PARTE II. MATERIALI E METODI
II.1. Gruppi sperimentali
Per la prima parte del lavoro, riguardante l’analisi delle spine dendritiche, sono stati utilizzati 20 ratti adulti (età superiore a P100) di razza Long-Evans (Charles River), mantenuti in stabularizzazione seguendo le disposizioni del Ministero della Salute italiano, con accesso a cibo e acqua ad libitum.
I ratti, sono stati suddivisi in 4 gruppi sperimentali:
1. Animali con visione binoculare, nei quali un emisfero è stato iniettato con Condroitinasi-ABC, mentre l’altro è stato utilizzato come controllo;
2. RS, animali deprivati a lungo termine (LMD), sui quali è stata praticata la riapertura dell’occhio deprivato e, contemporaneamente, la sutura inversa (reverse suture, RS) dell’occhio non deprivato;
3. RS-ChABC, costituisce l’insieme di animali LMD nei quali la RS è stata associata all’iniezione intracorticale di Condroitinasi-ABC;
4. RS-Pasi, comprende esemplari LMD sottoposti a RS e a iniezione con penicillinasi;
La seconda parte del lavoro, dedicata alla messa a punto di un metodo di valutazione degli effetti della deprivazione monoculare sul sistema inibitorio, ha richiesto l’uso di un totale di 6 ratti adulti (età superiore a P100), razza Long-Evans (Charles River).
Questo secondo gruppo di ratti è stato suddiviso in 2 gruppi sperimentali, composti ognuno da 3 individui:
1. RS-DR, ratti normali, che hanno subìto deprivazione monoculare sull’occhio sinistro; 2. LMD-DR, ratti sottoposti a deprivazione monoculare a P21, sui quali è stata praticata
la riapertura dell’occhio deprivato (destro) e contemporaneamente la RS dell’occhio non deprivato (sinistro);
Immediatamente dopo le operazioni appena descritte, gli esemplari appartenenti ad entrambi i gruppi sono stati sottoposti ad allevamento al buio per 3 giorni, in un’apposita camera ventilata a prova di luce.
II.2. Operazioni chirurgiche
Le operazioni sono state svolte sotto anestesia, ottenuta mediante iniezione intraperitoneale di avertina in soluzione fisiologica (dosaggio 1ml/hg di peso corporeo).
Per i gruppi sperimentali 2, 3, 4, della prima parte sperimentale, così come per il gruppo 2 della seconda, sono stati utilizzati animali deprivati a lungo termine (LMD), deprivati a P21 suturando l’occhio destro.
Il protocollo di deprivazione monoculare è largamente utilizzato negli studi sulla plasticità del sistema visivo (Hubel & Wiesel, 1970).
La chiusura della rima palpebrale, si effettua, con l’aiuto di un microscopio stereoscopico chirurgico, tramite l’asportazione dell’ispessimento fibroso che descrive la rima stessa, in modo da lasciare una stretta striscia di tessuto “vivo” che decorra lungo tutta la palpebra. Successivamente, le palpebre sono suturate insieme utilizzando filo sterile in polibutestere non assorbibile (misura 6/0, Novafil). È importante aver cura di lasciare il bulbo oculare il più possibile libero da corpi estranei e peli, poiché potrebbero causare reazioni infiammatorie dalle quali conseguirebbe l’opacizzazione della cornea. La cicatrice fresca è medicata con pomata antibiotica (Aureomicina).
A causa dell’asportazione precedentemente svolta, i due bordi palpebrali si saldano insieme nel processo di cicatrizzazione. In breve tempo, i movimenti dell’animale e la chiusura della ferita determinano caduta dei punti; l’occhio si trova così ricoperto da una membrana continua che impedisce il passaggio di gran parte della luce e, in ogni caso, non permette la visione definita.
La riapertura dell’occhio si svolge dopo il controllo preliminare dell’integrità della chiusura a carico dell’occhio deprivato: se si osservano aperture, anche minime, il soggetto è scartato.
