Università degli studi di Pisa
Corso di Laurea Specialistica in Farmacia
INIBITORI DELLA LRRK2:
UN NUOVO APPROCCIO TERAPEUTICO PER LA
MALATTIA DI PARKINSON
Relatore:
Prof.ssa Annalina Lapucci
Candidata:
Panagiota Pratsa
2
alla mia famiglia e mia nonna
3
INDICE
INTRODUZIONE
...4CAPITOLO 1 : La LRRK2 e la sua inibizione come
strategia terapeutica
1.1 La chinasi LRRK2...25 1.1.1. Struttura e dominii...26 1.1.2. Attività chinasica ...27 1.1.3. Attività GTPasica...28 1.1.4. Ipotetici substrati...301.2 Strategie terapeutiche per la malattia di Parkinson associata alla LRRK2...32
1.2.1. Attività chinasica e GTPasica come target...33
1.2.2. Strategie fuori dalle regioni enzimatiche...35
1.3 Problematiche nello sviluppo di farmaci per la LRRK2...36
CAPITOLO 2 : Inibitori della chinasi LRRK2 e loro
applicazione nel Parkinson
2.1 Inibitori della chinasi LRRK2 a piccola molecola...402.2 Altri inibitori della LRRK2 a piccola molecola, potenti, selettivi, che superano la barriera ematoencefalica ...52
CONCLUSIONI
...774
INTRODUZIONE
Storia
Il Parkinson è una malattia neurodegenerativa, ad evoluzione lenta ma progressiva, che coinvolge, principalmente, alcune funzioni quali il controllo dei movimenti e dell'equilibrio. La malattia fa parte di un gruppo di patologie definite "Disordini del Movimento" e tra queste è la più frequente. I sintomi del Parkinson sono forse noti da migliaia di anni: una prima descrizione sarebbe stata trovata in uno scritto di medicina indiana risalente al 5.000 a.C. ed un'altra in un documento cinese risalente a 2.500 anni fa. Il nome è legato però a James Parkinson, un farmacista chirurgo londinese del XIX secolo, che per primo descrisse gran parte dei sintomi della malattia, a cui più tardi fu dato il suo nome, in un famoso libretto, il "Trattato sulla paralisi agitante" [1].
L’insorgenza di segni motori nella malattia di Parkinson è tipicamente asimmetrica e il sintomo iniziale più comune è un tremore a riposo asimmetrico in un arto superiore. Nel corso del tempo, i pazienti notano sintomi correlati alla progressiva bradicinesia, rigidità e difficoltà di deambulazione [2].
Alcuni sintomi non motori precedono comunemente i sintomi motori nel morbo di Parkinson. La maggior parte dei pazienti hanno una riduzione sostanziale della funzione olfattiva (odore) prima che i segni motori emergano. Tuttavia, questo sintomo può non essere notato dai pazienti oppure i pazienti possono non rendersi conto che fa parte della malattia. Un altro sintomo non motorio comune è il disturbo comportamentale nel sonno REM (RBD). In questa circostanza, gli individui possono compiere dei movimenti durante il sonno REM che sono descritti spesso come colpire o moti di respinta. Ci sono inoltre una serie di fattori di rischio della mezza età per lo sviluppo successivo del morbo di Parkinson. Questi comprendono la costipazione e l'eccessiva sonnolenza diurna, sebbene siano lontani dallo specifico per il morbo di Parkinson.
5
I principali sintomi clinici nella malattia di Parkinson sono:
Tremore
Una diminuzione sottile nella coordinazione dei movimenti nelle varie attività come per esempio vestirsi o giocare a golf.
Diminuzione del movimento rotatorio del braccio sul primo lato coinvolto
Abbassamento della voce
Diminuzione della mimica facciale
Disturbi del sonno
RBD (REM Behaviour Disorders, o Disturbo Comportamentale nel sonno REM), in cui vi è una perdita della normale atonia durante il sonno REM: in uno studio, il 38% di uomini di 50 anni con RBD e nessun segno neurologico ha continuato a sviluppare parkinsonismo [3]; i pazienti "agiscono i loro sogni" e possono scalciare, colpire, parlare o gridare nel sonno.
Diminuzione dell’olfatto.
Disfunzioni dell’autonomia, compresa la costipazione, anomalie nella sudorazione, disfunzioni sessuali e dermatiti seborroiche.
Una sensazione generale di debolezza, malessere o stanchezza.
Depressione o anedonia.
Lentezza nel pensare
Comuni segni motori precoci della malattia di Parkinson sono il tremore, la bradicinesia, la rigidità, e la distonia.
