CAPITOLO
3
Risultati e discussione
1.1 Isolamento e identificazione dei composti
Le parti aeree di Clerodendrum splendens G. Don sono state essiccate e successivamente estratte, tramite una macerazione statica a temperatura ambiente, con solventi a polarità crescente: n-esano, CHCl3, miscela CHCl3-MeOH (9:1) e infine MeOH.
Dopo evaporazione del solvente si sono ottenuti i seguenti residui: 15.552 g (RE ), 13.474 g (RC),
21.747 g (RC-M), 26.759 g (RM).
Dopo un’analisi preliminare degli estratti, mediante cromatografia su strato sottile (TLC), sono stati presi come obiettivo di studio gli estratti: metanolico, cloroformio-metanolo e l’estratto cloroformico, i quali sono stati cromatografati e, successivamente, frazionati ed analizzati.
L’estratto esanico sarà oggetto di studio in un prossimo futuro.
Il processo di separazione ha portato all’isolamento e alla caretterizzazione di 14 composti. Tra i metaboliti secondari identificati troviamo:
3 flavonoidi (composti 2, 4, 7)
5 fenilpropanoidi (composti 1, 3, 5, 6, 8)
1 sesquiterpene (composto 9)
5 diterpeni (composti 10, 11, 12, 13, 14)
La determinazione strutturale di tutti i composti è stata effettuata mediante spettri NMR mono e bidimensionali (1H,13C,13C DEPT, HSQC, HMBC, DFQ-COSY, 1D-TOCSY) ed è stata confermata tramite spettrometria di massa ESI-MS.
L’analisi NMR è stata eseguita con due tipologie di strumenti operanti a campi diversi: 250 e 600 MHz, quest’ultimo è stato utilizzato soprattutto per piccole quantità di campione al fine di ottenere segnali ben risolti e separati.
Gli esperimenti monodimensionali 1H e 13C-NMR hanno fornito informazioni circa il numero di protoni presenti, il loro accoppiamento ed il valore delle costanti nonché sulnumero di atomi di carbonio. In particolare la tecnica multi-impulso DEPT (DistortionlessEnhancement by Polarization Transfer) ha permesso la determinazione del numero diprotoni legati a ciascun atomo di carbonio.
L’esperimento 1D-TOCSY (TOtal Correlation SpettroscopY) si è rivelato utile nell’ analisi spettroscopica degli zuccheri ed in generale in tutti i casi in cui il segnale di un singolo protone si rivelava molto complicato nel semplice spettro 1H-NMR. Questo esperimento, difatti, consente di costruire un sistema di spin eccitando selettivamente un protone che, grazie al trasferimento di magnetizzazione, consente di evidenziare tutti gli altri protoni appartenenti allo stesso sistema. Di fondamentale importanza nella determinazione strutturale dei composti sono sicuramente gli esperimenti di spettroscopia bidimensionale. Questi hanno consentito di ottenere un notevole incremento della risoluzione spettrale in quanto i segnali risultano dispersi in due dimensioni fornendo ulteriori informazioni sulle correlazioni degli stessi.
Tutti gli esperimenti NMR bidimensionali sono stati acquisiti in CD3OD in fase positiva con il
trasmettitore regolato sulla risonanza del solvente e sul TPPI (Incremento di Fase Proporzionale al Tempo) al fine di raggiungere una distinzione delle frequenze nella dimensione ω1 .
Gli esperimenti bidimensionali più utilizzati per la determinazione strutturale dei composti isolati sono stati:
HSQC (Heteronuclear Single Quantum Coherence), esperimento di correlazione eteronucleare che, impiegando il protone per la rivelazione del segnale, permette di ottenere l’assegnazione di uno spettro 13C a partire dal corrispondente spettro protonico e viceversa. Tutto ciò mediante la correlazione diretta 1H-13C.
HMBC (Heteronuclear Multiple Bond Coherence), esperimento di correlazione eteronucleare long range che consente di rivelare le correlazioni a più di un legame (2J e 3J)
tra un protone ed il carbonio non adiacente, consentendo in particolare di evidenziare correlazioni con i carboni quaternari.
DQF-COSY (Double Quantum Filtered COrrelated SpettroscopY), esperimento che consente di ottenere informazioni su protoni accoppiati attraverso due o tre legami e di correlare i chemical shifts mediante gli accoppiamenti scalari omonucleari. Inoltre tale metodica permette di assegnare i vari sistemi di spin presenti nella molecola anche laddove si abbiano costanti di accoppiamento molto piccole e, di conseguenza, segnali non ben risolti; infine l’esperimento difiltrato DQF permette di distinguere i segnali in prossimità del solvente, altrimenti coperti.
Ulteriori informazioni sui composti isolati sono state ottenute mediante analisi spettrometriche come ESI-MS che ne hanno confermato la struttura mediante il loro peso molecolare e le frammentazioni, espresse come rapporto massa/carica elettrica (m/z).
1.2 Frazionamento dell’estratto cloroformio-metanolo
Dall’estratto RC-M, attraverso varie tecniche cromatografiche, illustrate in Fig. 3.1, sono stati isolati
ed identificati 2 metaboliti (composti 1 e 2) . Entrambi i metaboliti sono dei derivati fenolici, il composto 1 è un fenilpropanoide, ed il composto 2 è un flavone glicoside.
Fig. 3.1 Schema di frazionamento dell’estratto CHCl3-MeOH, RC-M
Estratto
R
C-MR
C-M/16
RP-HPLC M-W (2:3)Composto
1
R
C-M/12
RP-HPLC M-W (35:65)Composto
2
SEPHADEX
LH 20
IN MeOH
1.3 Frazionamento dell’estratto metanolico
L’estratto metanolico RM è stato ripartito tramite un imbuto separatore in una porzione
n-butanolica, RBu , e in una acquosa, RW, rispettivamente di 5.7 g e di 15.3 g. Dal residuo
n-butanolico, RBu , attraverso varie tecniche cromatografiche, illustrate in Fig. 3.2, sono stati isolati 8
metaboliti, di cui 2 già caratterizzati nell’estratto CHCl3-MeOH (composti 1 e 2). I restanti composti
isolati sono anch’essi derivati fenolici, 4 fenilpropanoidi (composti 3, 5, 6 e 8), fra cui un nuovo composto naturale (3), e 2 flavoni glicosidi (composti 4, 7).
Fig.3.2 Schema di frazionamento dell’estratto n-butanolico, RBu
Estratto
R
Bu RBu/14 (RBu) RP-HPLC M-W (2:3) Composto 1 Composto 3 Composto 4 RBu/17 RP-HPLC M-W (2:3) Composto 3 Composto 4 Composto 5 RBu/19 RP-HPLC M-W (2:3) Composto 6 RBu/21 RP-HPLC M-W (2:3) Composto 2 Composto 7 RBu/24-25 RP-HPLC M-W (35:65) Composto 8SEPHADEX
LH 20
IN MeOH
1.4 Frazionamento dell’estratto cloroformico
Dall’estratto cloroformico RC attraverso varie tecniche cromatografiche, illustrate in Fig. 3.3, sono
stati identificati ed isolati 6 composti (composti 9, 10, 11, 12, 13, 14) tutti appartenenti alla classe dei terpeni. I composti 11, 12, 13, 14 son tutti diterpeni a nucleo neo-clerodanico, mai isolati in precedenti studi fitochimici. Il composto 9 è un sesquiterpene, ed il composto 10, un diterpene già isolato nella specie Clerodendrum inerme Gaertn.
Fig.3.3 Schema di frazionamento dell’estratto cloroformico, RC
Estratto
R
C RC/16-18 RP-HPLC M-W (7:3) Composto 9 RC/26 RP-HPLC M-W (3:2) Composto 10 Composto 11 RC/30 RP-HPLC M-W (2:3) Composto 12 RC33 RP-HPLC M-W (45:55) Composto 13 RC/35 RP-HPLC M-W (2:3) Composto 14FLASH
su silice a
gradiente
1.5 Flavonoidi, composti 2, 4, e 7
Il composto 2 è stato isolato a partire dall’estratto CHCl3-MeOH 9:1 RC-M, delle parti aeree di
Clerodendrum splendens G. Don. Una parte di residuo RC-M (10 g di 21.747 g) è stata sottoposta a
cromatografia su colonna Sephadex LH-20.