La cicatrice che descrive l’area suturata serve da guida per praticare un taglio, utilizzando una lama da bisturi sterile, che riproduce l’andamento originario della rima palpebrale, ponendo grande attenzione a non lesionare il sottostante bulbo oculare. È inoltre necessario eliminare le numerose membrane cicatriziali che si formano tra la cornea e l’epidermide. La bontà dell’operazione è verificata dal ripristino della capacità di estroflessione del bulbo oculare.
Nel caso in cui si osservino alterazioni della forma del bulbo oculare od opacizzazione della cornea, l’animale è escluso dal resto dell’esperimento, altrimenti esso è medicato con applicazione di gocce oftalmiche a base di cortisone (Tobradex) e pomata antibiotica (Aureomicina). Le applicazioni dei farmaci sono ripetute 2 volte il giorno per almeno 3 giorni seguenti l’operazione;
questo accorgimento è volto a evitare l’insorgere certo di una reazione infiammatoria che danneggerebbe l’occhio appena riaperto, portando all’opacizzazione della cornea e all’esclusione dell’animale dal resto dell’esperimento.
Gli animali che subiscono la RS sono sottoposti, contemporaneamente alla riapertura dell’occhio destro, a MD sull’occhio sinistro.
II.3. Microiniezione intracorticale
L’animale è anestetizzato come descritto nella precedente sezione e montato su un apparato stereotassico.
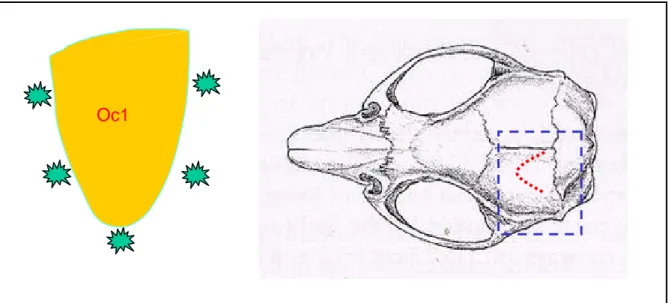
Dopo l’apertura dello scalpo, si pratica un assottigliamento dell’osso cranico mediante una fresa da dentista (diametro 1mm), descrivendo una “U rovesciata”, che si estende per circa 5mm, partendo da 1mm lateralmente rispetto alla sutura bregma.
All’interno dell’assottigliamento si praticano 5 fori, attraverso il quale passerà il capillare per l’iniezione, utilizzando un ago ipodermico rivestito quasi completamente da un tubetto di plastica, in modo che soltanto 1mm sia libero di perforare l’osso.
Per l’iniezione si utilizza un capillare in vetro al borosilicato, dal quale si ricava un ago per tiraggio a caldo, svolto da un apposito strumento (Sutter Instruments, co.). L’ago è montato sul supporto di un iniettore, collegato a un micromanipolatore (Sutter Instruments, co.).
Il micromanipolatore permette di misurare la distanza di penetrazione entro la corteccia cerebrale; la prima iniezione è svolta a 750µm, la seconda a 350µm. Per ognuno dei siti di iniezione, si rilasciano 250nl di soluzione, attendendo circa 1 minuto perché essa abbia tempo di diffondere entro il tessuto.
Al termine dell’operazione, lo scalpo è suturato con filo in polibutestere non assorbibile (Novafil).
Ciascun ciclo di iniezioni comprende due applicazioni, la prima effettuata in concomitanza della riapertura dell’occhio deprivato, la seconda dopo 3 giorni.
Il trattamento di degradazione della matrice extracellulare è costituito dall’applicazione di Condroitinasi ABC, 48 unità/ml (Seikagaku, Corp.), la quale provoca la degradazione delle catene laterali condroitin solfato dai CSPG, senza agire sulle proteine core. Il controllo negativo, riguardante la procedura di iniezione in sé, è rappresentato da Penicillinasi (Sigma-Aldrich), un enzima privo di substrati endogeni.