Tremore
Sebbene il tremore sia il più comune dei sintomi iniziali nella malattia di Parkinson, compare approssimativamente nel 70% dei pazienti, ma non deve essere presente per fare la diagnosi. Il tremore è abbastanza spesso descritto dai pazienti come debolezza o nervosismo e di solito inizia in un arto superiore e inizialmente può essere intermittente. Il tremore alle estremità superiori inizia generalmente nelle dita o nel pollice, ma può anche iniziare
6
all'avambraccio o al polso. Dopo parecchi mesi o anni, il tremore può diffondersi verso l'estremità inferiore isolaterale o controlaterale all'estremità superiore prima di diventare più generalizzato; tuttavia l’asimmetria è generalmente mantenuta.
Il tremore può variare considerevolmente, emergendo soltanto con lo sforzo, l'ansia, o l'affaticamento. Classicamente, il tremore del morbo di Parkinson è un tremore a riposo (che accade con l'arto in una posizione di riposo) e scompare con l’azione o l’uso dell'arto, ma questo non è stato osservato in tutti i pazienti. Inizialmente, il tremore può essere notato durante le attività come il mangiare o la lettura del giornale. Sebbene il morbo di Parkinson sia una causa rara del tremore che colpisce la testa o il collo, i tremori del mento, del labbro, o della lingua non sono rari. Come con altri tremori, l'ampiezza aumenta con lo sforzo e si risolve durante il sonno.
Bradicinesia
La bradicinesia, o lentezza dei movimenti, è particolarmente frustrante perché può rendere delle semplici azioni alquanto difficili. La persona non riesce ad eseguire rapidamente movimenti quotidiani; le attività che prima eseguiva rapidamente e facilmente, come lavarsi o vestirsi, potrebbero richiedere tempi molto più lunghi.
I sintomi della bradicinesia sono vari e possono essere descritti in modo diverso: possono includere un soggettivo senso di debolezza, senza debolezza vera all'esame fisico; perdita di destrezza, a volte descritto dai pazienti come "il messaggio a non raggiungere l'arto", affaticamento, o prurito durante l'esecuzione di azioni ripetute. La bradicinesia facciale è caratterizzata da una diminuita espressività del viso. Il discorso può diventare più morbido, meno distinto, o più monotonale. Nei casi più avanzati il discorso è impastato, poco articolato e di difficile comprensione.
La scialorrea (particolarmente la scialorrea notturna) è un sintomo iniziale raro da solo ma viene riferito successivamente nel corso della malattia.
7
Essa è associata alla bradicinesia in quanto dovuta ad un rallentamento dei muscoli coinvolti nella deglutizione.
La bradicinesia del tronco si deduce dalla lentezza o difficoltà del paziente di alzarsi da una sedia, girarsi nel letto, o camminare. Se la camminata è colpita, i pazienti possono intraprendere passi più piccoli e la cadenza di andatura è ridotta. Alcuni pazienti avvertono un'incapacità transitoria di camminare, come se i loro piedi fossero congelati al pavimento. Questo “congelamento” della marcia improvviso viene notato comunemente nei pazienti con la malattia più avanzata; è più pronunciato quando i pazienti tentano di attraversare le entrate o le aree strette e possono rimanere intrappolati dietro mobili o incapaci di oltrepassare la soglia di una porta.
Negli arti superiori, la bradicinesia può causare la piccola, faticosa scrittura (micrografia) e la difficoltà di usare le mani per le attività abili fini come l’uso di una chiave o degli utensili della cucina. Nelle estremità inferiori, la bradicinesia unilaterale causa comunemente lo strascicamento di quel piede sulla terra, poichè non è preso durante l'oscillazione della gamba. Questo fenomeno può anche essere descritto come il trascinamento di una gamba.
Rigidità
La rigidità può essere il primo sintomo della malattia di Parkinson e spesso si manifesta a un lato del corpo ma molti pazienti non lo avvertono. Un segno di rigidità, associata alla lentezza dei movimenti, è la riduzione dell’ oscillazione degli arti superiori durante il cammino. Il braccio colpito non può oscillare completamente durante il cammino ed il piede dello stesso lato può strusciare il pavimento. Col passare del tempo, la posizione assiale diventa flessa progressivamente e le andature diventano più brevi.
Alcuni pazienti possono descrivere una rigidità degli arti, ma questo può riflettere più la bradicinesia che la rigidità. Occasionalmente, gli individui descrivono una sensazione di rigidità quando muovono un arto, che può essere definita “a ruota dentata”, caratterizzata cioè dallo sviluppo di resistenza in punti diversi durante il movimento di una articolazione (rigidità a scatti).
