
15
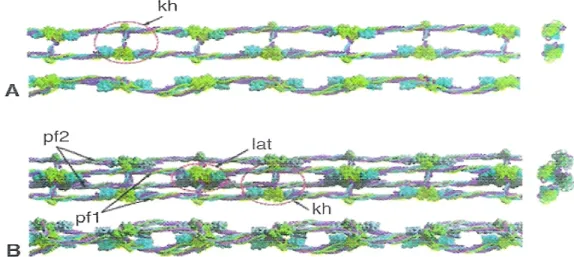
FIG 1.5 Rappresentazione schematica della struttura delle protofibrille; in viola catene α, in verde catene γ, in blu
catene β; kh: interazione knob-hole; pf: protofibrille; lat: contatti laterali end-to-end. (da Yang Z, Mochalkin I, Doolittle RF. A model of fibrin formation based on crystal structures of fibrinogen and fibrin fragments complexed with synthetic peptides. Proc Natl Acad Sci USA 2000; 97:14156)
L’associazione laterale delle protofibrille avviene quando queste hanno raggiunto una lunghezza di ca 600-800 nm e comporta la formazione di fibre più spesse di fibrina.(59)
FIG 1.6 Monomero di fibrina e polimerizzazione. A) schema del Fg con hole a e hole b, FpA e FpB. B)des AB fibrina con esposizione dei knobs A e B dopo il clivaggio rispettivamente di FpA e FpB. C) primi stadi di polimerizzaione, formazione di oligomeri, protofibrille e fibre. (da L Medved, Weisel JW, on behalf of fibrinogen and FXIII subcommittee of the scientific standardization committee of the international society on thrombosis and haemostasis. Recommendations for nomenclature on fibrinogen and fibrin J Thromb Haemost 2009;7:355-9)
16
Nella formazione del fibrin network sono coinvolti 2 tipi di branch junction. Nel primo caso (bilateral branch junction)un doppio strato di protofibrille si unisce ad un altro doppio strato di protofibrille formando così una struttura a 4 strati. Nel secondo caso invece (equilateral branch junction) grazie all’interazione di 3 molecole di fibrina si forma una struttura a 3 strati. La formazione di equilateral branch junctions avviene soprattutto quando il clivaggio dei Fp è relativamente lento: in queste condizioni il coagulo di fibrina che si forma è più denso (meno poroso) rispetto a quello che si forma in condizioni di alta concentrazione di trombina. (61)
Il dominio αC (porzione C terminale della catena α) gioca un ruolo importante nella stabilizzazione tridimensionale del coagulo di fibrina. Nel Fg tale dominio è legato al dominio centrale E con interazioni non covalenti, ma si dissocia in concomitanza con il clivaggio dei FpB . In questo modo i domini αC vengono resi disponibili per interagire tra loro, promuovendo così l’associazione laterale delle protofibrille e la formazione del network di fibrina.(62)
Anche i residui oligosaccaridici presenti nella molecola del Fg influenzano la formazione del coagulo di fibrina. Infatti il Fg completamente privo di glicosilazione forma coaguli di fibrina meno densi.(63)
Il ruolo centrale del doppio strato di protofibrille nella formazione del coagulo non è universalmente accettato. Infatti sono stati proposti modelli alternativi di polimerizzazione (polimerizzazione a singolo strato).(64)
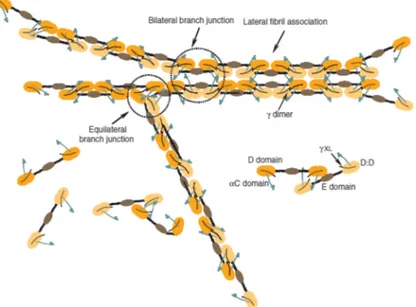
FIG 1.7 Diagramma schematico dell’assemblamento della fibrina, mostra branching, associazione laterale delle
fibrille e formazione di γ-γ dimeri trasversali. ( Mosesson MW, Fibrinogen and fibrin structure and functions. J Thromb Haemost 2005;3 1894-904)
1.4.4 Il fenomeno del Cross-linking
Il grado di resistenza alla trazione del coagulo dipende dall’entità dell’associazione laterale delle protofibrille, ma la resistenza del coagulo alla degradazione da parte della plasmina dipende dal fenomeno del cross linking. I legami covalenti alla base del cross linking si formano grazie all’azione del XIIIa, una transglutaminasi calcio dipendente, che viene attivata dalla trombina. (65,66)
17
FXIII è costituito da un tetramero A2B2 in cui la subunità A è attivata dalla trombina mediante il rilascio di un peptide NH2 terminale. I crosslinks si formano dapprima a livello delle porzioni C terminali delle catene γ, coinvolgendo residui di lisina e glutamina, e portano alla formazione dei cosiddetti γ-γ dimeri.(67) Due siti nella catena γ sono coinvolti nella formazione dei γ-γ dimeri,
rispettivamente indicati come ‘γXL’ e ‘D:D’. (68) Anche le catene α vanno incontro al fenomeno di
cross linking, ma a questo livello i legami si formano più lentamente .(69,70) I cross links si formano
inoltre anche tra catene α e γ (eterodimeri).(71) La disposizione dei cross links tra catene γ è ad oggi
oggetto di dibattito tra chi sostiene una disposizione longitudinale dei legami e chi sostiene piuttosto una disposizione trasversale.(72) La disposizione trasversale è tuttavia l’unica in grado di
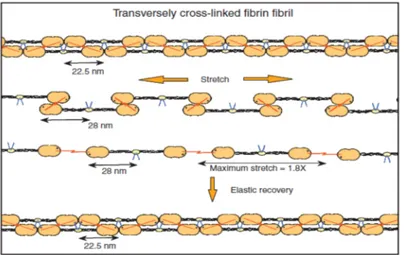
garantire importanti proprietà viscosoelastiche al coagulo di Fn, con ciò si intende la capacità delle fibrille di tornare alla loro lunghezza originale dopo trazione.(73)
FIG 1.8 Deformazione dei cross links trasversali a seguito trazione, e ritorno elastico alla normalità al termine
dell’applicazione della forza. Quando un doppio strato di fibrille viene stirato per 1.8’ diventa una struttura a singolo strato e le fibrille rimangono connesse tra loro grazie ai legami crociati γ-γ. (da Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost 2005;3:1894-904)
FIG 1.9 L’immagine mostra i legami tra i monomeri di fibrina all’interno di una fibrilla. I siti di legame γXL formano
sia legami longitudinali end to end sia trasversali. (da MW Mosesson, KR Siebenlist, DA Meh. The structure and biological features of fibrinogen and fibrin. Ann NY Acad Sci 2001;936:11-30)
18
Oltre ai γ-γ dimeri si formano lentamente anche γ trimeri e tetrameri. (71) Dal momento che c’è un
solo residuo di lisina disponibile a livello di γ406 (74) si ritiene che le strutture trimeriche e
tetrameriche si formino attraverso l’utilizzo ripetuto dello stesso residuo.