II.4. Preparazione del fissativo per la perfusione
Si utilizza una soluzione stock di paraformaldeide all’8%, preparata disciogliendo un’opportuna quantità del reagente solido in acqua distillata. L’operazione deve esser svolta sotto cappa aspirante, data la tossicità della sostanza.
Il discioglimento della paraformaldeide è facilitato dal riscaldamento ad una temperatura compresa tra 50 e 55°C, mantenendo la soluzione in agitazione.
Quando la soluzione da torbida comincia a divenire trasparente, il discioglimento è quasi avvenuto. Per terminare la preparazione, è necessario chiarificare mediante l’aggiunta di NaOH 10N (2,5 gocce ogni 100ml), poiché i gruppi aldeidici della paraformaldeide con il riscaldamento si acidificano.
La soluzione chiarificata è filtrata, lasciata raffreddare a temperatura ambiente e conservata in frigorifero a 4°C.
Al momento dell’utilizzo, la soluzione stock è portata ad una concentrazione del 4% per diluizione in tampone PB a pH 7,4.
II.5. Fissazione dei tessuti tramite perfusione vascolare
La procedura è preceduta da anestesia terminale, ottenuta con un sovradosaggio di cloralio idrato al 10,5% (peso/volume, dose normale 0,4ml/hg di peso corporeo) in soluzione fisiologica.
Dopo l’apertura della cassa toracica, il tratto discendente dell’arteria aorta è ostruito mediante pinza emostatica, in modo da dirigere la maggior parte del fissativo entro il circolo superiore. Per l’iniezione nel torrente circolatorio del fissativo, si utilizza un apposito ago a punta molata, inserito nel ventricolo cardiaco sinistro, mentre l’aumento dei liquidi circolanti è compensato da foratura dell’atrio cardiaco destro.
L’ago da perfusione è collegato a una pompa peristaltica, regolata su una velocità di flusso di 40ml/min, che si ritiene ottimale per la taglia degli animali adulti.
Prima del passaggio del fissativo, si effettua un “lavaggio” dei vasi sanguigni, facendo passare al loro interno almeno 150ml di tampone PBS 0,1M. A questo punto, si immettono nella circolazione sistemica almeno 250ml di paraformaldeide al 4%, che permettono la fissazione dei tessuti per le successive fasi sperimentali.
II.6. Isolamento del cervello
Una volta perfuso, l’animale è decapitato per facilitare l’estrazione del cervello dalla scatola cranica. L’osso cranico è frantumato con apposite pinze “ossivore”, partendo dalle narici e spostandosi via via posteriormente; quando si arriva presso l’encefalo si procede asportando piccoli frammenti, con le pinze mantenute il più possibile tangenti alla superficie. Si adottano questi accorgimenti pratici, perché in questa fase è importante evitare di intaccare l’encefalo con i becchi delle pinze ossivore o con i frammenti d’osso che si stanno asportando. Inoltre, si recidono i legamenti della dura madre con un paio di pinzette fini, poiché essi potrebbero letteralmente tagliare in due il cervello al momento dell’estrazione.
Il cervello così esposto è asportato dall’alloggiamento nella scatola cranica tramite una leva in metallo, inserita anteriormente tra l’organo e la parete ossea, scendendo fino a raggiungere il forame occipitale, per separare l’encefalo dal midollo spinale.
Per conservare al meglio il campione, si esegue una postfissazione di almeno 2 ore, per immersione in paraformaldeide al 4%. Successivamente si pone il cervello in una soluzione di saccarosio al 30% (peso/volume) in tampone PB pH 7,4, per almeno 12h; ciò permette di crioproteggere il tessuto dai danni derivanti dal congelamento con CO2, necessario per il suo
sezionamento.