8
Distonia
La distonia è un sintomo iniziale comune nel morbo di Parkinson giovanile, che è definito come sintomo iniziale prima dei 40 anni. La distonia nel morbo di Parkinson consiste comunemente in una rotazione involontaria del piede verso l’interno (inversione) o verso il basso (flessione plantare), spesso associata con crampi o dolore alla gamba. Si può verificare anche la flessione dorsale dell'alluce. Un'altra distonia comune nel morbo di Parkinson è l'adduzione del braccio e del gomito, portando la mano a riposare davanti all'addome o al torace. Le posture distoniche possono crescere e calare e sono legate a momenti di affaticamento o di sforzo.
Background
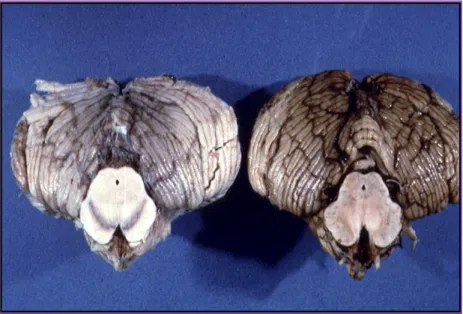
Il morbo di Parkinson colpisce approssimativamente l’1% degli individui al di sopra dei 60 anni. Ci sono due principali risultati neuropatologici: la perdita di neuroni dopaminergici pigmentati nella parte compatta della substantia nigra (SNpc) e la presenza di corpi di Lewy (Figura 1).
Figura 1. Confronto dell'aspetto della substantia nigra fra un cervello normale e un cervello colpito
dal morbo di Parkinson. Da notare come la substantia nigra sia ben pigmentata nell'esemplare normale di cervello, a sinistra. Nel cervello di un paziente affetto dal morbo di Parkinson, a destra, si osserva la perdita della substantia nigra pigmentata a causa dello spopolamento dei neuroni pigmentati [2].
9
La graduale e irreversibile perdita dei neuroni dopaminergici nella substantia nigra è la lesione principale del Parkinson. I sintomi clinici del morbo di Parkinson diventano evidenti quando il 50-60% dei neuroni nigrostriatali dopaminergici è perso. Il Parkinson progredisce insidioso per 5-7 anni (periodo preclinico) e poi continua a peggiorare anche nell'ambito del trattamento sintomatologico [4]. Sebbene i sintomi e la neuropatologia del Parkinson siano stati ben caratterizzati, i meccanismi e le cause della malattia non sono ancora del tutto chiariti.
Le mutazioni genetiche possono fornire degli indizi importanti al meccanismo della malattia, ma la maggior parte dei casi di Parkinson è sporadica piuttosto che familiare; i fattori ambientali sono stati sospettati a lungo per il loro contributo alla malattia (Figura 2). È stato ipotizzato che per la maggior parte dei casi del morbo di Parkinson idiopatico (IPD) la causa possa essere una combinazione di fattori genetici ed ambientali. Sebbene più del 90% dei casi di Parkinson accadano sporadicamente e siano probabilmente dovuti, in parte, allo stress ossidativo ed alla disfunzione mitocondriale, lo studio sulle mutazioni genetiche ha fornito una grande comprensione nei meccanismi molecolari di questa patologia [5].
Gli sviluppi recenti nella genetica molecolare hanno rivelato che il 5-20% dei pazienti provenienti di aree geografiche diverse ha forme monogeniche di Parkinson. I numerosi difetti genetici ( ~82% di mutazioni semplici e ~18% di variazioni del numero di copie) sono stati identificati in più di 13 loci e 9 geni nei pazienti con Parkinson ereditato e famigliare. Tra i geni individuati quelli più importanti sono: SNCA (PARK1) che codifica per l’α-sinucleina, parkin (PARK2) che codifica per la parkina, PINK1 (PARK6) che codifica per la chinasi putativa 1 PTEN-indotta (PINK1), DJ-1 (PARK7) che codifica per la DJ-1 e LRRK2 (PARK8) che codifica per la chinasi 2 con ripetizioni ricche di leucina.
10
Figura 2. Il Parkinson si sviluppa da interazioni complesse tra fattori ambientali e genetici e
comprende delle vie molecolari multiple che interferiscono conducendo alla neurodegenerazione [4].
Mutazioni ereditarie dominanti
Il gene SNCA (PARK1) codifica per la α-sinucleina che consiste di 140 amminoacidi ed è una proteina distesa nel suo stato nativo, solubile nel citoplasma o associata alle membrane lipidiche. Sebbene le funzioni fisiologiche della α-sinucleina non siano state ancora completamente comprese, evidenze sperimentali implicano la presenza di α-sinucleina nel rilascio del neurotrasmettitore, nel turnover vescicolare, nella plasticità sinaptica e nel traffico intracellulare all'interno del reticolo endoplasmatico e dell’apparato di Golgi.