La frazione RC-M/16 è stata poi sottoposta a RP-HPLC eluendo con una miscela MeOH-H2O (2:3),
ottenendo così il composto 2. Tale composto si presenta come un solido amorfo di colore giallo, di formula molecolare C22H22O11,determinata attraverso spettrometria ESI-MS registrata in modalità
negativa. Il picco quasi molecolare è a m/z 461 [M-H]- .
O O H3CO RO OH OH O HO HO OH CH2OH =R
Fig.3.4 Struttura del composto 2, ispidulina 7-O-β-D-glucopiranoside
L’analisi dello spettro monodimensionale 1H-NMR e dello spettro bidimensionale HSQC e il confronto di questi con i dati presenti in letteratura (Merfort e Wendisch, 1987), hanno permesso di caratterizzare il composto come ispidulina 7-O-β-D-glucopiranoside (Fig. 3.4), chiamata anche 6-metossiapigenina 7-O-β-D-glucopiranoside, un flavone già precedentemente isolato in altre specie vegetali. I dati NMR sono riportati in Tab. 3.1 .
I compostI 4 e 7 sono stati isolati entrambi a partire dall’estratto n-butanolico RBu,ottenuto dalla
ripartizione n-BuOH/H2O dell’estratto metanolico RM delle parti aeree di Clerodendrum splendens
G. Don. Il residuo RBu (5.7 g) è stato sottoposto a cromatografia su colonna Sephadex LH-20.
Dalla frazione RBu/14, in seguito a cromatografia RP-HPLC utilizzando come fase mobile una
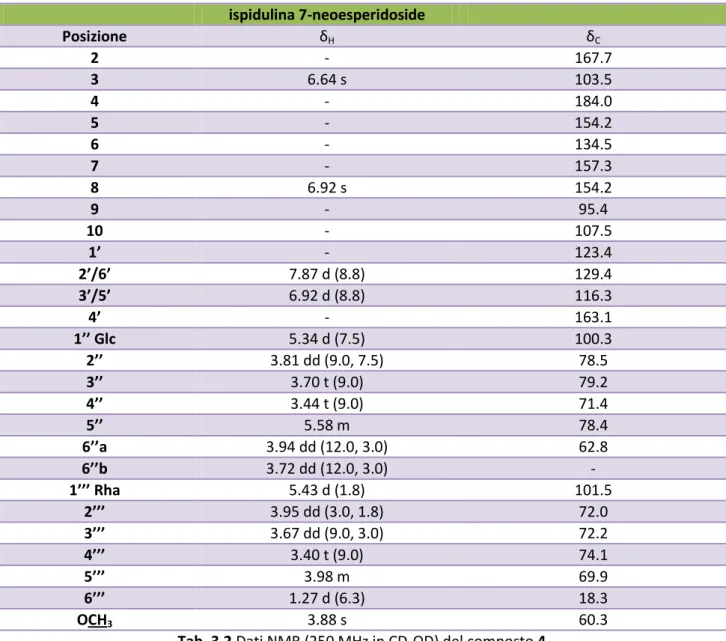
ispidulina 7-O-β-D-glucopiranoside Posizione δH δC 2 - 167.7 3 6.67 s 103.5 4 - 184.0 5 - 154.2 6 - 134.5 7 - 157.3 8 6.98 s 95.3 1’ - 123.4 2’/6’ 7.91 d (8.0) 129.2 3’/5’ 6.94 d (8.0) 116.8 1’’ Glc 5.13 d (7.8) 101.6 2’’ 3.54 dd (9.0, 7.8) 74.3 3’’ 3.55 t (9.0) 78.0 4’’ 3.43 t (9.0) 71.0 5’’ 3.47 m 77.8 6’’a 370 dd (12.0, 5.0) 62.1 6’’b 3.96 dd (12.0, 2.5) - OCH3 3.88 s 60.3
Tab. 3.1 Dati NMR (250 MHz in CD3OD) del composto 2
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
Il composto 4, isolato per la prima volta da Cirsium japonicum var. ussuriense DC (Compositae), si presenta come solido amorfo, di colore giallo, la cui formula molecolare C28H32O15 è stata
determinata attraverso spettrometria ESI-MS, registrata sia in modalità positiva che in modalità negativa. In particolare, nella modalità negativa è stato registrato il picco quasi molecolare a m/z 607 [M-H]-, e nella modalità positiva è stato ottenuto il picco equivalente al composto sodiato a
m/z 631 [M+Na]+, nonché i picchi a 485 [M+Na-146]+; 609 [M+H]+, 463 [M+H-146]+, 301 [M+H-146-162]+, 286 [M+H-146-162-15]+.
Dall’analisi degli spettri 1H-NMR ,COSY, 1D-TOCSY,HSQC e HMBC e il confronto con questi con i dati presenti in letteratura (Park et al., 1995), il composto 4 è stato caratterizzato come ispidulina 7-neoesperidoside (Fig. 3.5). I dati NMR sono riportati in Tab. 3.2
ispidulina 7-neoesperidoside Posizione δH δC 2 - 167.7 3 6.64 s 103.5 4 - 184.0 5 - 154.2 6 - 134.5 7 - 157.3 8 6.92 s 154.2 9 - 95.4 10 - 107.5 1’ - 123.4 2’/6’ 7.87 d (8.8) 129.4 3’/5’ 6.92 d (8.8) 116.3 4’ - 163.1 1’’ Glc 5.34 d (7.5) 100.3 2’’ 3.81 dd (9.0, 7.5) 78.5 3’’ 3.70 t (9.0) 79.2 4’’ 3.44 t (9.0) 71.4 5’’ 5.58 m 78.4 6’’a 3.94 dd (12.0, 3.0) 62.8 6’’b 3.72 dd (12.0, 3.0) - 1’’’ Rha 5.43 d (1.8) 101.5 2’’’ 3.95 dd (3.0, 1.8) 72.0 3’’’ 3.67 dd (9.0, 3.0) 72.2 4’’’ 3.40 t (9.0) 74.1 5’’’ 3.98 m 69.9 6’’’ 1.27 d (6.3) 18.3 OCH3 3.88 s 60.3
Tab. 3.2 Dati NMR (250 MHz in CD3OD) del composto 4.
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
O O H3CO RO OH OH O HO HO O CH2OH =R O CH3 OH OH OH
Lo spettro 1H-NMR mostra due singoletti e due doppietti orto-accoppiati a δ 6.64 e a 7.02 appartenenti rispettivamente al C-3 e al C-8, indicando che l’unità del flavone è ossigenata ai C-5, C-6, C-7 e C-4’. Inoltre il singoletto a δ 3.88 indica la presenza del gruppo OCH3. I segnali di due
protoni anomerici a δ 5.34 e a δ 5.43 indicano la presenza di due zuccheri di cui uno è il ramnosio essendo presente il segnale del CH3 a δ 1.27.
La frazione RBu/21sottoposta a RP-HPLC, eluita con una miscela MeOH-H2O (2:3), ha permesso
l’isolamento del composto 7.
Il composto 7 si presenta come solido amorfo di colore giallo, di formula molecolare C27H30O15,
determinata attraverso spettrometria ESI-MS registrata in modalità negativa e positiva, dando rispettivamente il picco quasi molecolare a m/z 593 [M-H]- e a m/z 595 [M-H]+.