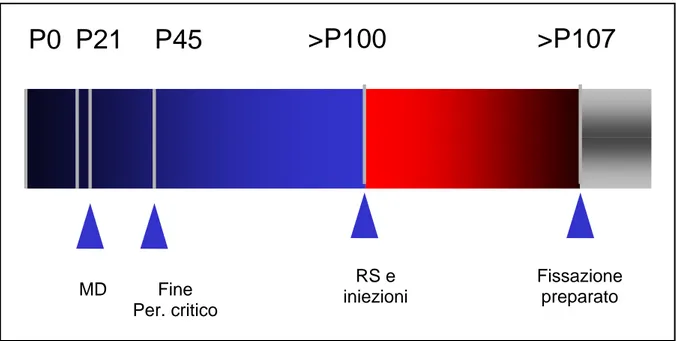
MD iniezioniRS e Fissazione preparato
P0 P21 P45
>P107
Fine Per. critico
>P100
Oc1
Figura 2.2 - Localizzazione dei siti d'iniezione in riferimento al cranio di ratto.
II.7. Colorazione immunoistochimica con WFA
L’emisfero destro di ogni campione è contrassegnato con un’incisione sulla parte inferiore del lobo temporale; ciò lo rende riconoscibile dall’altro, senza danneggiare l’area d’interesse, ossia la corteccia visiva.
Le sezioni, ottenute per taglio al microtomo congelatore, hanno uno spessore di 50µm. Il taglio è effettuato sul piano coronale, estendendosi dal lobo occipitale in direzione anteriore per comprendere tutta la corteccia visiva.
La colorazione si avvale della lectina WFA, estratta dalla Wisteria floribunda, coniugata a biotina (Sigma-Aldrich).
I determinanti antigenici aspecifici sono mascherati lavando le sezioni per 45 minuti (su bascula a temperatura ambiente) in un’apposita soluzione bloccante, composta di albumina di siero bovino (BSA) al 3% (volume/volume, da stock al 20%) disciolta in tampone PBS.
La WFA è utilizzata in rapporto di diluizione 1:100, disciolta in PBS; il tempo di incubazione è almeno 12 ore, su bascula a 4°C. Al termine dell’incubazione, l’eccesso di anticorpo è rimosso lavando le sezioni per 3 volte in PBS, mantenendole ad ogni passaggio per 5 minuti in agitazione su bascula.
La rivelazione della reazione si avvale di un fluroforo, in questo caso Fluoresceina IsoTioCianato (FITC), coniugata ad avidina (Molecular Probes), che quindi legherà selettivamente la biotina coniugata alla WFA. La diluizione è di 1:300 in PBS, il tempo di
incubazione è di almeno 2 ore (su bascula a temperatura ambiente). Al termine della reazione, le sezioni sono nuovamente lavate per 3 volte in PBS.
Infine, i campioni sono preparati per l’osservazione al microscopio, montandoli su vetrini portaoggetto. Tra vetrino portaoggetto e coprioggetto si interpone un sottile strato di liquido Vectashield (Vector Laboratories, Inc.), destinato a preservare la fluorescenza nel tempo. Il preparato è sigillato con vernice a smalto, anche per evitare la fuoriuscita del Vectashield, che non essicca mai a causa della base costituita da glicerolo.
I vetrini sono conservati al buio e a 4°C.
II.8. Colorazione immunoistochimica cFOS/parvalbumina
La preparazione delle sezioni è stata svolta in maniera analoga al paragrafo 2.7, mentre le fasi successive presentano delle particolarità che saranno qui descritte in dettaglio. Tra i particolari salienti è l’utilizzo di un agente detergente, il Triton X-100, destinato alla parziale degradazione delle membrane plasmatiche, in modo da renderle permeabili al passaggio degli anticorpi, che devono infatti rivelare antigeni intracellulari. Inoltre, la rivelazione contemporanea di due elementi richiede l’utilizzo di due anticorpi secondari recanti fluorofori differenti e, di conseguenza, di anticorpi primari prodotti in animali di specie diverse, in modo che la loro porzione costante sia differente.