L’interazione knobs-holes favorisce la disposizione antiparallela delle catene γ e quindi la formazione dei γ-γ dimeri. (68)
I cross links che si formano nella variante del Fg con catena γ’ conferiscono una maggiore resistenza del coagulo rispetto a quanto si verifica nella forma comune di Fg, ciò è probabilmente legato alla maggiore affinità della catena γ’ per il FXIIIa. (75)
La formazione di polimeri di Fn accelera il rilascio del FXIIIa, tuttavia quando si raggiunge la formazione del 40% dei dimeri γ-γ possibili, questo effetto viene meno, evitando così un’eccessiva formazione di legami crociati. Il FXIIIa non rimane attivo a lungo all’interno del coagulo: probabilmente esistono meccanismi di inattivazione come monossido di azoto delle cellule endoteliali (76) ,plasmina, trombina e alte proteasi.(77)
Mentre nel processo di degradazione nel Fg (fibrinogenolisi), la plasmina produce la formazione in successione dei frammenti solubili denominati X,Y,D e E (FDP, prodotti di degradazione del Fg), nel processo di degradazione del polimero stabilizzato di Fn si ha la formazione di frammenti caratterizzati dal legame crociato (cross-linking) D-D: si formano quindi i D-dimeri, indice del processo di fibrinolisi.(40)
FIG 1.10 Formazione dei dimeri-D (da G.Carulli Manuale di ematologia per gli studenti di Medicina)
Il rilascio del FpB non è un prerequisito per la formazione dei γ-γ cross-linking, dal momento che questi possono formarsi già prima del clivaggio di tale Fp. Infatti l’attivazione del FXIII va di pari passo con il rilascio del FpA. (57)
Il FpA è rilasciato ad una velocità maggiore rispetto all’attivazione del FXIIIa, in ragione della differenza tra le rispettive concentrazioni dei 2 substrati della trombina. (78)
1.5
Siti attivi di legame sul fibrinogeno e fibrinaIl Fg e la Fn hanno ruoli che in parte si sovrappongono, in contesti diversi: coagulazione del sangue, fibrinolisi, interazioni cellule-matrice, risposta infiammatoria, guarigione delle ferite, neoplasie. Queste funzioni sono regolate da siti presenti sul Fg, alcuni dei quali sono mascherati o non accessibili, se non dopo la formazione della Fn o l’interazione del Fg con altre molecole. (79)
Numerosi sono i siti attivi presenti a livello della molecola del Fg/fibrina, i più importanti dal punto di vista funzionale sono:
19
- Legame del complesso A2B2 e soppressione dell’attività di cross linking del FXIIIa nel sangue. Secondariamente anche un eccesso di subunità B nel plasma può costituire un elemento soppressivo di tale attività. (80)
- Il Fg si lega anche al recettore piastrinico αIIbβ3 (GP IIb/IIIa, CD41/CD61), esposto nelle PLT, favorendo il reclutamento di queste all’interno del trombo in via di formazione. (81,82)Tale
recettore è un’integrina che si lega alle sequenze RGD arg-gly-asp-X a livello della catena Aα572-575 (RGDS) e Aα95-98 (RGDF). Il crosslinking dei domini αC promuove il signalling integrina mediato.(83) L’integrina αIIbβ3 delle PLT si lega anche alla sequenza γA 400-411(84)
e alla sequenza γA370-383 presente esclusivamente sul Fg.(85)
Il legame dell’integrina al sito sulla catena Aα o al sito sulla catena γ è mutuamente esclusivo. Questo può essere dovuto al fatto che entrambe le sequenze amminoacidiche sulla catena Aα e γ si legano allo stesso sito o su siti sovrapposti dell’integrina, oppure può essere dovuto al fatto che il legame di una sequenza amminoacidica comporta una modifica conformazionale dell’integrina che impedisce il legame dell’altra sequenza amminoacidica del Fg. I risultati degli studi svolti per chiarire la modalità di interazione ha portato alla conclusione che esistano 2 distinti siti di legame e che una modifica conformazionale conseguente al legame di una delle due sequenze amminoacidiche sia alla base del legame mutuamente esclusivo delle catene γ e Aα.(86)
La GP IIb/IIIa esposta dalle PLT non attivate lega esclusivamente il Fg, mentre nelle PLT attivate da agonisti (ADP, epinefrina, trombina), mostra invece uguale capacità di legame con ben 4 molecole diverse: Fg (87), Fn (88), vitronectina (89,90), vWF.(91) Inoltre l’integrina delle
PLT inattivate presenta elevata affinità di legame esclusivamente per la molecola intatta del Fg, mentre il legame ai frammenti X derivati dalla fibrinolisi è meno efficiente: questo costituisce un sistema di regolazione, fibrinolisi-mediato, del legame delle PLT non stimolate al Fg. (92) Sappiamo che GP Ib-IX e vWF sono coinvolti nell’adesione delle PLT non attivate al
subendotelio (93,94), questo potrebbe rappresentare il principale meccanismo di adesione
piastrinica in condizioni di elevato shear stress. Invece il meccanismo di adesione delle PLT non attivate, mediato da GP IIb-IIIa, potrebbe diventare importante in condizioni di basso shear stress.(95)
Nei soggetti con tromboastenia di Glanzmann sono presenti difetti quantitativi o qualitativi di tale integrina ed è alterata l’aggregazione piastrinica. (81,82) Anche mutazioni o delezioni
della sequenza γ316-322 comportano un deficit di aggregazione delle PLT.(96)
GP IIb-IIIa interagisce anche con la fibrina ed ha un ruolo nella retrazione del coagulo (cessione di acqua da parte del polimero con conseguente accorciamento dello stesso).(97) I
siti di legame dell’integrina alla Fn sono diversi da quelli presenti a livello del Fg. I siti di legame della Fn rimangono ad oggi indeterminati. Si è ipotizzato un possibile coinvolgimento delle catene β.(98)
La trombospondina, una GP rilasciata dagli α granuli delle PLT attivate, stabilizza l’aggregazione delle PLT, formando un legame non covalente con il Fg.(99) La
trombospondina è inoltre incorporata nel coagulo di fibrina in formazione (copolimerizzazione), dove favorisce la polimerizzazione della fibrina.(100)
- Studi recenti hanno dimostrato che l’interazione delle PLT con la fibrina può essere mediata anche da vWF e GP Ib. Questo legame incrementa l’entità della generazione di trombina in plasma ricco di PLT.(101) Si possono formare legami crociati tra vWF e Fn. Un’altra molecola
20
- È stato rilevato che il FXa si lega alla Fn, ai FDP e ai D dimeri. Infatti riconosce un sito di legame localizzato tra i domini D e E.(103)