11
Studi genetici hanno dimostrato che le mutazioni puntiformi o l'alterazione genetica (duplicazioni o triplicazioni) che aumenta il numero delle copie del gene dell’ α-sinucleina (SCNA) possono causare la malattia di Parkinson o la demenza da corpi di Lewy, strettamente correlata con il Parkinson. La α-sinucleina può polimerizzare in maniera aberrante nelle fibrille con proprietà tipiche dell'amiloide. Queste fibrille sono la componente principale di molti tipi di inclusioni patologiche, compresi i corpi di Lewy, che sono associate con le malattie neurodegenerative, quale la malattia di Parkinson. Sebbene ci siano delle prove sostanziali che sostengono la natura tossica delle inclusioni dell’ α-sinucleina, sono stati proposti anche altri meccanismi di tossicità come la conversione in oligomeri [6].
Le mutazioni nel gene LRRK2, la causa più conosciuta del Parkinson, sono associate con le forme autosomiche dominanti e sporadiche di Parkinson. Finora sono state identificate più di 80 mutazioni di senso attraverso l'intera LRRK2 e colpiscono tutti i dominii funzionali della proteina.
Il trascritto di LRRK2 (PARK8) contiene 51 esoni e codifica per una proteina di 2527 amminoacidi, la LRRK2 (conosciuta anche come Dardarina). Parecchi dominii funzionali in LRRK2 (per esempio Roc GTPasi, Roc-COR e dominii della proteina chinasi) suggeriscono che questa proteina abbia funzioni multiple, comprese le interazioni proteina-proteina, il binding a substrato e la fosforilazione proteica. Έ stato osservato che la LRRK2 in condizioni fisiologiche, forma dei dimeri e può svolgere un ruolo nello sviluppo e nell'orientamento di un neurone. La Wildtype LRRK2, ma non i suoi mutanti, attenua la morte cellulare indotta da stress ossidativo, indicando un ruolo protettivo per la LRRK2.
Mutazioni ereditarie recessive
Le mutazioni con perdita della funzione nei geni che codificano per la parkina, la PINK1 e la DJ-1, causano il Parkinson autosomico recessivo. Tutte queste forme di Parkinson sono relativamente rare e rispondono alla terapia con la levodopa (L-DOPA). Le mutazioni semplici (per esempio mutazioni di
12
senso, di non senso e splice site), i riarrangiamenti esonici, le piccole inserzioni e le delezioni come pure le variazioni di numero di copia della regione promotrice e degli esoni del promotore sono stati identificati in questi geni recessivi.
Le proteine PINK1, Parkina e DJ-1 sono coinvolte dal punto di vista funzionale nel mantenimento dell'integrità e della funzionalità mitocondriale e proteggono le cellule dagli effetti negativi dei fattori di stress multipli. Le mutazioni in questi geni causano la disfunzione mitocondriale ed il declino successivo nella produzione dell’ATP ed aumentano la produzione dei radicali liberi, come risultato dello stress ossidativo e della carenza energetica. I mitocondri alterati possono liberare il citocromo c ed altri fattori pro-apoptotici che avviano le cascate apoptotiche e la morte cellulare. L'eccessiva produzione di radicali liberi può danneggiare le proteine (per esempio, modificazione anormale della α-sinucleina ed inattivazione della Parkina), i lipidi, il DNA o l’RNA, conducendo alla disfunzione delle cellule ed alla morte finale.
Fattori ambientali
Recenti studi hanno dimostrato una stretta interazione tra cause genetiche e ambientali della neurodegenerazione della malattia di Parkinson. In particolare, è stato suggerito che le esposizioni ambientali a determinate sostanze neurotossiche (metalli pesanti, pesticidi e fungicidi) possano svolgere un certo ruolo nello sviluppo dei disordini neurodegenerativi del movimento, quale la malattia di Parkinson.
L'uso accidentale di 1-metil-4-fenil-1,2,3,6-tetraidropiridina (MPTP), un sottoprodotto della sintesi illecita dell'eroina, conduce ad una sindrome di Parkinson che è clinicamente indistinguibile dal Parkinson vero e proprio. La similarità strutturale fra il paraquat (N,N’-dimetil-4,4’-bipiridinio dicloruro), un diserbante comune e lo ione di 1-metil-4-fenilpiridinio (MPP+), il metabolita attivo di MPTP, conferma l’ipotesi che il paraquat potrebbe essere un neurotossico dopaminergico e l'esposizione al paraquat può essere collegata
13
allo sviluppo di Parkinson. Negli studi sperimentali in cui il paraquat è stato somministrato agli animali, i ricercatori hanno osservato una perdita di neuroni dopaminergici della substantia nigra, deplezione della dopamina e apoptosi. L'esposizione sperimentale dei modelli animali a parecchie classi di pesticidi, quale il paraquat, a fungicidi a base di ditiocarbammato (per esempio maneb), all'insetticida rotenone e a pesticidi organoclorurati (per esempio la dieldrina) conduce a neurotossicità dopaminergica. L'insetticida rotenone induce nei ratti delle caratteristiche cliniche e patologiche simili a quelli indotti dal Parkinson, compresi la degenerazione selettiva del sistema dopaminergico nigrostriatale e disordini di movimento.