La soluzione bloccante è costituita dal 10% (volume/volume) di Normal Goat Serum (NGS), che ha il compito di mascherare i determinanti antigenici aspecifici del preparato; si aggiunge anche uno 0,5% di detergente Triton X-100 (volume/volume, stock al 10%) e si porta a volume con tampone PBS 0,1M. Il lavaggio si protrae per 3 ore, su bascula e a temperatura ambiente.
La soluzione contenente gli anticorpi primari è composta dallo 0,5% di NGS, 0,3% Triton X-100, disciolti in PBS 0,1M. Gli anticorpi primari utilizzati sono anti-cFOS (Calbiochem), prodotto in coniglio (1:3000, volume/volume) e anti-parvalbumina (Chemicon), prodotto in topo (1:1000, volume/volume). L’incubazione è di 36 ore su bascula a 4°C; la bassa temperatura, se da un lato allunga sensibilmente il tempo d’incubazione, dall’altro permette di conservare in maniera ottimale le sezioni, diminuendo la possibilità che insorgano processi di degradazione e marcescenza.
Prima della rivelazione mediante anticorpi secondari coniugati a fluorofori, si sono effettuati 3 lavaggi in PBS 0,1M puro, separati da almeno 5 minuti di agitazione su bascula, per rimuovere l’eccesso di anticorpo non legato al campione.
La soluzione contenente gli anticorpi secondari è composta di 1% NGS; 0,3% Triton X-100; anticorpo anti-coniglio coniugato al fluoroforo Alexa-488 (Molecular Probes), prodotto in capra (1:400, volume/volume); anticorpo anti-topo coniugato al fluoroforo Alexa-568 (Molecular Probes, prodotto in capra (1:400, volume/volume). Il tempo di incubazione è di 2 ore su bascula a temperatura ambiente.
Al termine del procedimento, si effettuano altri 3 lavaggi in PBS.
I preparati sono montati su vetrino portaoggetto e ricoperti con uno strato uniforme di liquido per montaggio VectaShield (Vector Labs., Inc.). Il vetrino coprioggetto è bloccato in posizione tramite vernice a smalto, in modo da evitare fuoriuscite del liquido di montaggio.
I preparati sono conservati al buio e a 4°C.
II.9. Colorazione immunoistochimica cFOS/GAD67
Il procedimento seguito è del tutto analogo a quello mostrato nel paragrafo 2.8, con la sola differenza che al posto dell’anticorpo primario anti-parvalbumina, si è scelto un anti-GAD67 (Chemicon), prodotto in topo (1:1000, volume/volume). Poiché la specie animale di produzione dei due anticorpi (anti-GAD67 e anti-PV) è lo stesso, si è potuto usare lo stesso anticorpo secondario per la rivelazione. Allo stesso modo, è identico alla sezione precedente il protocollo di immunoistochimica per cFOS.
II.10. Acquisizione immagini delle colorazioni immunoistochimiche al
microscopio confocale
Immagini provenienti dalla corteccia visiva binoculare di entrambi gli emisferi cerebrali sono state acquisite usando un microscopio confocale (Olympus) e un obiettivo a secco 20X (Olympus).
Per ognuno dei campi d’interesse sono stati acquisiti 3 piani focali; per aumentare il rapporto segnale/rumore, ciascun piano è stato acuisito 3 volte, in modo da ottenere un immagine finale sottoposta a un processo di filtrazione selettiva dei pixel mediamente più luminosi. Il processo di elaborazione è stato svolto dal software FluoView (Olympus), abbinato al microscopio confocale.
La doppia colorazione è stata acquisita su due canali separati (rosso e verde), per facilitare le successive operazioni.
Le immagini sono state memorizzate in formato single-TIFF 24-bit, che ha il vantaggio di mantenere separati sia i canali del colore (rosso, verde e blu), sia i piani focali.