- Anche TFPI (inibitore della via di attivazione del TF: inibisce l’attività del TF e del FVIIa e viene attivato molto precocemente nella fase iniziale della coagulazione) si lega alla Fn a livello delle estremità C terminali anche se l’esatto sito di legame non è stato ancora precisato.(104)
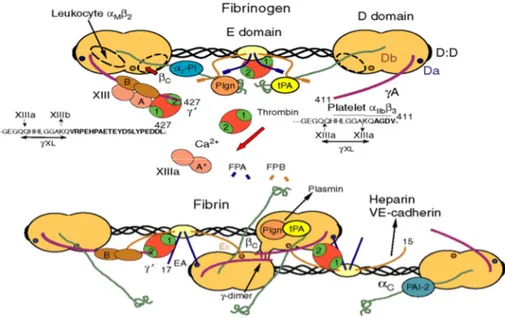
FIG 1.11 Diagramma schematico della struttura del fibrinogeno, della sua conversione in fibrina, e della
conversione mediata dalla trombina del fattore XIII native a f. XIII a. Sono illustrati anche siti di legame per proteine, enzimi, recettori ed altre molecole che partecipano alle funzioni del fibrinogeno/fibrina (M. W. MOSESSON, JTH 2005)
TAB 1C
LIGANDI DEL Fg SITO DI LEGAME FUNZIONE
FXIIIa Catena γ’ Stabilizzazione del coagulo
Regolazione dell’attività del XIIIa
Trombina Dominio E
Catena γ’, Dominio E
Rilascio Fp Attività AT- I (105)
Calcio Catene Bβ e γ Conservazione della struttura e
stabilità del Fg e della Fn (106)
Promuove la polimerizzazione
(107)
Plasminogeno tPA
Catena Aα (dominio αC) Catena Aα (dominio αC) e γ
Promuove la fibrinolisi(108)
α 2 antiplasmina
PAI-2 Lp(a)
Catena Aα
Catena Aα (dominio αC) Catena Aα (dominio αC)
Resistenza alla fibrinolisi
(109,110,111)
Fibronectina Catena Aα Migliora le caratteristiche
meccaniche del coagulo (112)
Integrina αIIbβ3 Catena Aα e γ Aggregazione delle PLT
21
2 Disfibrinogenemie
Le disfibrinogenemie sono disordini della coagulazione causate da anormalità strutturali e/o funzionali del fibrinogeno, che interferiscono con il ruolo emostatico della molecola. Possono essere congenite/ereditarie o acquisite. Queste ultime, pur nelle loro rarità, sono molto più frequenti rispetto alle forme ereditarie.
2.1
Disfibrinogenemie congeniteLe disfibrinogenemie congenite sono conseguenti a mutazioni a carico dei geni posti sul cromosoma 4 che codificano per le catene del fibrinogeno. Sinora sono state identificate fino a 245 alterazioni geniche, la maggior parte delle quali vengono ereditate con meccanismo autosomico dominante. Generalmente non sono associate a storia di sanguinamento o trombosi (55% dei casi è asintomatico con sole alterazioni dei test di laboratorio, allungamento del tempo di trombina-TT, dell’ aPTT e del PT; il 25% presenta sanguinamento e il 20% trombosi). È interessante notare come storia di sanguinamento e trombosi possano coesistere nello stesso pz (27% dei casi). Le manifestazioni emorragiche riscontrate nei pz sono variabili per sede ed entità: epistassi, sanguinamento gengivale, menorragia, ematomi, emartri, sanguinamento a seguito di procedure chirurgiche o parto, ritardo nella guarigione delle ferite. Le manifestazioni trombotiche includono: TVP degli arti inferiori ed embolia polmonare, tromboflebiti, trombosi arteriose.
Le manifestazioni emorragiche sono dovute alla mancata formazione del coagulo di fibrina. La patogenesi delle manifestazioni trombotiche è invece meno chiara: coesistono due ipotesi.
- prima ipotesi: difettoso legame della trombina alla fibrina (legame poco stabile) che favorirebbe il rilascio in circolo di una maggiore quantità di trombina responsabile di un patologica attivazione della coagulazione con conseguente trombosi.
- seconda ipotesi: il difetto risiede nelle proteine profibrinolitiche (attivatore tissutale del plasminogeno, plasminogeno) o in un’aumentata resistenza della fibrina all’azione della plasmina.(114-118)
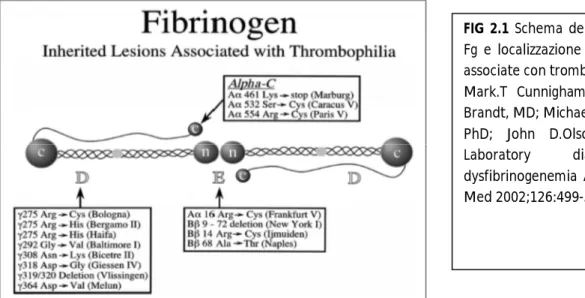
FIG 2.1 Schema della molecola del
Fg e localizzazione delle mutazioni associate con trombofilia. (tratto da Mark.T Cunnigham, MD; John T. Brandt, MD; Michael Laposata, MD, PhD; John D.Olson, MD, PhD. Laboratory diagnosis of dysfibrinogenemia Arch Pathol Lab Med 2002;126:499-505)
22
2.2
Disfibrinogenemie acquisite- Disfibrinogenemie secondarie o acquisite spesso si associano ad alterazioni della funzionalità
epatica(119)(cirrosi, epatite cronica, insufficienza epatica acuta, tossicità da paracetamolo, cisti del
coledoco, ittero ostruttivo). Un aumento dei residui di acido sialico a livello delle catene Bβ e γ altera la carica elettrica della molecola, ostacolando la polimerizzazione della fibrina.(120) È difficile
stabilire se e in che grado la disfibrinogenemia contribuisca alle manifestazioni emorragiche e/o trombotiche, dal momento che i pz epatopatici presentano multiple anomalie della coagulazione (deficit dei fattori vitamina K dipendenti, piastrinopenia e piastrinopatia).(121)
- Una disfibrinogenemia acquisita può essere una manifestazione paraneoplastica: presenza di tumori quali epatomi e tumori a cellule renali (le cellule neoplastiche secernono molecole anomale di fibrinogeno). In questi casi la remissione della patologia neoplastica è accompagnata da risoluzione del quadro coagulativo. (122,123)
- La disfibrinogenemia acquisita può essere dovuta alla presenza di autoanticorpi diretti contro la molecola del fibrinogeno, che bloccano il clivaggio dei FpA e FpB(124,125) ,interferiscono con la
polimerizzazione della fibrina(126,127) o con il processo di cross-linking mediato dal fattore XIIIa,
sebbene le manifestazioni cliniche siano spesso assenti. Ciò può avvenire, ad esempio, in corso di malattie del connettivo (LES)(128)o paraproteinemie (MM)(129-131), insufficienza renale(132),
tromboflebite migrante(133), sarcoidosi trattata con isoniazide(134), malattie croniche del fegato(135),
sindrome di Down.(136)
Sono stati descritti casi nei quali gli autoanticorpi si formano in soggetti in apparente buona salute.(137) In letteratura è riportato un interessante caso di disfibrinogenemia su base autoimmune
che andava incontro a remissione spontanea a seguito della formazione di nuovi autoanticorpi che bloccavano gli stessi anticorpi leganti il fibrinogeno. Poiché erano assenti IC in circolo, si è ipotizzata la possibilità che questi autoanticorpi agissero inibendo la sintesi degli anticorpi leganti il fibrinogeno.(138) Le anomalie della coagulazione risultano direttamente proporzionali alla quantità di
autoanticorpi presenti in circolo. La plasmaferesi è una possibilità terapeutica in alcuni di questi pz.(139)
Raramente possono riscontrarsi alloanticorpi contro il fibrinogeno, in particolare nei pz affetti da afibrinogenemia in terapia sostitutiva con fibrinogeno esogeno.(140)