Oltre ai prodotti agricoli, gli agenti inquinanti industriali stanno cominciando a ricevere il riconoscimento come fattori di rischio potenziali per lo sviluppo di Parkinson. I tossici industriali sono una vasta e diversa classe di composti utilizzati nella fabbricazione e nella produzione di vari prodotti domestici e commerciali. Come con i pesticidi, la popolazione umana è esposta ordinariamente ai tossici industriali, sul luogo di lavoro, attraverso l’alimentazione o per la loro presenza in alcuni prodotti domestici [7].
I composti organo-alogenati sono una classe di composti organici con vari livelli di sostituzione dell'alogeno (prevalentemente cloro, bromo o fluoro) sugli atomi di carbonio. L'uso di questi composti nell’industria può variare dai rivestimenti isolanti elettrici, oli antifiamma e in adesivi e plastica. Le stesse proprietà che hanno reso questi composti così attraenti per l’uso industriale, come il fatto di essere altamente stabili e resistenti alla degradazione, li hanno resi anche estremamente pericolosi per l’ambiente e per la popolazione umana.
I policlorobifenili (PCB) sono stati introdotti nell'industria e nell'uso commerciale verso il 1930 e sono costituiti dagli anelli del difenile i cui atomi di idrogeno sono sostituiti da uno fino a dieci atomi di cloro. Un pericolo di PCB è il loro alto livello di liposolubilità, che permette loro di depositarsi preferenzialmente nelle regioni del corpo ricche di lipidi, quali il tessuto
14
adiposo ed il cervello. Questa caratteristica dei PCB abbinata con la difficoltà nella degradazione, metabolizzazione ed eliminazione, permette loro di accumularsi all'interno dell’organismo e di creare intossicazione per esposizione cronica. Parecchi studi hanno identificato i potenziali meccanismi cellulari e molecolari responsabili dell'associazione fra l'esposizione ai PCB ed il Parkinson. Le alterazioni della neurotrasmissione della dopamina sono state studiate più estesamente, sia in vitro che in vivo. Questi studi hanno evidenziato che l’esposizione ai PCB causa una riduzione di dopamina in linee di cellule dopaminergiche o porzioni del sistema nigrostriatale dei topi, dei ratti e dei primati non umani. I meccanismi che conducono alla riduzione dei livelli della dopamina sono vari poichè è stato dimostrato che l'esposizione ai PCB inibisce la tirosina idrossilasi e la DOPA decarbossilasi, entrambi enzimi coinvolti nella sintesi di dopamina.
I solventi rappresentano una vasta gamma di prodotti chimici con l'utilità comune di dissoluzione di una sostanza in un’altra. I solventi più comuni includono il tricloroetilene (TCE), il toluene, l'acetone, l'esano, il solfuro di carbonio, tutti usati a scopi multipli a livello domestico e industriale. Sono generalmente lipofili, e ciò permette loro di essere assorbiti rapidamente dall’organismo e dagli organi bersaglio dopo l'esposizione. Parecchi deficit neurologici, quali il tremore e altri deficit motori, sono stati associati all'esposizione ai solventi. Tuttavia, è poco chiaro se questi sintomi rappresentano il Parkinson o un disordine simile al Parkinson. Sebbene molti studi epidemiologici abbiano tentato di mettere in luce un'associazione fra l'esposizione ai solventi ed il Parkinson, non è stato ancora definito un collegamento valido.
È stato suggerito che, come con altri fattori ambientali, l'esposizione ai metalli può essere un fattore di rischio per lo sviluppo del morbo di Parkinson ed altri disordini del movimento relativi al Parkinson. I metalli, in particolare i metalli di transizione svolgono un ruolo essenziale nel funzionamento normale di numerosi processi biologici, specialmente come cofattori enzimatici.