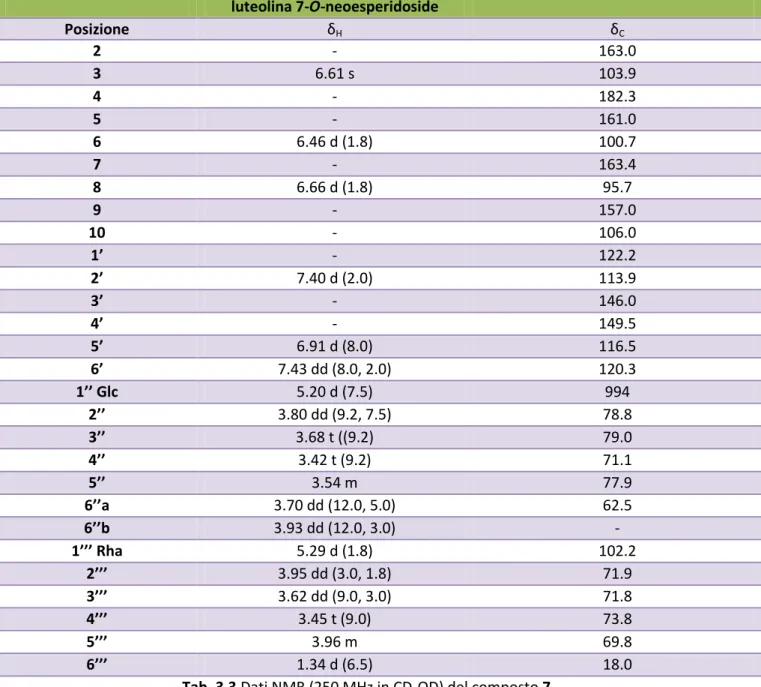
L’analisi dello spettro monodimensionale 1H-NMR e dello spettro bidimensionale HSQC e il confronto di questi con i dati presenti in letteratura (Agrawal, 1989) hanno permesso di caratterizzare il composto 7 come luteolina 7-O-neoesperidoside (Fig. 3.6). I dati NMR sono riportati in Tab. 3.3. O O RO OH OH O HO HO O CH2OH =R O CH3 OH OH OH OH
luteolina 7-O-neoesperidoside Posizione δH δC 2 - 163.0 3 6.61 s 103.9 4 - 182.3 5 - 161.0 6 6.46 d (1.8) 100.7 7 - 163.4 8 6.66 d (1.8) 95.7 9 - 157.0 10 - 106.0 1’ - 122.2 2’ 7.40 d (2.0) 113.9 3’ - 146.0 4’ - 149.5 5’ 6.91 d (8.0) 116.5 6’ 7.43 dd (8.0, 2.0) 120.3 1’’ Glc 5.20 d (7.5) 994 2’’ 3.80 dd (9.2, 7.5) 78.8 3’’ 3.68 t ((9.2) 79.0 4’’ 3.42 t (9.2) 71.1 5’’ 3.54 m 77.9 6’’a 3.70 dd (12.0, 5.0) 62.5 6’’b 3.93 dd (12.0, 3.0) - 1’’’ Rha 5.29 d (1.8) 102.2 2’’’ 3.95 dd (3.0, 1.8) 71.9 3’’’ 3.62 dd (9.0, 3.0) 71.8 4’’’ 3.45 t (9.0) 73.8 5’’’ 3.96 m 69.8 6’’’ 1.34 d (6.5) 18.0
Tab. 3.3 Dati NMR (250 MHz in CD3OD) del composto 7
1.6 Fenilpropanoidi, composti 1, 3, 5, 6, 8
Il composto 1 è stato isolato a partire dall’estratto CHCl3-MeOH (9:1) RC-M, delle parti aeree di
Clerodendrum splendens G. Don. Una parte di residuo RC-M (10 g di 21.747 g) è stata sottoposta a
cromatografia su colonna Sephadex LH-20.
Dalla frazione RC-M/12, sottoposta a RP-HPLC, eluita con una miscela MeOH-H2O (35:65), è stato
ottenuto il composto 1 che si presenta come solido amorfo bruno, di formula molecolare C34H44O19 determinata attraverso spettrometria ESI-MS registrata sia nella modalità positiva che
negativa.
I picchi quasi molecolare sono stati registrati a m/z 755 [M-H]- e 779 [M+Na]+.
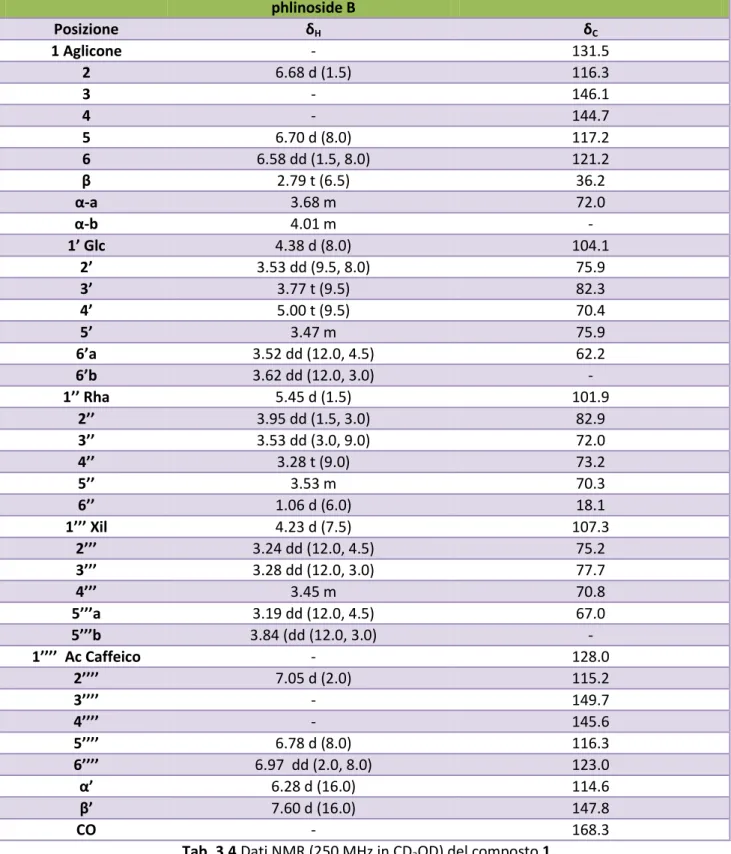
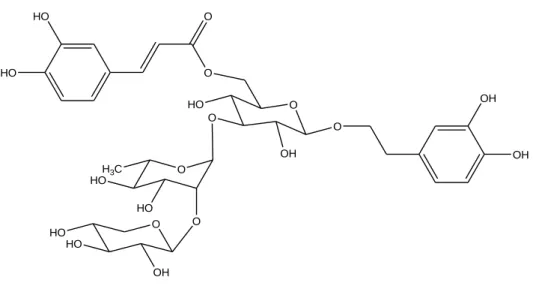
La struttura riportata in Fig. 3.7, è stata stabilita per mezzo del confronto con i dati riportati in letteratura (Calis et al., 1990) e dall’analisi degli spettri monodimensionali 1H-NMR, 13C-NMR e bidimensionale HSQC, arrivando alla caratterizzazione del composto 1 come phlinoside B.
I dati NMR sono riportati in Tab. 3.4.
O OH O HOH2C O CH3 OH HO O O HO HO OH O OH OH RO R= HO HO O R
Tab. 3.4 Dati NMR (250 MHz in CD3OD) del composto 1
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
I composti 3, 5, 6, e 8 sono stati tutti ottenuti a partire dall’estratto n-butanolico RBu ottenuto dalla
ripartizione n-BuOH/H2O dell’estratto metanolico RM delle parti aeree di Clerodendrum splendens
G. Don.
Il residuo RBu (5.7 g) è stato sottoposto a cromatografia su colonna Sephadex LH-20.