- Una diversa causa di disfibrinogenemia è stata riscontrata in un pz con MGUS: la catena leggera λ in forma monomerica legava la molecola di fibrinogeno. Tale legame non alterava il rilascio dei FpA e FpB, ma andava a interferire con le fasi di formazione del coagulo subito a valle dell’azione catalitica della trombina, cioè agiva come inibitore della polimerizzazione dei monomeri di fibrina. Essendo la catena λ legata al fibrinogeno, essa non veniva filtrata a livello renale, perciò si riscontravano solo bassi livelli di proteinuria di BJ. All’elettroforesi (sia su plasma che con fibrinogeno purificato) la molecola di fibrinogeno del pz migrava più velocemente dei controlli, inoltre si evidenziavano due bande di migrazione per la catena λ, di cui una si trovava alla stessa altezza della banda del fibrinogeno. I livelli di IgG, IgA,IgM e di catene leggere k libere erano bassi. Solo le catene leggere λ libere mostravano valori elevati con un basso rapporto k:λ, suggerendo una produzione monoclonale. L’alterata migrazione della molecola del fibrinogeno all’elettroforesi era imputabile al legame con la catena λ. All’elettroforesi si rilevavano due bande di migrazione delle catene λ, perché una corrispondeva alle catene libere, mentre l’altra, posta alla stessa
23
altezza della banda del fibrinogeno, corrispondeva alle catene legate al fibrinogeno. La proprietà di interferire con la molecola di fibrinogeno era peculiare della forma monomerica della catena λ e la natura del legame tra le due molecole era di tipo non covalente. Infatti le catene λ purificate ottenute dalle urine del pz non legavano il fibrinogeno, poiché nelle urine tali catene sono in forma dimerica. Lo studio di questo particolare caso di disfibrinogenemia è stato inoltre completato con l’analisi dell’mRNA delle catene λ del pz, ma non è tutt’ora chiaro quale sia il sito di legame delle catene λ sul fibrinogeno (regione D, E, siti multipli?). Le alterazioni emocoagulative non avevano riscontro clinico.
- Un ulteriore caso di disfibrinogenemia acquisita causata dalle catene λ si ritrova in letteratura.
Donna giapponese affetta da sindrome nefrosica che presentava anomalie del profilo coagulativo, muta dal punto di vista clinico e con negatività della BJP. All’autopsia sono stati rilevati importanti depositi di amiloide AL. Probabilmente la scarsa proteinuria era dovuta sia al legame delle catene λ al fibrinogeno sia all’incorporazione di queste catene nella formazione di fibrille di amiloide. (141) - Casi di disfibrinogenemia sono indotti da farmaci quali mitramicina(142), glucocorticoidi,
isotretinoina.(143) La glicazione delle proteine nei soggetti con diabete mellito, altera la struttura
del fibrinogeno ed è anch’essa una possibile causa di disfibrinogenemia.(144)
2.3
Test di laboratorioI test di screening sono il tempo di trombina e il tempo di reptilase. La conferma diagnostica è data dalla misurazione dei livelli di fibrinogeno nel plasma.(145)
2.3.1
Tempo di trombina TTÈ il tempo in cui una certa quantità di trombina calcica induce la formazione di un coagulo in un campione di plasma citrato. La trombina agisce direttamente sul fibrinogeno e il tempo è indipendente da tutti gli altri fattori della coagulazione. Un allungamento del TT si verifica nelle condizioni di ipo-afibrinogenemia, disfibrinogenemia congenitae acquisita (ad esempio in pz con mieloma(146)) , presenza di eparina, presenza di prodotti di degradazione della fibrina e del
fibrinogeno . In un solo caso è stato riscontrato un accorciamento piuttosto che un allungamento del TT in corso di disfibrinogenemia.(115)
2.3.2
Tempo di reptilaseTempo in cui una certa quantità di reptilase (enzima ottenuto dal veleno del serpente Bathrops) induce la formazione di un coagulo in un campione di plasma citrato. L’enzima in questione stacca solo il FpA e non il FpB a differenza della trombina. Anche questo tempo risulta allungato in caso di disfibrinogenemia. Il test ha il vantaggio di non essere influenzato dalla presenza di eparina o da altri fattori inibenti la trombina (fattori eparin-like, paraproteine).
Se i test di screening di cui sopra risultano allungati, si prosegue con i test di conferma. (In un caso è stata documentato un aumento del TT ma non del tempo di reptilase, gli autoanticorpi del pz andavano ad interferire con il solo rilascio del FpB, ma non del FpA).(124)
24
2.3.3
Misurazione dei livelli di fibrinogeno nel plasma(147,148)- Metodo di Clauss: il dosaggio del fibrinogeno nel tempo di coagulazione con trombina si basa sul metodo originariamente descritto da Clauss; in presenza di un eccesso di trombina, il fibrinogeno si trasforma in fibrina e il tempo di formazione del coagulo è inversamente proporzionale alla concentrazione di fibrinogeno presente nel campione di plasma. Il plasma povero di piastrine è diluito (1:10) per minimizzare l’effetto di sostanze inibitorie come eparina e FDP. Si aggiunge trombina ad alte concentrazioni (100U/ml) così da ottenere tempi di coagulazione indipendenti dalla concentrazione di trombina. Dal confronto del tempo necessario alla formazione del coagulo con i valori di una curva di calibrazione si ricava la concentrazione del fibrinogeno nel plasma.
- Derivazione dei livelli di fibrinogeno sulla base del PT: si valuta il PT del pz sulla base della variazione della densità ottica del campione di plasma povero di piastrine e si confronta il valore ottenuto con la curva di calibrazione, derivando così la concentrazione del fibrinogeno.
- Metodo immunologico: misurazione della concentrazione del fibrinogeno presente, mediante tecnica ELISA (enzyme linked immunoabsorbant assays) o immunodiffusione radiale. Non valuta l’attività funzionale del fibrinogeno, perciò è particolarmente utile per evidenziare le disfibrinogenemie, dove è presente una discrepanza tra la quantità di fibrinogeno nel plasma e la sua attività funzionale.
- Metodo gravimetrico: si basa sul peso del coagulo di fibrina. È analogo al metodo di Clauss, con la differenza che invece di considerare il tempo di formazione del coagulo, viene valutato il peso del coagulo. È un test che presenta difficoltà tecniche perciò è poco usato.