15
Mentre gli effetti tossici derivanti dall'esposizione ai metalli sul corpo umano sono stati ben documentati, soltanto negli ultimi 20-30 anni i risultati neurotossicologici sono stati apprezzati. Effettivamente, il cervello sembra essere particolarmente vulnerabile alla tossicità dei metalli in quanto essi colpiscono vari aspetti dei neuroni, compresa la generazione di stress ossidativo e l’interruzione della neurotrasmissione. Questi effetti sono particolarmente nocivi quando avvengono nei gangli basali. Per esempio, l'esposizione professionale cronica dei minatori ad alti livelli di manganese, un cofattore essenziale di parecchi enzimi, come la superossido dismutasi (SOD), causa l'accumulo di questo metallo nei gangli basali, con conseguente tremore, rigidità e psicosi che somigliano al Parkinson.
Numerosi casi clinici hanno associato gli episodi di traumi cranici con l'avvenimento di un Parkinsonismo di fase acuta o con lo sviluppo di Parkinson più tardi nella vita. Parecchi studi epidemiologici compresa un'analisi delle anamnesi di un grande gruppo di veterani della Seconda guerra mondiale hanno identificato le lesioni alla testa antecedenti come un fattore di rischio importante per lo sviluppo di Parkinson in età più avanzata. Tuttavia, i risultati epidemiologici sono contradditori e non indicano una tale correlazione.
Le tossine ambientali ed il trauma cranico possono provocare delle lesioni neuronali danneggiando i mitocondri, causando lo stress ossidativo, inducendo l'infiammazione nel sistema nervoso centrale (SNC) e compromettendo i meccanismi di difesa delle cellule. Alcuni fattori di rischio ambientali possono attivare la microglia direttamente (le cellule immunitarie residenti nel SNC) o causare un'infiammazione sistemica, che a sua volta colpisce il SNC. La variazione genetica ed i polimorfismi nella regione di HLA (Human Leucocyte Antigen) e in parecchie citochine infiammatorie possono trasformarsi in fattori di rischio per il Parkinson. Le cellule della microglia attivate producono e secernono una gamma di molecole infiammatorie e citotossiche, quali le citochine, le chemochine, i radicali liberi
16
reattivi, gli eicosanoidi e le proteasi. Oltre a modulare l'attività microgliale, queste molecole influenzano il destino dei neuroni circostanti. L'eccessiva reazione infiammatoria diventa solitamente esagerata e distruttiva e si trasforma in infiammazione cronica che determina il processo neurodegenerativo progressivo.
Gli studi epidemiologici rilevano in modo consistente la riduzione dei rischi per il morbo di Parkinson in relazione al fumo di sigarette. La maggior parte degli studi indica che la riduzione supplementare è conferita dal fumo aumentato, valutato dai pacchetti per giorno, dagli anni di tabagismo o dall’ incapacità evidente di smettere di fumare. Questa associazione può riflettere un effetto neuroprotettivo di nicotina o altre componenti di tabacco. Alternativamente, persone predisposte al Parkinson possono avere risposte differenti alla nicotina, quali il metabolismo alterato della nicotina o le differenze precliniche nei neuroni dopaminergici, che sono centrali sia alla patogenesi del Parkinson che alla dipendenza di nicotina [8].
Interazione gene-ambiente
Il concetto che la predisposizione al Parkinson viene influenzata da interazioni gene-ambiente è stata proposta per più di una decade e molti studi hanno descritto le associazioni positive fra i polimorfismi genetici ed il rischio aumentato per il Parkinson. Fino ad ora, dozzine di polimorfismi a singolo nucleotide (SNP) in parecchi geni hanno indicato le interazioni gene-ambiente relativamente al Parkinson. Gli effetti cumulativi ed interattivi del tabagismo e dei polimorfismi genici in NOS1 (NO sintetasi neuronale), in NOS2A (NO sintetasi inducibile) o in SNCA (sinucleina) possono modulare il rischio per Parkinson. L'esposizione professionale ai pesticidi come pure l'alta esposizione al paraquat ed al maneb in trasportatori delle varianti genetiche di DAT (trasportatore della dopamina) hanno aumentato il rischio per il Parkinson. Polimorfismi a singolo nucleotide in NOS1 e GSTP1 (la glutatione S-Transferasi 1) sono stati collegati ad un rischio aumentato per il Parkinson in individui esposti ai pesticidi.