phlinoside B Posizione δH δC 1 Aglicone - 131.5 2 6.68 d (1.5) 116.3 3 - 146.1 4 - 144.7 5 6.70 d (8.0) 117.2 6 6.58 dd (1.5, 8.0) 121.2 β 2.79 t (6.5) 36.2 α-a 3.68 m 72.0 α-b 4.01 m - 1’ Glc 4.38 d (8.0) 104.1 2’ 3.53 dd (9.5, 8.0) 75.9 3’ 3.77 t (9.5) 82.3 4’ 5.00 t (9.5) 70.4 5’ 3.47 m 75.9 6’a 3.52 dd (12.0, 4.5) 62.2 6’b 3.62 dd (12.0, 3.0) - 1’’ Rha 5.45 d (1.5) 101.9 2’’ 3.95 dd (1.5, 3.0) 82.9 3’’ 3.53 dd (3.0, 9.0) 72.0 4’’ 3.28 t (9.0) 73.2 5’’ 3.53 m 70.3 6’’ 1.06 d (6.0) 18.1 1’’’ Xil 4.23 d (7.5) 107.3 2’’’ 3.24 dd (12.0, 4.5) 75.2 3’’’ 3.28 dd (12.0, 3.0) 77.7 4’’’ 3.45 m 70.8 5’’’a 3.19 dd (12.0, 4.5) 67.0 5’’’b 3.84 (dd (12.0, 3.0) - 1’’’’ Ac Caffeico - 128.0 2’’’’ 7.05 d (2.0) 115.2 3’’’’ - 149.7 4’’’’ - 145.6 5’’’’ 6.78 d (8.0) 116.3 6’’’’ 6.97 dd (2.0, 8.0) 123.0 α’ 6.28 d (16.0) 114.6 β’ 7.60 d (16.0) 147.8 CO - 168.3
Dallo studio della frazione RBu/14, per mezzo di RP-HPLC, eluendo con una miscela MeOH-H2O
(2:3), è stato separato il composto 3. Tale composto si presenta come un solido amorfo di colore bruno, otticamente attivo con [α]25= - 10.7 (c=-0.5, MeOH). La formula molecolare C34H44O19 è
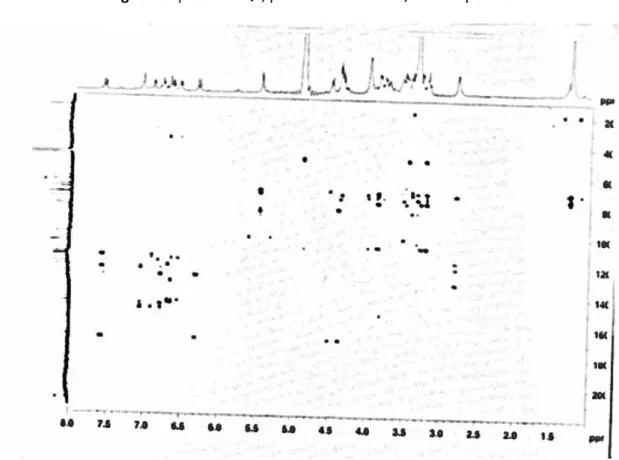
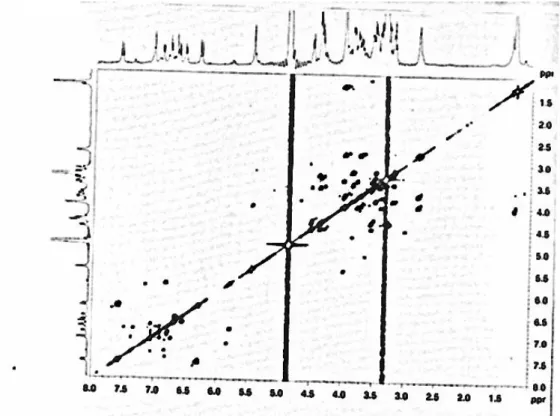
stata determinata mediante spettrometria di massa a ionizzazione elettrospray ad alta risoluzione (HR-ESI) (m/z 757.2637 [M+H]+). Lo spettro ESIMS mostra il picco quasi molecolare a m/z 779 [M+Na]+ (Fig. 3.9); sono stati inoltre osservate due frammentazioni importanti a m/z 647 [M+Na-132]+ e 501 [M+Na-132-146]+ dovute alla perdita di un pentoso e di un desossiesoso (Fig. 3.10). Lo spettro 13C NMR (Fig. 3.11) del composto 3 mostra 34 segnali, di cui 17 appartenenti all’aglicone, inclusi due anelli aromatici ed un gruppo idrossietilico; i rimanenti 17 segnali sono attribuibili ai residui zuccherini di due esosi e di un pentoso. Lo spettro 1H NMR (Fig. 3.12) di 3 ha evidenziato i segnali caratteristici dei protoni di un sistema ABX di un gruppo E-caffeoile [3 protoni aromatici che risuonano a δ 6.80 (1H, d, J = 8.0 Hz), 6.90 (1H, dd, J = 8.0, 2.0 Hz], 7.05 (1H, d, J = 2.0 Hz) e di due protoni olefinici trans come sistema AB a δ 6.30, e 7.58 (J = 16.0 Hz) e di un gruppo 3,4-diidrossifeniletilico [3 protoni aromatici come sistema ABX a δ 6.56 (1H, dd, J = 8.0, 2.0 Hz), 6.64 (1H, d, J = 2.0 Hz), 6.70 (1H, d, J = 8.0 Hz) e un sistema A2B2 assegnato a un gruppo idrossietilico a δ
2.78 (2H, m), 3.76 (1H, m), 3.96 (1H, m)] (Karioti et al. 2003). In aggiunta, tre segnali relativi a protoni anomerici indicano la presenza di tre zucccheri: un doppietto a δ 4.36 (1H, J = 7.8 Hz, H-1 di un glucosio), un ampio singoletto a δ 5.45 (1H, J = 1.8 Hz, H-1 di un ramnosio) e un doppietto a δ 4.34 (1H, J = 7.5 Hz, H-1 di un xilosio), coerente con la seguente configurazione al C-1: β per glucosio, α per il ramnosio, e β per lo xilosio. Queste conclusioni trovano conferma attraverso gli spettri HSQC/13C NMR (Fig. 3.13), dove i corrispondenti tre carboni anomerici risuonano rispettivamente a δ 104.1, 101.6, e 107.3. Gli altri segnali 1H e 13C NMR sono stati assegnati con l’ausilio di spettri 2D NMR, 1D-TOCSY, DQF-COSY (Fig. 3.15), HSQC, e HMBC (Fig. 3.14). Il chemical shift del C-3’ del glucosio a campi bassi (83.8 ppm) indica che è questa la posizione del sito di glicosilazione. Questa ipotesi ha trovato conferma a seguito di esperimenti HMBC, nei quali si è osservato il picco di correlazione tra H-1rha (δ 5.45) e C-3glc (83.8 ppm). I valori di chemical shifts dei
carboni dell’unità del β-xilosio si sono ossevati a valori usuali, suggerendo la sua posizione terminale. Sulla base degli spettri 1H e 13C NMR, un altro sito di interconnessione è stato trovato al C-2 del ramnosio, e attraverso lo spettro HMBC si sono osservate le correlazioni tra H-1xil (δ 4.34) e
C-2rha (82.3 ppm). Il sito di acilazione è stato assegnato al C-6 del glucosio come evidenza del forte
deschermo di H-6aglc e H-6bglc che risuonano a δ 4.36 e 4.52 e anche la correlazione fra H-6aglc e
caratterizzato come
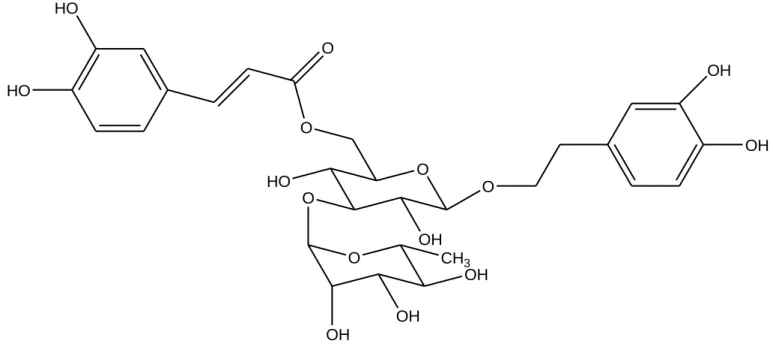
(3,4-diidrossifenil)etil-O-β-D-xilopiranosil-(1→2)-α-L-ramnopiranosil-(1→3)-6-O-t-caffeoil-β-D-glucopiranoside (Fig. 3.8), un nuovo glicoside fenilpropanoidico.
I dati dello spettro 1H e 13C-NMR sono riportati in Tab. 3.5.
HO HO O O O OH O OH OH HO O O O O HO HO OH HO H3C HO
Fig. 3.8 Struttura del composto 3,
(3,4-diidrossifenil)etil-O-β-D-xilopiranosil-(1→2)-α-L-ramnopiranosil-(1→3)-6-O-t-caffeoil-β-D-glucopiranoside.