2.3.4
TT 1:1 mixing studyIn alcuni casi il TT risulta allungato, in presenza di normali livelli di fibrinogeno nel plasma.(119,142) Per
stabilire se la causa dell’allungamento del TT risiede in un’anomalia funzionale del fibrinogeno si esegue questo test. Il test prevede un’ iniziale determinazione del TT su plasma del pz a cui è stato aggiunto plasma normale (il rapporto tra i due tipi di plasma deve essere 1:1). In un secondo tempo si determina il TT su plasma defibrinato del pz a cui viene aggiunto plasma normale (anche in questo caso in un rapporto 1:1). Nelle disfibrinogenemie il TT risulta allungato solo nel primo test, mentre nel secondo risulterà normale, dal momento che non si ha più fibrinogeno interferente.
Nel sospetto di forme congenite (funzionalità epatica nella norma, familiarità per disturbi della coagulazione clinici o laboratoristici) può risultare utile il test di elettroforesi per lo studio della proteina del fibrinogeno e l’analisi genetica.(116,149)
25
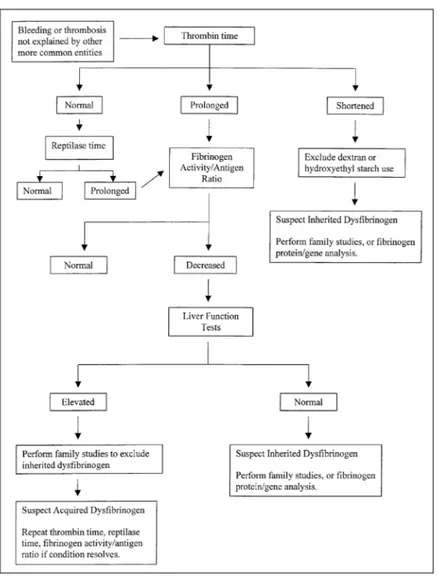
FIG 2.3 Algoritmo diagnostico per le disfibrinogenemie (tratto da Mark.T Cunnigham, MD; John T. Brandt, MD;
Michael Laposata, MD, PhD; John D.Olson, MD, PhD. Laboratory diagnosis of dysfibrinogenemia Arch Pathol Lab Med 2002;126:499-505
26
3 Gammopatie monoclonali
Le gammopatie monoclonali (GM) sono patologie che interessano la linea linfoide B/ plasmacellule e sono caratterizzate dalla produzione di quantità variabili di Ig prodotte dallo stesso clone cellulare (componente monoclonale ,CM)con restrizione per una delle due catene leggere (κ o λ). Il termine paraproteinemie dovrebbe essere limitato a casi di dimostrata alterazione della struttura immunoglobulinica. Il termine discrasie plasmacellulari si riferisce ai casi caratterizzati da infiltrazione plasmacellulare significativa a carico del midollo osseo o di altri distretti.
3.1
Immunoglobuline (150,151)3.1.1 Struttura delle Ig
Le immunoglobuline sono proteine tetrameriche costituite da due catene polipeptidiche pesanti (H) identiche e da due catene polipeptidiche leggere (L) identiche. Interazioni non covalenti e ponti di-solfuro uniscono tra loro le due catene pesanti e ciascuna catena leggera alla catena pesante, generando la classica forma ad Y dell’immunoglobulina.
Le catene leggere e quelle pesanti hanno in comune lo stesso tipo generale di organizzazione, in cui ciascuna catena consiste di due regioni principali:
- la regione variabile (regione V) ammino-terminale - la regione costante (regione C) carbossi-terminale
Regioni corrispondenti delle catene pesanti e leggere si associano per generare domini. Il dominio localizzato all’estremità N-terminale delle catene pesanti e delle catene leggere è chiamato dominio variabile o dominio V poiché è costituito da una sequenza amminoacidica che differisce significativamente tra le singole immunoglobuline conferendo loro un’enorme versatilità; tale dominio è responsabile del riconoscimento dell’antigene. All’interno di ciascun dominio variabile (VH e VL ) la variabilità non è distribuita uniformemente, ma è concentrata in tre regioni ipervariabili, dette regioni di complementarietà o CDRs (Complementarity Determining Regions: CDR1, CDR2 e CDR3) poichè formano una superficie complementare rispetto alla struttura tridimensionale dell’antigene destinato a legarvisi. Tra le tre regioni ipervariabili CDR sono intercalate altre quattro regioni di minore variabilità amminoacidica denominate regioni strutturali o Framework Regions (FRI, FRII, FRIII e FRIV).
Nelle rimanenti regioni della catena leggera e della catena pesante, le sequenze amminoacidiche sono sostanzialmente identiche tra le immunoglobuline appartenenti alla stessa classe o isotipo; per questa ragione si parla di domini costanti (C). Vi è un dominio costante della catena leggera (CL ) e 3 domini costanti della catena pesante (CH1, CH2, CH3), numerati a partire dall’estremità amminica; le IgM e le IgE, che hanno catene pesanti più lunghe, presentano un ulteriore dominio CH4.
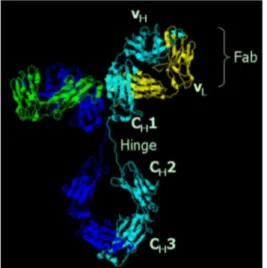
Mediante clivaggio enzimatico (pepsina, papaina) un anticorpo viene tagliato a livello del punto di congiunzione tra i tre bracci e si divide in tre frammenti: due frammenti Fab, o "frammento legante l'antigene",un frammento Fc, o "frammento cristallizzabile". Le catene leggere rimangono all'interno del Fab; il Fc comprende parte delle regioni costanti delle catene pesanti. Viene così chiamato per la sua capacità a cristallizzare, ovvero a formare complessi con altre strutture se immerso in un liquido proteico.
27
Ogni classe immunoglobulinica può presentare catene leggere di tipo κ (kappa) oppure di tipo λ (lamba) ma le singole immunoglobuline non presentano mai contemporaneamente una catena di un tipo ed una di un altro. L’isotipo è determinato dal tipo di catena pesante utilizzato nella sintesi della Ig (G,M,A,D e E).
FIG 3.1 Struttura cristallografica di un’immunoglobulina con la classica forma a Y (Da Wikipedia, 2006.)
3.1.2 Sintesi delle Ig
Le immunoglobuline sono codificate in 3 loci disposti su 3 cromosomi diversi (catene pesanti:cromosoma 14; catene leggere di tipo κ: cromosoma 2 ; catene leggere di tipo λ: cromosoma 22)
Durante la maturazione delle cellule B, la diversità della regione variabile delle Ig è generata da una serie di riarrangiamenti successivi e programmati dei geni per le catene pesanti (il segmento D-diversity- viene avvicinato a quello J-joining- e viene inserito in prossimità del segmento V-variable). Successsivamente i 3 segmenti, avvicinati a quello C-constant-, che codifica per la regione costante, formano un gene funzionale per la catena pesante dell’Ig (V-D-J-C) e per la catena leggera (il riarrangiamento è analogo a quello per la catena pesante, ma manca il segmento D).