II.11. Conta di cellule
La quantificazione dei risultati delle colorazioni immunoistochimiche è stata svolta tramite conta manuale, avvalendosi del software MetaMorph (Molecular Devices, Inc.).
Ciascun piano focale è stato analizzato separatamente, un canale alla volta (rosso per la marcatura PV o GAD67, verde per la marcatura cFOS). Le sovrapposizioni tra le cellule contrassegnate sia in un canale sia nell’altro, hanno fornito un’indicazione per l’attribuzione della colocalizzazione cFOS/GAD67 o cFOS/parvalbumina, ma in ogni caso è stato effettuato un controllo visivo con entrambi i canali attivi.
I risultati delle conte, divisi in cFOS, parvalbumina o GAD67, cellule con marcatura doppia, sono stati riportati su foglio di lavoro di Microsoft Excel.
Il conteggio delle cellule è stato trasformato in un valore di densità, dividendolo per l’area in mm2 del campo d’acquisizione, ottenuta attraverso la conversione dell’area in pixel, grazie al
rapporto pixel/µm relativo all’obiettivo usato.
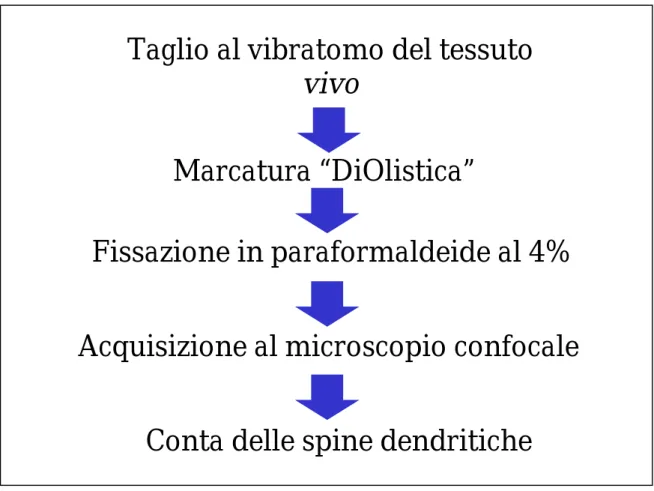
II.12. Marcatura “DiOlistica” delle spine dendritiche
Le membrane biologiche sono costituite in prevalenza da lipidi, sostanze non polari si disciolgono in essa con grande facilità, mentre scarsa è la loro affinità per l’ambiente citoplasmatico ed extracellulare. Su questa proprietà si basa l’utilizzo dei coloranti lipofilici della classe delle carbocianine (DiI, DiO, DiD).
Uno dei metodi che si possono scegliere per veicolare i coloranti nel tessuto d’interesse è il
gene gun, una vera e propria pistola propulsa da elio compresso. I “proiettili” sono costituiti da
sferule di tungsteno od oro, recanti adsorbito sulla superficie il colorante che, una volta nel campione diffonderà da solo nelle sue strutture lipidiche.
Come già descritto da Gan e collaboratori (Lichtman et al., 2000; Gan et al., 2003), le fasi cruciali per l’applicazione di questo protocollo sono la preparazione dei proiettili, il loro “sparo” e la procedura di fissazione del preparato.
II.12.1. Preparazione dei proiettili
Il colorante lipofilico DiI (1,1-dioctadecil-3,3,3,3-tetrametilindocarbocianina; Molecular Probes) è stato adsorbito su sferule di tungsteno del diametro di 1,3µm (Bio-Rad), in accordo con quanto descritto in Gan et al. (2000). La soluzione stock si ottiene disciogliendo 4mg di colorante
in 200µl di diclorometano, operazione da eseguire sotto cappa aspirante a causa della tossicità di quest’ultimo reagente.