Fig. 3.9 Spettro ESI-MS in modalità positiva
Fig. 3.11 Spettro 13C-NMR del composto 3
Fig. 3.13 Spettro HSQC, porzione zuccherina, del composto 3
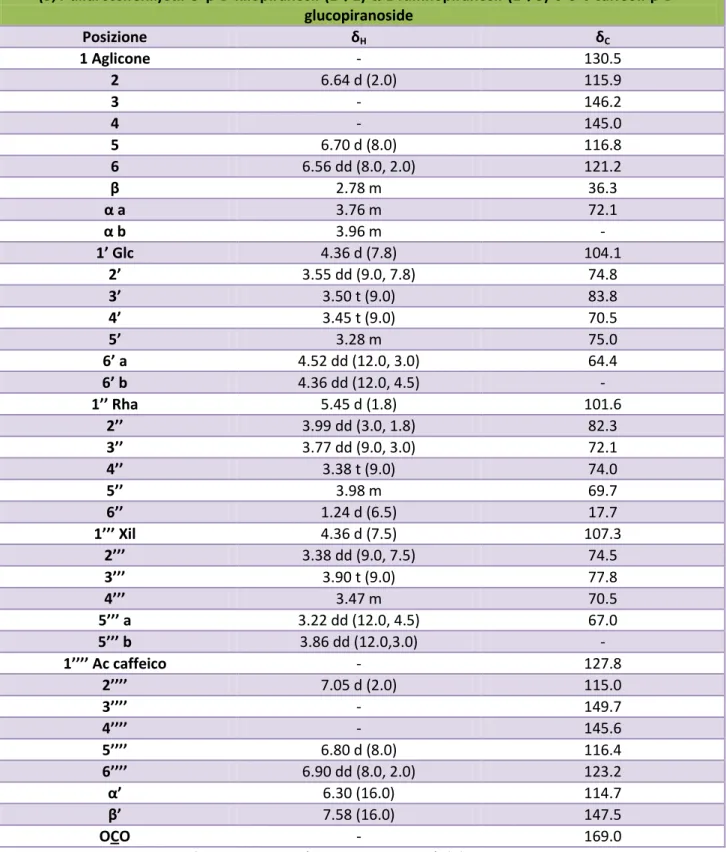
(3,4-diidrossifenil)etil-O-β-D-xilopiranosil-(1→2)-α-L-ramnopiranosil-(1→3)-6-O-t-caffeoil-β-D-glucopiranoside Posizione δH δC 1 Aglicone - 130.5 2 6.64 d (2.0) 115.9 3 - 146.2 4 - 145.0 5 6.70 d (8.0) 116.8 6 6.56 dd (8.0, 2.0) 121.2 β 2.78 m 36.3 α a 3.76 m 72.1 α b 3.96 m - 1’ Glc 4.36 d (7.8) 104.1 2’ 3.55 dd (9.0, 7.8) 74.8 3’ 3.50 t (9.0) 83.8 4’ 3.45 t (9.0) 70.5 5’ 3.28 m 75.0 6’ a 4.52 dd (12.0, 3.0) 64.4 6’ b 4.36 dd (12.0, 4.5) - 1’’ Rha 5.45 d (1.8) 101.6 2’’ 3.99 dd (3.0, 1.8) 82.3 3’’ 3.77 dd (9.0, 3.0) 72.1 4’’ 3.38 t (9.0) 74.0 5’’ 3.98 m 69.7 6’’ 1.24 d (6.5) 17.7 1’’’ Xil 4.36 d (7.5) 107.3 2’’’ 3.38 dd (9.0, 7.5) 74.5 3’’’ 3.90 t (9.0) 77.8 4’’’ 3.47 m 70.5 5’’’ a 3.22 dd (12.0, 4.5) 67.0 5’’’ b 3.86 dd (12.0,3.0) - 1’’’’ Ac caffeico - 127.8 2’’’’ 7.05 d (2.0) 115.0 3’’’’ - 149.7 4’’’’ - 145.6 5’’’’ 6.80 d (8.0) 116.4 6’’’’ 6.90 dd (8.0, 2.0) 123.2 α’ 6.30 (16.0) 114.7 β’ 7.58 (16.0) 147.5 OCO - 169.0
Tab. 3.5 Dati NMR (250 MHz in CD3OD) del composto 3
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
Il composto 5 è stato isolato dalla frazione RBu/17 sottoposta a RP-HPLC, utilizzando come fase
mobile una miscela MeOH-H2O (2:3). Tale composto si presenta come una polvere amorfa di
colore giallo pallido, la cui formula molecolare C29O15H36 è stata determinata mediante
Il picco quasi molecolare si presenta a m/z 625 [M+H]+. La struttura, riportata in Fig. 3.16, è stata stabilita per mezzo del confronto con i dati riportati in letteratura (Liu et al., 1998) e mediante l’ausilio degli spettri monodimensionali 1H-NMR e 13C-NMR e bidimensionale HSQC, si è identificato il composto come verbascoside, un fenilpropanoide isolato per la prima volta da
Lysionotus pauciflorus (Gesneriaceae). I dati NMR sono riportati in Tab. 3.6.
HO HO O O O HO O O OH O CH3 OH OH OH OH OH
verbascoside Posizione δH δC 1 A - 127.8 2 7.07 d (2.0) 116.3 3 - 146.6 4 - 149.1 5 6.71 d (8.0) 117.1 6 6.99 dd (2.0) 123.1 α a 3.78 m 72.0 α b 4.05 m β a 2.84 m 36.2 β b 2.84 m - 1’ Glc 4.39 d (7.9) 104.0 2’ a 3.29 dd (9.0, 7.9) 75.6 2’ b 3.90 t (9.0) - 3’ 3.74 t (9.0) 81.5 4’ 4.90 t (9.0) 70.1 5’ 3.56 m 75.7 6’ a 3.65 dd (12.0, 4.5) 62.1 6’ b 3.90 dd (12.0, 3.0) - 1’’ Rha 5.20 d (1.8) 102.6 2’’ 3.62 dd (3.0, 1.8) 72.2 3’’ 3.50 dd (9.0, 3.0) 72.1 4’’ 3.40 t (9.0) 73.7 5’’ 3.95 m 70.2 6’’ 1.10 d (6.0) 18.2 1’’’’ Ac caffeico - 127.6 2’’’’ 6.67 d (2.0) 115.1 3’’’’ - 146.6 4’’’’ - 149.1 5’’’’ 6.78 d (8.0) 116.3 6’’’’ 6.58 dd (8.0, 2.0) 123.1 α’ 6.28 d (15.9) 115.3 β’ 7.57 d (15.9) 147.8 COO - 168.5
Tab. 3.6 Dati NMR (250 MHz in CD3OD) del composto 5
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
Dallo studio della frazione RBu/19 sottoposta a RP-HPLC con una miscela eluente MeOH-H2O (2:3),
è stato isolato il composto 6. Tale composto si presenta come un solido amorfo bruno e di formula molecolare C29H36O15,determinata mediante esperimenti ESI-MS, registrati in modalità positiva,
ottenendo il picco quasi molecolare corrispondente al composto sodiato a m/z 647 [M+Na]+, e in modalità negativa il corrispondente picco quasi molecolare a m/z 623 [M-H]-.
L’analisi dello spettro monodimensionale 1H-NMR e degli spettri bidimensionali HSQC e HMBC e il confronto di questi con dati presenti in letteratura (Miyase et al., 1982), hanno permesso dicaratterizzare il composto 6 (Fig. 3.17) come isoacteoside, composto isolato per la prima volta da Leucosceptum japonicum (Miq.) Kitamura e Murata, appartenente alla famiglia delle Labiateae. I dati NMR sono riportati in Tab. 3.7.
O O HO O OH O O HO HO O OH OH OH CH3 OH OH
Isoacteoside Posizione δH δC 1 Aglicone - 131.4 2 6.64 d (2.0) 115.9 3 - 146.0 4 - 145.0 5 6.67 d (8.0) 116.7 6 6.54 dd (8.0, 2.0) 121.0 α a 3.72 m 72.2 α b 3.97 m - β 2.78 t 36.2 1’ Glc 4.34 d (8.0) 104.2 2’ 3.57 dd (9.0, 8.0) 75.0 3’ 3.55 t (9.0) 83.5 4’ 3.44 t (9.0) 70.2-69.7 5’ 3.30 m 75.3 6’ a 4.36 dd (12.0, 5.0) 64.2 6 ’b 4.51 dd (12.0, 3.0) - 1’’ Rha 5.19 d (1.5) 102.3 2’’ 3.62 dd (3.0, 1.5) 72.2 3’’ 3.52 dd (9.0, 3.0) 72.2 4’’ 3.43 t (9.0) 73.6 5’’ 4.00 m 69.7 6’’ 1.25 d (6.5) 17.5 α’ 6.29 d (16.0) 114.5 β’ 7.57 d (16.0) 147.0 1’’’ Ac caffeico - 127.7 2’’’ 7.05 d (2.0) 114.8 3’’’ - 149.5 4’’’ - 145.5 5’’’ 6.78 d (8.0) 116.3 6’’’ 6.91 dd (8.0, 2.0) 123.0 COO - 169.0
Tab. 3.7 Dati NMR (250 MHz in CD3OD) del composto 6
La frazione RBu/24-25, analizzata mediante RP-HPLC, utilizzando come fase mobile una miscela
MeOH-H2O (35:65), ha permesso la separazione del composto 8. Esso si presenta come un solido
amorfo di colore giallo pallido, e di formula molecolare C18H16O8, determinata tramite
spettroscopia ESI-MS, in modalità negativa.