I linfociti B presentano sulla superficie cellulare un recettore, chiamato B Cell Receptor (BCR), con le stesse caratteristiche strutturali delle immunoglobuline che verranno prodotte dalla cellula dopo il riconoscimento dell’antigene da parte di questo recettore. La maturazione antigene-dipendente, che avviene negli organi linfoidi secondari -milza, linfonodi, placche del Peyer- , porta allo sviluppo di cellule B memoria e di plasmacellule e consiste nel processo di ipermutazione somatica, nel quale mutazioni somatiche puntiformi nei geni riarrangiati di H e L danno origine a Ig mutate caratterizzate da una maggiore affinità anticorpale (maturazione dell’affinità). Le Ig di superficie (BCR) e le prime Ig prodotte appartengono alle classi IgM e IgD, le altre Ig (IgG, IgA, IgE) vengono prodotte a seguito di switching isotipico.
28
3.2
Classificazione e diagnosi delle GM3.2.1
Inquadramento nosologico delle GM(152,153)TAB 3A
Tipo di
gammopatia monoclonale
Caratteristiche generali Criteri diagnostici
gammopatie “preneoplastiche” con basso rischio di progressione: MGUS (GM di incerto significato)
-Le MGUS rappresentano il tipo più frequente di GM (50-55% di tutte le GM). La diagnosi di MGUS aumenta con l’età (la prevalenza della MGUS è pari a circa il 3% nei soggetti >50 anni).
-Si distinguono 3 tipi di MGUS:
Non-IgM MGUS (IgG 69%, IgA 11%,biclonali 3%, occasionalmente IgD e IgE) forma più frequente, caratterizzata da tendenza a progressione verso MM. IgM-MGUS (17% delle MGUS) mostrano tendenza alla
progressione in disordini linfoproliferativi (macroglobulinemia di Waldenstrom e altri linfomi) piuttosto che all’evoluzione in MM IgM.
Light-chain MGUS, FLC-MGUS, mostra tendenza ad evoluzione in mieloma micromolecolare, che rappresenta il 20% dei nuovi casi di mieloma diagnosticati.
-Le MGUS sono gammopatie “preneoplastiche” con basso rischio di progressione in patologia maligna, stimato intorno all’1% per anno (rischio maggiore nel primo anno dalla diagnosi); tuttavia non è possibile all’esordio di una MGUS identificare sicuri criteri prognostici relativi ad una eventuale progressione, pertanto è necessaria un’osservazione clinica periodica.
Diagnosi di non-IgM MGUS e IgM MGUS Tutti i criteri IMWG 2010 devono essere soddisfatti:
1. CM<3g/dl
2. Infiltrazione linfoplasmocitica clonale del midollo<10% 3. Assenza di danno d’organo
attribuibile a disordine proliferativo plasmacellulare (secondo i criteri CRAB)* o in caso di MGUS IgM assenza di anemia, sintomi costituzionali, sindrome da iperviscosità, linfoadenopatia o epatosplenomegalia attribuibili a disordine linfoproliferativo sottostante. Diagnosi di FLC-MGUS
Tutti i criteri devono essere soddisfatti: 1. κ/λ ratio alterato (<0.26 o
>1.65)
2. aumentata concentrazione di una delle due catene leggere ( aumento κ-FLC se ratio>1.65; aumento λ-FLC se ratio<0.26) 3. Assenza di Ig monoclonali
complete all’immunofissazione 4. Infiltrazione linfoplasmocitica
clonale del midollo<10% 5. Assenza di danno d’organo
attribuibile a disordine proliferativo plasmacellulare (secondo i criteri CRAB)* Gammopatie
“preneoplastiche” ad alto rischio di progressione
-Si distinguono 3 tipi di gammopatie “preneoplastiche” ad alto rischio di progressione in patologia maligna, stimato intorno al 10% annuo nei primi 5 anni:
SMM: Smouldering Multiple Myeloma o mieloma multiplo asintomatico.
Smoldering Waldenstrom macroglobulinemia o macroglobulinemia di Waldenstrom asintomatica
BJP idiopatica
Diagnosi di SMM e macroglobulinemia di Waldenstrom asintomatica
Tutti i criteri devono essere soddisfatti: 1. CM (IgG o IgA)>3g/dl (IgM nel
caso di macroglobulinemia) e/o Infiltrazione linfoplasmocitica clonale del midollo >10% 2. Assenza di danno d’organo
attribuibile a disordine proliferativo plasmacellulare
FIG 3.3 Spettro clinico delle gammopatie
monoclonali (Mayo clinic 1510 casi, 2005 DA Kyle RA et al, BHJ 2006)
29
(secondo i criteri CRAB)* o in caso di smoldering Waldenstrom
macroglobulinemia assenza di anemia, sintomi costituzionali, sindrome da iperviscosità, linfoadenopatia o epatosplenomegalia attribuibili a disordine linfoproliferativo sottostante. Diagnosi di BJP idiopatica
Tutti i criteri devono essere soddisfatti: 1. Proteine monoclonali nelle
urine (rilevate mediante elettroforesi) >500 mg/24h e/o plasmacellule clonali infiltranti il midollo>10%
2. Assenza di Ig monoclonali complete all’immunofissazione. 3. Assenza di danno d’organo attribuibile a disordine proliferativo plasmacellulare (secondo i criteri CRAB)*
Gammopatie monoclonali maligne
-Si distinguono 3 tipi di gammopatie monoclonali maligne:
MM: mieloma multiplo: 5-6 casi /100.000 abitanti/anno, età mediana 70 anni, raro sotto i 40 anni, 15% di tutt le emopatie maligne. 65% dei casi IgG 25% IgA.
Macroglobulinemia di Waldenstrom
Light- chain myeloma o mieloma micromolecolare (CM costituita da sole catene leggere, in assenza di Ig monoclonali complete circolanti) 10% dei casi di MM.
ISS: International Staging System
Diagnosi di MM
Tutti i criteri devono essere soddisfatti: 1. Plasmacellule clonali infiltranti il
midollo>10%
2. Presenza nel siero e/o nelle urine di CM (ad eccezione del MM non secernente)
3. Danno d’organo attribuibile a disordine proliferativo plasmacellulare (secondo i criteri CRAB)*; in assenza di danno d’organo infiltrazione linfoplasmocitica clonale del midollo >=60%
Diagnosi di macroglobulinemia di Waldenstrom
1. CM IgM
2. Infiltrazione linfoplasmocitica clonale del midollo>10% con cellule caretterizzate da immunofenotipo tipico 3. anemia, sintomi costituzionali,
sindrome da iperviscosità, linfoadenopatia o epatosplenomegalia attribuibili a disordine linfoproliferativo sottostante *criteri CRAB: Calcemia> 11.5 mg/dl;
Insufficienza Renale (creatininemia >2.0 mg/dl o clearance della creatinina<40ml/min);
Anemia normocromica-normocitica (Hb<10g/dl o un valore di Hb più di 2 g/dl al di sotto del limite inferiore di normalità); Lesioni ossee-Bone lesions (lesioni litiche, osteopenia grave, fratture patologiche);
Altri: iperviscosità sintomatica, amiloidosi, infezioni batteriche ricorrenti (>2 in 12 mesi). FIG 3.4 Stadiazione del mieloma da Durie BG, Salmon SE, Cancer 1975. FIG 3.5 ISS da Greipp P et al, JCO 2005
30
MGUS e SMM sono gammopatie monoclonali “pre- neoplastiche” asintomatiche, con tendenza ad evolvere in patologie francamente maligne e sintomatiche.