Un piccolo quantitativo (circa 150mg) di sferule in tungsteno è stato sparso su di un vetrino per microscopia ed è stato ricoperto con circa 100µl di soluzione. L’evaporazione del solvente fa sì che il DiI si adsorba sulla superficie delle sferule; il compimento del processo è dimostrato dal cambiamento progressivo di colore del colorante, che in soluzione è fuxia e diviene antracite.
Le sferule così preparate sono delicatamente raschiate dalla superficie del vetrino e sono sospese per sonicazione in 3,750ml di acqua distillata.
I “proiettili” per il gene gun si ottengono da un tubo in materiale plastico (Bio-Rad), inserito su un apposito strumento denominato “tubing prep station” (Bio-Rad).
L’aderenza delle sferule alle pareti del tubo è facilitata dal suo trattamento preventivo con PoliVinilPirrolidone (PVP), 0,1mg/ml in etanolo assoluto (stock: 200mg/ml in etanolo assoluto). Il PVP è aspirato nel tubo mediante una siringa applicata all’estremità opposta e mantenuto al suo interno per 8 minuti, passati i quali è espulso. Il tubo deve esser lasciato asciugare finché non scompaiono le gocce di PVP visibili al suo interno, tuttavia un’eccessiva evaporazione del PVP è da evitarsi, poiché riduce l’adesività del tubo.
A questo punto si aspira la sospensione delle sferette, che si è avuta cura di sonicare per facilitare la dispersione nel solvente. Azionando l’asciugatore, si fa roteare su se stesso il tubo per 2-3 minuti, per facilitare la dispersione delle sferule sulle sue pareti.
L’acqua distillata è rimossa aspirandola con delicatezza; il peso del tungsteno e l’azione del PVP fanno sì che le sferule restino nel tubo. L’asciugatura del tubo si ottiene facendo passare al suo interno una corrente di azoto gassoso: è importante mantenere bassa la pressione, altrimenti le particelle sarebbero dilavate dalle pareti.
Completata l’asciugatura, il tubo è sezionato in frammenti della lunghezza di circa 1,5-2cm, che costituiscono le “cartucce” con cui caricare il gene gun. I proiettili si conservano per svariati mesi, se inseriti in una provetta assieme a un agente essiccante, come il gel di silice, che mantenga bassa l’umidità.
II.12.2. Preparazione delle sezioni di tessuto cerebrale
Il prelievo dei campioni è stato svolto in maniera simile a quanto descritto da Hensch e collaboratori (Mataga et al., 2004).
I ratti sono stati sottoposti ad anestesia terminale con cloralio idrato (in soluzione fisiologica al 10,4% peso/volume, 0,6ml/hg peso corporeo) e successivamente decapitati. Il cervello è stato estratto dal cranio in maniera analoga a quanto descritto nella sezione “isolamento del cervello” e immerso in una soluzione di taglio contenente NaCl 130mM, KCl 3.1mM, K2HPO4 1.0mM,
NaHCO3 4.0mM, destrosio 5.0mM, MgCl2 2.0mM, CaCl2 1.0mM, Hepes 10mM, acido ascorbico 1.0mM, mio-inositolo 0.5mM, acido piruvico 2mM (pH 7.3), opportunamente ossigenata.
Ognuno dei cervelli è stato sezionato in vivo in fettine dello spessore di 300µm, utilizzando un vibratomo (Leica). Le sezioni così ottenute sono state immerse nuovamente nella soluzione di taglio, all’interno della quale si fa gorgogliare ossigeno, per mantenere il tessuto vivo. In questo modo si evita che il tessuto vada incontro a processi di degradazione che si evidenziano sotto forma di “blebbing” del dendrite, che apparirà marcato in maniera irregolare, ostacolando le successive fasi sperimentali.
II.12.3. Marcatura dei tessuti
Le “cartucce” contenenti le sferule di tungsteno sono inserite nell’apposito tamburo contenuto nel gene gun (Bio-Rad).