L’analisi !H-NMR e 13C-NMR ha consentito l’immediata identificazione del composto 8 con l’acido rosmarinico (Fig. 3.18) in accordo con i dati riportati in letteratura (Tezuka et al., 1998).
HO HO O O OH OH COOH H
Fig. 3.18 Struttura del composto 8, acido rosmarinico acido rosmarinico Posizione δH δC 1 - 127.7 2 7.04 d (2.0) 115.2 3 - 146.8 4 - 149.7 5 6.80 d (8.0) 116.5 6 6.94 dd (2.0) 123.1 7 7.55 d (16.0) - 8 6.27 d (16.0) 114.6 1’ - 128.7 2’ 6.76 d (1.8) 117.6 3’ - 116.7 4’ - 143.8 5’ 6.68 d (8.0) 116.3 6’ 6.64 d (8.0) 121.8 7’ 3.03 dd (10.8) 38.0 8’ 5.70 dd ( 8.3) 74.9
Tab. 3.8 Dati NMR (250 MHz in CD3OD) del composto 8
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
3.7 Sesquiterpene, composto 9
Il composto 9 è stato ottenuto a partire dall’estratto cloroformico RC delle parti aeree di
Clerodendrum splendens G. Don. Una parte del residuo cloroformico (10 g) è stata sottoposta a
cromatografia flash su gel di silice, ed il composto 9 è stato isolato dalla frazione RC/16-18, in
seguito a RP-HPLC, eluendo con una miscela MeOH-H2O (7:3). Tale composto si presenta come un
solido amorfo incolore e di formula molecolare C20H30O4, determinata mediante esperimenti
ESI-MS in modalità positiva, ottenendo il picco quasi molecolare corrispondente al composto sodiato a
m/z 357 [M+Na]+.
Gli spettri monodimensionali 1H-NMR e 1D-TOCSY e quelli bidimensionali HSQC, HMBC e DFQ-COSY, ed il confronto con i dati presenti in letteratura, hanno permesso di identificare e caratterizzare il composto 9 come 2β-angeloilossi-5β-idrossi-7αH,10β-metil-eudesm-3-en-1-one
(Fig. 3.19) già precedentemente isolato da Seseli vayredanum (Umbelliferae) (Barrero et al., 1994). I dati NMR sono presenti nella Tab. 3.9.
O
O
OH O
Fig. 3.19 Struttura del composto 9, 2β-angeloilossi-5β-idrossi-7αH,10β-metil-eudesm-3-en-1-one 2β-angeloilossi-5β-idrossi-7αH,10β-metil-eudesm-3-en-1-one Posizione δH δC 1 - 223.8 2 5.25 d (7.6) 77.3 3 5.60 d (7.7) 119.8 4 - 148.6 5 - 82.9 6 a 2.12 ma 30.0 6 b 2.56 br dd (13.7) - 7 2.26 m 52.4 8 a 2.19 ma 39.8 8 b 2.43 m - 9 a 2.20 ma 37.7 9 b 2.34 m - 10 - 61.4 11 2.12 ma 27.0 12 1.00 d (6.7) 21.9 13 1.08 d (6.7) 25.0 14 1.05 s 19.0 15 1.80 s 26.0 1’ - 167.8 2’ - 127.9 3’ 6.07 qq (1.4, 7.1) 140.0 4’ 1.95 dq (1.5,7.1) 15.0 5’ 1.86 dq (1.4) 20.9
Tab. 3.9 Dati NMR (600 MHz in CD3OD) del composto 9
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
a
3.8 Diterpeni, composti 10, 11, 12, 13, 14
I composti 10, 11, 12, 13, 14 sono stati tutti isolati a partire da dall’estratto cloroformico RC delle
parti aeree di Clerodendrum splendens G. Don. Una parte del residuo cloroformico (10 g) è stata sottoposta a cromatografia flash su gel di silice seguita da analisi RP-HPLC.
Lo studio della frazione RC/26 sottoposta a RP-HPLC , eluita con una miscela MeOH-H2O (3:2), ha
permesso l’isolamento del composto 10 e del composto 11.
Il composto 10, precedentemente isolato dalle parti aeree di Clerodendrum inerme (L.) Gaertn. (Pandey et al., 2005), si presenta come solido amorfo incolore, di formula molecolare C26H38O10,
determinata mediante analisi spettrometrica ESI-MS, registrata sia in modalità negativa che positiva. Il picco quasi molecolare è a m/z 509 [M-H]- mentre in modalità positiva si è registrato il picco a m/z 533 [M+Na]+.
Dall’analisi degli spettri monodimensionali 1H-NMR e 13C-NMR e bidimensionale HSQC, e dal confronto con i dati riportati in letteratura, è stato possibile identificare il composto 10 come 14,15-diidro-15-idrossi-3-epicarioptina (Fig. 3.20). I dati NMR sono riportati in Tab. 3.10.
O O OH AcO O CH2OAc OAc
14,15-diidro-15-idrossi-3-epicarioptina Posizione δH δC 1 a 1.64 m 21.0 1 b 1.81 m - 2 a 2.03 m 31.2 2 b 1.54 m - 3 5.27 d (6.5) 68.3 4 - 64.0 5 - 47.0 6 4.78 dd (11.1, 4.59) 72.5 7 a 1.84 m 32.7 7 b 1.62 m - 8 1.54 m 36.8 9 - 41.6 10 1.79 m 48.5 11 4.63 dd (11.0, 6.0) 4.00 dd (12.0,5.0) 84.3 e 84.8 12 a 1.60 m 32.5 12 b 1.83 m - 13 3.10 m 41.0 14 a 2.08 m 40.0 14 b 1.78 m - 15 5.42 br d (5.5) e 5.53 br d (3.0) 99.0 e 99.4 16 5.73 d (5.2) e 5.70 d (4.5) 108.2 e 109.8 17 a 2.88 d (3.5) 43.2 17 b 2.60 d (3.5) . 18 a 4.49 d (12.0) 62.7 18 b 4.76 d (12.0) - 19 0.93 s 13.9 20 0.90 d (6.5) 16.2 OCOCH3 1.91, 2.09, 1.96 s 20.8, 20.8, 20.4
Tab. 3.10 Dati NMR (600 MHz in CD3OD) del composto 10
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
Come possiamo osservare dai risultati degli spettri NMR, il composto 10 è una miscela di epimeri al C-15. Infatti i segnali dei protoni H-16, H-15 e H-11 appaiono come coppie di segnali.
Il composto 11, mai precedentemente isolato in natura, si presenta come un solido amorfo incolore, otticamente attivo con [α]25=- 9,4 (c=0.5, MeOH).
La formula molecolare è stata determinata come C35H50O16 grazie ai risultati degli spettri HR-ESIMS
(m/z 727.3204 [M+H]+) e 13C NMR. Lo spettro ESIMS (Fig. 3.22) del composto 11 mostra il picco quasi molecolare a m/z 749 [M+Na]+ oltre ai picchi a m/z 689 [M+Na-60]+, 531 [M+Na-218]+, 471 [M+Na-218-60]+ e 411 [M+Na-218-60-60]+ dovuti alla perdita di due gruppi acetilici e di una catena laterale acetilata (Fig. 3.23). Gli spettri 1H e 13C NMR (Fig. 3.24, 3.25) rivelano la presenza di cinque
segnali attribuibili a dei gruppi acetilici,un doppietto di un metile (δ 1.20, d, J = 6.5 Hz) accoppiato ad un quartetto di un metino (δ 5.10, q, J = 6.5 Hz), e un singoletto di un metile (δ 1.58) derivanti da un gruppo 2,3-diacetossi-2-metilbutanoilossidico, insieme ai segnali caratteristici di un diterpene neo-clerodanico avente un gruppo 4α,17-ossiranico [δH 2.63, 2.91 (H2-17), δC 41.8
(C-17); δC 64.0 (C-4)]. L’analisi degli spettri 2D NMR mostrano che il composto 11 possiede un anello
esaidrofurofuranico, tipico delle 14-idroclerodine (Ohno et al., 1996). Infatti, i protoni H-11, H-15, and 16 appaiono come coppie di segnali: 11 dd a δ 4.04 e 4.65 (dd, J = 11.0, 6.2 Hz, 0.5 H); H-15 δ 5.33 e 5.45 (br d, J = 5.7 Hz, 0.5 H), e H-16 δ 5.61 (d, J=5.2 Hz, 0.5 H) e 5.65 (d, J = 4.3 Hz, 0.5 H ciascuno). Lo spettro 13C NMR di 11 è in completo accordo con la struttura ipotizzata e con i risultati degli spettri 13C NMR di altri diterpeni neo-clerodanici isolati dal genere Clerodendrum contenenti lo stesso gruppo ai C-11/C-16 (Pandey et al., 2005). Il chiarimento completo della struttura è stato ottenuto tramite le correlazioni degli spettri 1D-TOCSY, DQF-COSY (Fig. 3.26), HSQC (Fig. 3.27), e HMBC (Fig. 3.28), le quali hanno anche permesso di assegnare nello spettro 13C NMR tutti i valori di risonanza pertinenti ad ogni carbonio. Le correlazioni HMBC tra H-1 e C-2, C-5 e C-10, tra H-3 e C-4 e C-17, tra H-16 e C-11, C-13, e C-15, tra H-12 e C-13, C-14, e C-16, tra H-18 e C-4, C-5, C-6, e C-10, hanno confermato l’assegnazione dei carboni ad un’anello neo-clerodanico. I segnali relativi ai picchi di correlazione tra δ 5.46 (H-3) e 169.2 ppm e δ 4.90 (H-2) e 172.2 ppm rivelano che un gruppo acetile e la funzione 2,3-diacetossi-2-metilbutanoilossi sono legate rispettivamente al C-2 e C-3. L’elucidazione della stereochimica relativa è stata ottenuta soprattutto sulla base delle strette somiglianze di 11 con i valori NMR di altri composti simili (Pandey et al., 2005; Jannet et al., 1999). In questo modo, il composto 11 è stato caratterizzato come miscela epimera al C-15 di 2α-acetossi-3β-(2’,3’-diacetossi-2’-metil)-butanoilossi-14-idro-15-idrossiclerodina (Fig. 3.21). I dati NMR sono riportati in Tab. 3.11.