FIG 3.7 La trasformazione biologica di plasmacellule normali verso plasmacellule neoplastiche (clonalità) e poi in
MM franco è caratterizzata dall’accumularsi di eventi oncogenetici. I 2 principali eventi oncogenetici precoci che intervengono sono: traslocazioni che coinvolgono il gene che codifica per IgH -t(4;14), t(14;16), t(6;14), t(11;14), t(14;20)- e iperdiploidia. I due tipi di eventi sono mutuamente esclusivi, cioè una cellula presenta generalmente uno solo dei 2 eventi. Altri eventi oncogenetici iniziali sono la disregolazione dell’espressione della ciclina D, la mutazione di NRAS, la delezione del crom 13. Nella progressione verso la forma maligna avvengono mutazioni di KRAS e FGFR3, attivazione costitutiva di NF-kB, traslocazioni secondarie di IgH. Eventi tardivi sono invece la up-regulation del protooncogene MYC e l’inattivazione dei geni oncosoppressori p18, p53, Rb. La progressione di malattia è caratterizzata da cambiamenti inerenti il microambiente midollare (attivazione degli osteoclasti, promozione dell’angiogenesi, inibizione degli osteoblasti, alterata espressione di citochine, fattori di crescita e molecole di adesione). (da Neha Korde, Sigurdur Y, Kristinsson, Ola Landgren. MGUS e SMM: novel biological insights and development of early treatment strategies. Blood 2011; 117(21):5573-5581).
FIG 3.6 Differente pattern di
avoluzione delle MGUS a seconda dell’isotipo Ig (da Rajkumar SV et al, Mayo Clinic Proc 2010)
31 Altre GM (154):
GM associate a patologie non ematologiche (neoplasie, infezioni croniche, epatopatie croniche e cirrosi, malattie autoimmuni, malattie parassitarie, patologie cutanee croniche, iperparatiroidismo).
leucemia plasmacellulare (plasmacellule nel sangue periferico >1,5x109/l).
plasmocitoma solitario dell’osso o extramidollare
sindrome POEMS (polineuropatia, organomegalia, endocrinopatia, componente monoclonale, skin changes).
malattie delle catene pesanti γ,α,μ.
Amiloidosi AL: accumulo in sede extracellulare di sostanza amiloide di natura proteica (frammento N-terminale della catena leggera monoclonale, comprendente la regione variabile e parte della regione costante) evidenziabile con colorazione Rosso Congo. I depositi di amiloide sono responsabili di sintomi neurologici (neuropatie), intestinali (macroglossia,epatomegalia, malassorbimento), renali (sindrome nefrosica, insufficienza renale), cardiaci (cardiomiopatia). L’età media alla diagnosi è di 60 anni, con lieve prevalenza nel sesso maschile. I fenomeni di aggregazione delle proteine monoclonali possono dare origine anche a sostanze diverse dall’amiloide: aggregati amorfi di tipo puntiforme (deposito di catene leggere e/o pesanti), cristalli (sindrome di Fanconi acquisita), microtubuli (crioglobulinemia tipo I con glomerulonefrite), cilindri (myeloma kidney in particolare da catene lambda).
Crioglobulinemie (presenza nel plasma di una proteina anomala che precipita quando esposta a una temperatura inferiore a quella corporea e che ridiventa solubile quando esposta alla temperatura di 37°C).
3.2.2 Iter diagnostico delle GM
(155,156)- Dosaggio delle proteine totali: permette di verificare se la CM si accompagna a un
incremento della quantità delle proteine plasmatiche.
- Protidogramma: il tracciato elettroforetico delle proteine sieriche (SPEP Serum Protein
ElctroPhoresis) evidenzia nel contesto della zona gamma, talora della zona beta, la presenza di un picco ristretto rappresentativo della componente monoclonale (CM). È possibile una valutazione quantitativa della CM.
- Immunofissazione sierica (e urinaria): la CM sospetta viene confermata e caratterizzata
mediante immunofissazione: le proteine separate per elettroforesi su gel vengono fatte reagire con antisieri specifici e questo permette di identificare l’isotipo della Ig e la restrizione per una delle due catene leggere.
- Dosaggio delle Ig nel plasma e delle catene leggere libere nel plasma (calcolo κ/λ ratio) e
nelle urine (positività della proteinuria di Bence-Jones BJP). È possibile riscontrare una CM limitata alle sole catene leggere (mieloma micromolecolare, amiloidosi AL).
Il rapporto κ/λ (κ/λ ratio) è il parametro più importante per distinguere una crescita monoclonale in corso di discrasie plasmacellulari da una crescita policlonale delle sFCLs (per esempio dovuta: a- aumentata sintesi in corso di malattie infiammatorie, b- ridotta clearance renale in caso di malattia renale cronica -è utile modificare gli intervalli di riferimento del rapporto κ/λ in base alla funzionalità renale del pz in analisi, così da migliorare la specificità diagnostica, c- alla combinazione di entrambi i meccanismi, p.es. nel LES). Nel primo caso il rapporto risulta alterato dal momento che la produzione
32
monoclonale vede la crescita di un solo tipo di catena leggera, con contemporanea soppressione della catena leggera non coinvolta nella crescita monoclonale, nel secondo caso la crescita interessa sia le κFLC che le λFLC perciò il rapporto κ/λ rimane costante. In realtà è possibile riscontrare valori di κ/λ ratio lievemente alterati (borderline) anche in corso di crescita policlonale delle FLCs.
I valori delle FCLs possono essere normali pur in presenza di κ/λ alterato: ciò si spiega con la presenza di una patologia monoclonale caratterizzata da un midollo ipoplastico e quindi iposecernente.(157)
Nel 2001 è stato messo a punto un sistema di misurazione delle sFLC basato sull’utilizzo di Ab policlonali che legano epitopi nascosti delle FLC. Tali epitopi sono localizzati tra catena leggera e pesante e diventano evidenti solo quando la catena leggera è libera dal legame con la catena pesante(Freelite sFLCs ®).(158)
FIG 3.8 Una molecola Ab costituita da catena L e H, a dx catene leggere libere κ e λ. (Da Wikilite.com the binding site)
Vantaggi dell’utilizzo del dosaggio immunologico delle FLCs sieriche: (159-161)
Migliore sensibilità e precisione (>100 volte) rispetto a protidogramma (SPEP Serum Protein ElectroPhoresis) e immunofissazione sierica (IFE immunofixation electrophoresis).