17
Strutture coinvolte nella malattia del Parkinson
Lo striato è il punto di ingresso principale di informazioni corticali ai gangli basali e riceve afferenze da aree anatomicamente e funzionalmente differenti della corteccia cerebrale. Secondo il modello corrente della funzione dei gangli basali, le informazioni corticali sono elaborate nei nuclei dei gangli basali e rimandate alla corteccia tramite il talamo (circuito cortico-basale) [9]. Gli studi anatomici sperimentali dei tracciati nelle scimmie hanno identificato tre circuiti paralleli dei collegamenti corticostriatali:
1) il circuito limbico, che è coinvolto nel comportamento emozionale, motivazionale e stereotipato ed è implicato nel disturbo da deficit di attenzione, nel disordine di iperattività, nei disordini compulsivi e nella sindrome di Tourette. Esso include lo striato ventromediale, il nucleo accumbens, il nucleo caudato rostrale/ventrale e i putamen, che ricevono degli input dalla corteccia prefrontale orbitale e mediale;
2) il circuito associativo, implicato nelle funzioni cognitive, come l’attenzione e il monitoraggio delle informazioni all'interno della memoria di lavoro (Working memory). Esso coinvolge la testa del nucleo caudato e le aree del putamen rostrali, che ricevono degli input dalla corteccia prefrontale laterale, dall'area motoria pre-supplementare (SMA) e dalla corteccia parietale posteriore;
3) il circuito senso-motorio, implicato nelle funzioni motorie, che comprende il nucleo caudato e le parti laterali del putamen, che ricevono degli input dalla corteccia somatosensoriale primaria e l’area motoria supplementare.
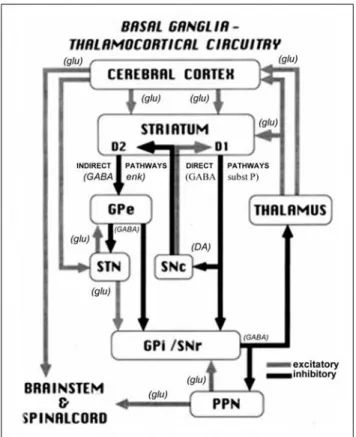
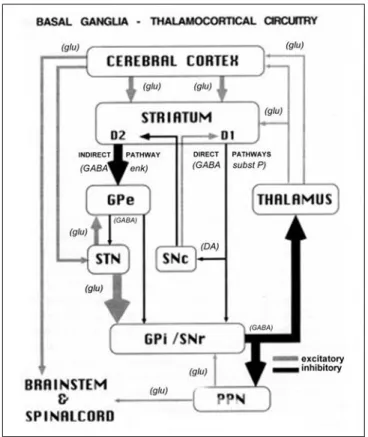
La dopamina ha un ruolo fondamentale in quasi tutte le funzioni normali dei gangli basali. In condizioni fisiologiche, il rilascio della dopamina nello striato sembra essere fortemente coinvolta nel processo di elaborazione della ricompensa [10]. Inoltre, il livello di dopamina nello striato può regolare il flusso generale di informazioni corticali attraverso le vie dirette e indirette nello striato (Figura 3).
18
Figura 3. Rappresentazione schematica del circuito motorio dei gangli basali talamocorticali ed i suoi
neurotrasmettitori nello stato normale. (Da Vitek J. Stereotaxic surgery and deep brain stimulation for Parkinson disease and movement disorders. In: Watts RL, Koller WC, eds. Movement Disorders: Neurologic Principles and Practice. New York: McGraw-Hill, 1997:240. Copyright, McGraw-Hill Companies, Inc. Used with permission.)
La disfunzione di questo sistema è stata collegata tradizionalmente allo sviluppo di disturbi motori in assenza di dopamina (parkinsonismo), o alle anomalie nella trasmissione dopaminergica (come può accadere nelle discinesie indotte dalla levodopa ed in certe forme di distonia). Le proiezioni dopaminergiche possono anche avere funzioni all'infuori della modulazione della trasmissione sinaptica. Esiste un considerevole numero di studi che comprovano come l'assenza della trasmissione dopaminergica possa avviare dei cambiamenti nella densità e nella morfologia delle spine dendritiche sui neuroni della proiezione striatale che, a loro volta, possono influenzare la trasmissione corticostriatale (Figura 4).
Recenti ricerche hanno inoltre evidenziato che tutti gli altri nuclei dei gangli basali ricevono gli input dopaminergici, sebbene queste proiezioni e le concentrazioni di dopamina in questi nuclei siano molto più piccole. L’importanza dell’input dopaminergico per la funzione normale di queste aree
19
Figura 4. Rappresentazione schematica del circuito motorio dei gangli basali talamocorticali e il
relativo cambiamento nell’attività neuronale nel morbo di Parkinson. (Da Vitek J. Stereotaxic surgery and deep brain stimulation for Parkinson disease and movement disorders. In: Watts RL, Koller WC, eds. Movement Disorders: Neurologic Principles and Practice. New York: McGraw-Hill, 1997:241. Used with kind permission. Copyright, McGraw-Hill Companies, Inc.)
extrastriatali rimane poco chiara, ma è stato dimostrato che l'attivazione o il blocco del recettore della dopamina in queste regioni altera profondamente l'accensione di un neurone. Risultati recenti hanno suggerito che la diminuzione dei livelli di dopamina possa interferire seriamente con i meccanismi della plasticità sinaptica nello striato e nella SNr (parte reticolata della substantia nigra) ed è possibile che questi cambiamenti contribuiscano allo sviluppo dei sintomi motori nel Parkinson.