La sezione da marcare è adagiata sul fondo di una piastra Petri; tra la sezione e il gene gun si interpone un supporto (vedi fig.2.3b) su cui si appoggia un filtro in policarbonato con pori da 3µm (Molecular Probes). Inoltre, al di sopra si aggiunge una rete metallica con maglie da 1mm di lato. Questi accorgimenti evitano che aggregati grossolani di tungsteno raggiungano il preparato, danneggiandolo. Il filtro deve essere sostituito al massimo ogni 3 spari.
La densità della marcatura e la penetrazione all’interno della sezione si regolano agendo sulla distanza dal preparato e sulla pressione del gas; nel nostro caso sono stati scelti una distanza di 1cm e una pressione di 80psi.
La diffusione del DiI nel tessuto è abbastanza veloce ed entro un’ora la sezione è pronta per l’osservazione. .
Al termine della procedura per la marcatura, è necessario fissare i preparati, immergendoli per almeno 2 ore in paraformaldeide al 4%; questo passaggio impedisce la diffusione aspecifica del colorante, che tenderebbe ad avvenire col passare del tempo.
Il montaggio avviene per immersione in PBS, utilizzando un vetrino sul quale è stata ricavato un pozzetto con vernice a smalto, dato l’elevato spessore del campione.
a)
b)
Figura 2.3 - a) gene gun; b) dispositivo di filtraggio dei proiettili (v. testo).
II.13. Acquisizione immagini delle spine dendritiche al microscopio
confocale
La zona d’interesse è costituita dai dendriti basali dei neuroni degli strati 2/3 della corteccia visiva binoculare. La correttezza dell’area inquadrata dall’oculare è stata verificata osservando un atlante stereotassico del cervello di ratto (Paxinos & Watson, 1998). Per ogni animale, sono stati selezionati dai 5 ai 15 neuroni per ogni emisfero cerebrale. L’acquisizione è stata effettuata “in cieco”, ossia senza che lo sperimentatore conoscesse il tipo di trattamento praticato su ogni esemplare.
I preparati sono stati esaminati con un microscopio confocale Olympus Fluoview, equipaggiato con un obiettivo 60X con apertura numerica pari a 0,9. Un ulteriore ingrandimento delle immagini è operato dallo zoom digitale (1,5X) incluso nel software Fluoview (Olympus), abbinato al microscopio.
La ricostruzione della struttura dei dendriti si basa sull’acquisizione di piani focali consecutivi (almeno 30), separati da 0,8µm di spessore.
II.14. Conta delle spine dendritiche
Le immagini delle spine dendritiche sono state analizzate avvalendosi del software di imaging MetaMorph (Molecular Devices).
Ognuna delle immagini è memorizzata in formato Multi-TIFF, nel quale ciascun piano focale è registrato come immagine singola; tutti i piani focali relativi allo stesso dendrite sono state sovrapposti in registro, senza fonderli tra loro. La conta delle spine è, quindi, stata effettuata separatamente per ogni piano focale, per evitare sovrapposizioni tra oggetti appartenenti a piani focali diiferenti; tale procedimento richiede di prestare attenzione a non contare più volte lo stesso oggetto, che può eventualmente estendersi su più di un’immagine.
Tutto il procedimento è stato svolto manualmente, contrassegnando individualmente ogni spina dendritica; i risultati sono stati annotati su foglio di lavoro di Microsoft Excel.
Per risalire al valore della densità delle spine dendritiche, la lunghezza del tratto di dendrite analizzato è stata calcolata grazie al fattore di conversione pixel/µm caratteristico dell’obiettivo utilizzato. Il numero delle spine contate è stato quindi diviso per la lunghezza del dendrite, ottenendo la densità in spine/µm.
Questa fase è stata, inoltre, eseguita “in cieco”, ossia lo sperimentatore ignorava il tipo di trattamento cui era stato sottoposto ciascun campione, in modo da evitare distorsioni dei risultati.