Risultati quantitativi, utili nel monitoraggio vs misurazione semiquantitativa di SPE e IFE.
Accurato e precoce marker di remissione, risposta al trattamento, malattia residua, ricaduta, in ragione della breve emivita (2-6 ore) delle FLCs rispetto alle Ig (giorni-settimane)
Valore prognostico.
Importanza nella stadiazione del MM (valore indipendente da albumina e β2-microglobulina).
- Il range di normalità delle concentrazioni sieriche delle FLCs ( e il κ/λ ratio) è meno variabile rispetto al range delle concentrazioni urinarie, questo depone a favore di una misurazione su siero piuttosto che sulle urine.
-La misurazione su siero è più sensibile rispetto a quella urinaria dal momento che per avere un aumento delle uFCLs si deve superare la capacità di riassorbimento del tubulo prossimale (le FLCs vengono liberamente filtrate a livello glomerulare e poi riassorbite a livello del TP).
33
-A favore della misurazione su siero depone anche la difficoltà nel raccogliere campioni urinari in pz con danno renale importante e anuria.
-Lo studio delle FLCs urinarie (immunofissazione delle proteine nelle urine delle 24 h) rimane importante tuttavia nella valutazione iniziale dell’amiloidosi AL (una piccola minoranza di pz con amiloidosi possono avere uFCLs aumentate in assenza di un alterazione del κ/λ ratio, questo è legato alla presenza di sindrome nefrosica con perdita di albumina nelle urine che compete con il riassorbimento prossimale delle FLCs).
Le concentrazioni delle Ig sono influenzate da cambiamenti dell’Ht e del volume
ematico. Invece le FLCs sono maggiormente distribuite nel compartimento extravascolare, perciò risentono meno di questi cambiamenti.
Un nuovo metodo di misurazione che permette di valutare contemporaneamente catene pesanti e leggere (Hevilyte®).(162)
FIG 3.9 Gli epitopi riconosciuti dagli Ab HLC nella tecnica Hevylite® sono
presenti a livello delle regioni costanti (CH1 e CL) tra la catena pesante e leggera dell’Ig.(da Wikilite.com the binding site)
Dal momento che questa tecnica si basa su Ab policlonali che riconoscono contemporaneamente epitopi presenti sulla catena pesante e su quella leggera, è possibile identificare il tipo di catena leggera presente per ciascuna classe di Ig, ad es posso stimare la concentrazione di IgGκ e IgGλ e calcolare il rapporto IgGκ/IgGλ. È interessante notare che il rapporto κ/λ medio varia a seconda della classe di Ig considerata. Con questa nuova metodica è possibile valutare l’entità del fenomeno di immunoparesi (riduzione della sintesi delle normali Ig), attraverso una misurazione precisa della riduzione della concentrazione delle Ig normali, non solo appartenenti a classi diverse da quella della CM (componente monoclonale), ma anche delle Ig normali della stessa classe della CM, ma con diversa catena leggera (HLC pair suppression). L’entità di tale fenomeno ha valore prognostico in quanto una maggiore riduzione delle Ig policlonali si associa a peggior prognosi.
- Biopsia osteomidollare (BOM): permette di stimare il grado di infiltrazione plasmacellulare
a carico del midollo osseo e il pattern infiltrativo (interstiziale, nodulare, diffuso, con fibrosi)
- Mieloaspirato (MA): osservazione della morfologia delle plasmacellule, le cui anomalie
hanno rilievo prognostico. Le plasmacellule patologiche possono essere classificate come: mature, intermedie, immature, plasmoblasti. Su sangue midollare, ottenuto con MA, è possibile condurre studio citofluorimetrico dell’immunofenotipo delle plasmacellule e
34
calcolare il rapporto κ/λ (rapporto tra la percentuale di plasmacellule che esprimono nel citoplasma le catene leggere κ e quelle che esprimono le catene leggere λ).
- Indagini di biologia molecolare (PCR): studio del riarrangiamento monoclonale IgH
(cromosoma 14).
- Studio del cariotipo.
3.3
Gestione clinica del pz con GM (MGUS e MM)(152,163) 1. Nella gestione dei pz con MGUS è necessario:- eseguire la diagnosi secondo i criteri aggiornati; - eseguire una corretta stratificazione prognostica;
- eseguire un follow-up mirato sulla base del rischio di evoluzione. La suddivisione in base al rischio si basa sulla valutazione di 3 fattori (Mayo Clinic Scheme):
- Entità CM >1.5 g/dL 1 punto - Isotipo non IgG 1 punto - FLC ratio alterato 1 punto
Si distinguono 3 classi di rischio nell’ambito delle MGUS:
- Basso rischio: 0 punti CM<1.5 g/dl, IgG, FLC ratio normale follow-up
SPEP (protidogramma), ematocrito, calcemia e funzione renale (creatininemia e GFR più sensibile) dopo 6 mesi, se i valori sono stabili ripetere ogni 2-3 anni o in presenza di sintomi-segni suggestivi di MM. MA, BOM, Rx scheletro non sono indicati se emocromo, calcemia, creatininemia nella norma.
- Rischio intermedio (1-2 punti) e rischio alto (3 punti): CM>1.5 g/dl e/o IgA o IgM e/o FLC ratio alterato eseguire mieloaspirato e BOM, studio del cariotipo mediante citogenetica in metafase e FISH (su PC isolate), citofluorimetria, Rx scheletro (IgA e IgG), eco o TC addome (IgM), LDH, beta2microglobulina e PCR (se dati sospetti per MM o macroglobulinemia di Waldenstrom).
Follow-up: SPEP (protidogramma), dosaggio delle proteine totali, dosaggio IgG, IgA, IgM, FLC su siero o plasma; emocromo; LDH, beta2-microglobulina, PCR; calcemia, funzione renale (creatininemia, GFR, proteinuria/24 h, azotemia) ogni 6 mesi nel primo anno; se stabili ripetere annualmente per tutta la vita.
Non iniziare terapia in assenza di segni CRAB. 2. Nella gestione clinica del pz con SMM sono previsti:
- Alla diagnosi e dopo 2-3 mesi SPEP, emocromo, creatininemia, calcemia, BJP quantitativa nelle urine delle 24 h, FLC, dosaggio Ig.
- Alla diagnosi: BOM/MA (+FISH), Rx scheletro completo, RMN colonna e pelvi per ricerca lesioni occulte predittive di malattia più aggressiva.
- Se gli esami sono stabili ripetere ogni 4-6 mesi nel primo anno, poi ogni 6-12 mesi. Non ci sono indicazioni a terapia al di fuori di trial clinici.
35
FIG 3.10 Flow-chart
monitoraggio pz con proteina M (da IMWG 2010)