O O OH O O O O O O O O O OAc OAc
Fig. 3.21 Struttura di 2α-acetossi-3β-(2’,3’-diacetossi-2’-metil)-butanoilossi-14-idro-15-idrossiclerodina,
composto 11
Tab. 3.11 Dati NMR (600 MHz in CD3OD) del composto 11
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
a
segnale sotto il solvente
2α-acetossi-3β-(2’,3’-diacetossi-2’-metil)-butanoilossi-14-idro-15-idrossiclerodina Posizione δH δC 1 a 2.75 m 28.0 1 b 1.85 m - 2 4.90 a 71.6 3 5.46 d (10.5) 68.3 4 - 64.0 5 - 47.0 6 4.87 dd (11.0, 4.5) 72.0 7 a 1.60 m 32.4 7 b 1.92 m - 8 1.55 m 37.0 9 - 41.6 10 1.92 m 43.0 11 4.65 dd (11.0, 6.2) 4.04 dd (12.0, 5.0) 84.0 e 85.1 12 a 1.70 m 33.7 12 b 1.48 m - 13 3.11 m 41.0 14 a 2.26 m 40.6 14 b 1.78 m - 15 5.45 br d (5.7) e 5.33 br d (3.0) 99.3 e 99.6 16 5.61 d (5.2) e 5.65 d (4.3) 108.2 e 109.8 17 a 2.63 (4.0) 41.8 17 b 2.91 (4.0) . 18 a 4.52 d (12.3) 62.7 18 b 4.82 d (12.3) - 19 0.96 s 15.0 20 0.94 d 15.2 OCOCH3 2.10, 2.10, 1.97 s 21.3, 21.3, 20.3 OCOCH3 - 172.2, 171.8, 170.0 1’ - 169.2 2’ - 83.5 3’ 5.10 q (6.5) 73.0 4’ 1.20 d (6.5) 15.0 5’ 1.58 s 15.2 COCH3 2.10, 1.92 s 21.3, 21.2 COCH3 - 171.9
Fig. 3.24 Spettro 1H-NMR del composto 11
Fig. 3.26 Spettro DQF-COSY del composto 11
Fig. 3.27 Spettro HMBC del composto 11
Dall’analisi della frazione RC/30, sottoposta a RP-HPLC usando come fase mobile una miscela
MeOH-H2O (2:3), si è ottenuto il composto 12, otticamente attivo con [α]25=+ 5.7 (c=.015, MeOH),
composto mai precedentemente isolato in natura. La formula molecolare del composto 12 C24H36O9 è stata stabilita tramite analisi degli spettri 13C NMR e HRESIMS (m/z 469.2500 per
[M+H]+). Nello spettro ESIMS si sono osservate due frammentazioni principali a m/z 425 [M-H-42] -e 383 [M-H-42-42]- dovute alla perdita di due gruppi acetili. I dati NMR (Tab. 3.12) suggeriscono che la struttura di 12 è simile a quella di 11, ma differisce per i sostituenti sull’anello A. Sono state osservate differenze negli spettri NMR di 12 e 11 quali la presenza in 12 di un gruppo idrossilico legato al C-3 al posto del gruppo 2,3-diacetossi-2-metil-butanoilico e di un’acetile al C-2 del composto 11. In questo modo, la struttura di 12 è stata identificata come miscela epimera al C-15 di 3β,15-diidrossi-14-idro-clerodina (Fig. 3.28).
HO O O O O O O O HO
3β,15-diidrossi-14-idro-clerodina Posizione δH δC 1 a 1.75 m 21.1 1 b 2.28 m - 2 a 1.30 m 31.3 2 b 1.82 m - 3 3.98 m 65.4 4 - 68.0 5 - 49.4 6 4.80 dd (11.0, 4.5) 71.7 7 a 1.70 m 33.0 7 b 1.42 m - 8 1.54 m 36.8 9 - 42.0 10 1.78 47.6 11 4.63 dd (11.0, 6.0) 3.99 dd (12.0, 5.0) 84.6 e 84.7 12 a 1.61 m 32.8 12 b 2.06 m - 13 2.97 m 40.2 14 a 2.07 m 49.7 14 b 1.78 m - 15 5.45 br d (5.0) e 5.54 br d (4.6) 97.09 e 98.5 16 5.71 d (4.7) e 5.67 d (6.2) 106.8 e 108.7 17 a 2.84 d (3.5) 41.6 17 b 2.72 d (3.5) . 18 a 4.91 d (12.5) 61.6 18 b 4.33 d (12.5) - 19 0.92 d (6.5) 13.0 20 0.95 s 15.9 OCOCH3 2.10, 1.93 s 19.7, 20.2 OCOCH3 - 172.8, 171.2
Tab. 3.12 Dati NMR (600 MHz in CD3OD) del composto 12.
Il composto 13 è stato isolato dalla frazione RC/33 in seguito a RP-HPLC, eluendo con una miscela
MeOH-H2O (45:55). Tale composto, mai isolato in precedenza in natura, si presenta come un
solido amorfo incolore, otticamente attivo con [α]25=- 5.7 (c=0.11, MeOH).
Il composto 13 (C31H46O14) mostra il picco quasi molecolare a m/z 643.2899 [M+H]+ nello spettro
HRESIMS in modalità positiva. L’importante frammentazione ionica osservata nello spettro ESIMS a m/z 486 [M+Na-176]+ ha suggerito la presenza di un gruppo 2’-idrossi-3’-acetossi-2’-metilbutanoile nel composto 13. L’analisisi degli spettri 1H e 13C NMR di tale composto ha chiaramente indicato che anch’esso appartiene alla classe dei diterpeni neo-clerodanici. Inoltre, attraverso il confronto dei dati NMR dello spettro di 13 con quelli dei derivati neo-clerodanici già isolati dal genere Clerodendrum, è stato evidenziato che il composto contiene delle funzionalità in comune. Lo spettro 1H NMR mostra i segnali dovuti alla risonanza di un metile terziario (δ 1.02, 3H, s), di un metile secondario (δ 0.93, 3H, J = 6.0 Hz), di tre residui acetilici (δ 1.91. 1.99, e 2.11. ciascuno 3H, s), e di un metile doppietto (δ 1.27, d, J = 6.5 Hz) accoppiato a un metino quartetto (δ 5.12, q, J = 6.5 Hz) e un metile singoletto (δ 1.29, s) attribuibile ad un sistema 2’-idrossi-3’-acetossi-2’-metilbutanoilossidico nella posizione C-3β. Lo spettro 1H-NMR inoltre mostra due quartetti AB , tipici di un CH2OH a δ 4.51 e 4.75 (2H, J = 11.5 Hz ciascuno, H-18), un metilene di un gruppo
epossidico a δ 2.64 e 2.90 (2H, J = 3.5 Hz ciascuno, H-17), un doppio doppietto a δ 4.82 (1H, J = 11.6, 4.5 Hz, H-6) dovuto al protone al C-6, un multipletto a δ 3.64 (1H, m, H-2), e un doppietto a δ 5.15 (1H, J =6.6 Hz, 3) dovuto al protone al C-3. I segnali a δ 1.80 (1H, m, 14a), 1.95 (1H, m, H-14b), 2.06 (1H, m, H-12a), 2.37 (1H, m, H-12b), 2.86 (1H, m, H-13), 3.67 (1H, m, H-15a), 3.72 (1H, m, H-14b) indicano la presenza di un sostituente in posizione 9β avente un sistema lattonico a cinque termini. La struttura del composto 13 è stata confermata grazie allo spettro 13C NMR. La risonanza a campi bassi nello spettro 13C NMR a 181.0 ppm suggerisce la presenza di un 11,16-lattone. Inoltre lo spettro contiene i segnali a δ 13.8 e 16.2 dovuti rispettivamente ai metili al C-19 e C-20. Segnali attribuibili ai tre gruppi acetato appaiono a δ 171.5, 171.7, 172.6, e a δ 21.2 (2 segnali sovrapposti) e 21.3, dove i segnali dei carboni portanti i gruppi acetato appaiono a δ 62.5 18) e 72.2 6). I carboni implicati nella formazione dell’anello ossiranico risuonano a δ 43.2 17) e 64.0 4). Altri segnali significativi sono stati osservati a δ 34.3 14), 39.9 13), e 60.0 (C-15). Il chiarimento dell’intera struttura è stato ottenuto soprattutto grazie all’analisi delle correlazioni negli spettri 1D-TOCSY, DQF-COSY, HSQC, e HMBC, i quali hanno anche permesso di assegnare tutti i valori di risonanza pertinenti ad ogni carbonio nello spettro 13C NMR. L’anello 11,16-lattonico è stato confermato grazie alle correlazioni HMBC tra H-12, H-13 e H-14 e C-16. In
base a tali risultati, il composto 13 è stato caratterizzato come 2α,15-diidrossi-3β-(2’-idrossi-3’-acetossi-2’-metil)-butanoilossi-6α,18-diacetossi-4α,17-epossi-clerodan-11,16-lattone (Fig. 3.29). I dati NMR sono riportati in Tab. 3.13.
OAc O O O OH HO O AcO O OAc OH
Fig. 3.29 Struttura del composto 13,
Tab. 3.13 Dati NMR (600 MHz in CD3OD) del composto 13
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in Hz
2α,15-diidrossi-3β-(2’-idrossi-2’-metil-3’-acetossi)-butanoilossi-6α,18-diacetossi-4α,17-epossi-clerodan 11,16-lattone Posizione δH δC 1 a 1.83 m 32.0 1 b 2.19 m - 2 3.64 m 71.0 3 5.15 d (6.6) 74.4 4 - 64.0 5 - 47.2 6 4.82 dd (11.6, 4.5) 72.2 7 a 1.70 m 33.7 7 b 1.50 m - 8 1.67 m 35.6 9 - 42.5 10 1.95 dd 43.1 11 4.70 t (9.0) 89.9 12 a 2.37 br dd (13.0, 9.0) 28.4 12 b 2.06 m - 13 2.86 m 39.9 14 a 1.80 m 49.7 14 b 1.95 m 34.3 15 a 3.72 m 60.0 15 b 3.67 m 16 - 181.0 17 a 2.64 d (3.5) 43.2 17 b 2.90 d (3.5) . 18 a 4.51 d (11.5) 62.5 18 b 4.65 - 19 1.02 s 13.8 20 0.93 d (6.0) 16.2 OCOCH3 2.11, 1.99 s 21.3, 21.2 OCOCH3 - 172.6, 171.5 1’ - 172.0 2’ - 77.9 3’ 5.12 q (6.5) 75.0 4’ 1.27 d (6.5) 13.6 5’ 1.29 s 22.0 OCOCH3’ 1.91 s 21.2 OCOCH3’ - 171.7
Dallo studio della frazione RC/35 analizzata mediante RP-HPLC, utilizzando come fase mobile una
miscela eluente MeOH-H2O (45:55) si è isolato il composto 14, composto mai precedentemente
isolato in natura.
Esso si presenta come un solido incolore, otticamente attivo con [α]25= + 2.5 (c=0.13, MeOH)
Lo spettro HRESIMS di 14 mostra lo ione quasi molecolare a m/z 485.2423 [M+H]+, in accordo con la formula molecolare C24H36O10. Nello spettro ESIMS due frammentazioni (Fig. 3.31) a m/z 447
[M+Na-60]+ e 387 [M+Na-60-60]+ indicano la perdita del residuo acetossilico. I dati NMR di 14 sono simili a quelli del composto 13 differendo nei valori dell’anello A e nei valori di 13/C-13, H-14/C-14, H-15/C-15 (2.90/45.0 in 14 vs 2.86/39.9 in 13, 4.06/72.6 in 14 vs 1.95, 1.89/34.3 in 13, 3.49, 3.60/64.9 in 14 vs 3.67, 3.72/60.0 in 13, rispettivamente). Le differenze osservate negli spettri NMR di 14 (Fig. 3.32, 3.33, 3.34, 3.35, 3.36, 3.37) e 13 sono coerenti con la presenza in 14 di un gruppo idrossilico legato al C-3 invece del gruppo 2-idrossi-3-acetossi-2-metil-butanoilossilico e un gruppo acetossilico al C-2 in 13. Un’altra differenza è la presenza in 14 di un gruppo 14,15-diolo. La configurazione assoluta del gruppo 14,15-diolico di 14 è stata determinata tramite dicroismo circolare (CD) dopo complessazione in-situ con dimolibdeno tetracetato in soluzione di DMSO (Di Bari et al., 2001). In accordo con la regola di Snatzke (Frelek et al., 1999), il segno della banda diagnostica a circa 305 nm è correlata con la configurazione assoluta dei centri chirali nei gruppi 1,2-diolici. In particolare i glicoli S-monosostituiti danno effetto Cotton positivo a 305 nm, mentre un effetto negativo è dato dalla configurazione-R. Di conseguenza al segno positivo dello spettro CD di 14 segue l’assegnazione della configurazione-S al C-14. Conseguentemente il composto 14 è stato identificato come 3β,14S,15-tridrossi-6α,18-diacetossi-4α,17-epossi-clerodan-11,16-lattone (Fig. 3.38).
I dati NMR sono riportati in Tab. 3.14.
Fig. 3.31 Frammentazioni dovute alla perdita del gruppo acetossilico nel composto 14
Tab. 3.14 Dati NMR (250 MHz in CD3OD) del composto 14
I valori di chemical shift sono indicati in ppm e le costanti di accoppiamento sono in parentesi e riportati in H
3β,14S,15-triidrossi-6α,18-diacetossi-4α,17-epossi-clerodan-11,16-lattone Posizione δH δC 1 1.85 m 22.7 2 a 1.40 m 34.3 2 b 2.12 br dd (11.5, 4.5) 3 4.02 dd (11.5, 5.43) 66.3 4 - 68.4 5 - 47.9 6 4.84 t (9.0) 73.0 7 a 1.75 m 34.3 7 b 1.50 m - 8 1.65 m 35.5 9 - 48.0 10 1.92 dd 43.1 11 4.72 t (9.0) 88.5 12 a 2.31 m 23.0 12 b 2.22 m - 13 2.90 m 45.0 14 4.06 m 72.6 15 a 3.49 dd (7.2, 12.0) 64.9 15 b 3.60 dd (6.2, 11.2) - 16 - 181.0 17 a 2.75 d (4.5) 43.2 17 b 2.90 d (4.5) . 18 a 4.36 d (12.0) 62.8 18 b 4.94 d (12.0) - 19 1.03 s 14.0 20 0.96 d (6.6) 16.4 OCOCH3 2.12, 1.96 s 21.2, 21.4 OCOCH3 - 172.7, 171.4
Fig. 3.33 Spettro HSQC del composto 14
Fig. 3.35 Spettro 1D-TOCSY dell’anello B del composto 14
Fig. 3.37 Spettro HMBC del composto 14 OAc O O OH HO O AcO HO HO
Fig. 3.38 Struttura del composto 14,