Attualmente non esiste una cura per il morbo di Parkinson. Il trattamento farmacologico e la chirurgia alleviano temporaneamente soltanto i sintomi della malattia. I farmaci principalmente utilizzati nel trattamento dei sintomi motori sono la levodopa, di solito in combinazione con un inibitore della dopa decarbossilasi e un inibitore delle COMT, gli inibitori MAO-B e gli agonisti della dopamina [11a].
20
trattamento del Parkinson. Inizialmente la levodopa si somministrava per infusione, poi per via orale senza l’inibizione degli enzimi di degradazione. Successivamente, la somministrazione orale della levodopa è stata associata con gli inibitori della dopa decarbossilasi (Figura 5). L’inibizione enzimatica del metabolismo della levodopa riduce la sua degradazione periferica a dopamina e aumenta l'emivita del composto, migliorando cosi la sua efficacia. Gli inibitori della dopa decarbossilasi, quali il benserazide e la carbidopa non attraversano la barriera ematoencefalica. La combinazione della levodopa con gli inibitori della dopa-decarbossilasi permette la riduzione della dose orale, limitando gli effetti secondari periferici correlati con la levodopa, come la nausea e il vomito [12].
L-DOPA Benserazide Carbidopa
Figura 5. Strutture chimiche della levodopa e degli inibitori della dopa-decarbossilasi [11b].
L'inibizione della degradazione della levodopa tramite la combinazione con un inibitore della dopa decarbossilasi sostiene il metabolismo della levodopa attraverso le COMT (catecolo-O-metiltrasferasi). Il blocco dell'attività delle COMT riduce ulteriormente la degradazione periferica della levodopa, prolunga la sua emivita ed aumenta la sua concentrazione a livello cerebrale. I risultati sperimentali e clinici hanno confermato l'efficacia di questo principio terapeutico con la doppia inibizione periferica di entrambi gli enzimi che metabolizzano la levodopa.
Gli inibitori delle MAO-B (monoamino ossidasi B) stabilizzano i livelli della dopamina nello spazio sinaptico (Figura 6). Le MAO-B catalizzano la deaminazione ossidativa delle ammine attive come la dopamina e quindi la loro inibizione causa un prolungamento dell'attività dopaminergica. La selegilina e la rasagilina, due composti del gruppo delle propargilamine, sono inibitori irreversibili e relativamente specifici delle MAO-B. Tuttavia la
21
selettività di questi composti si perde a dosi elevate perché possono inibire anche le MAO-A, enzimi che convertono altre ammine, come la noradrenalina. Di conseguenza, c'è un rischio, anche se basso, di ipertensione indotta da tiramina, chiamata “cheese effect”.
Selegilina Rasagilina
Figura 6. Inibitori MAO-B [11c].
Gli agonisti dopaminergici sono composti che agiscono direttamente sui recettori dopaminergici postsinaptici senza la necessità di conversione metabolica a dopamina (Figura 7 e 8). Essi si dividono in agonisti dopaminergici ergolinici (bromocriptina, pergolide, lisuride, cabergolina) e non ergolinici (pramipexolo, ropinirolo, apomorfina).
Bromocriptina Pergolide Lisuride Cabergolina
Figura 7. Agonisti dopaminergici ergolinici [11d].
Pramipexolo Ropinirolo Apomorfina
Figura 8. Agonisti dopaminergici non ergolinici [11d].
22
NMDA (recettore postsinaptico dell’acido glutammico), come l’amantadina (Figura 9). L’amantadina, un farmaco inizialmente sviluppato come antivirale, può migliorare i sintomi motori attraverso una stimolazione dopaminergica indiretta, aumentando la sintesi e la liberazione della dopamina.
Amantadina
Figura 9. La struttura chimica dell’amantadina [11d].
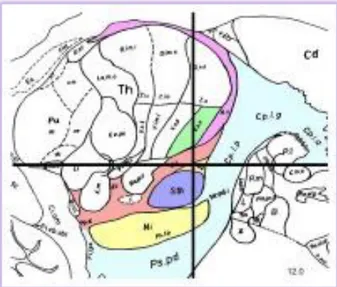
La stimolazione cerebrale profonda (DBS) dei nuclei subtalamici è un trattamento chirurgico che può portare alla riduzione del dosaggio dei farmaci dopaminergici e migliorare alcuni dei sintomi motori. (Figura 10). Questo metodo è raccomandato per i pazienti con Parkinson che soffrono di forte tremore che non viene adeguatamente controllato da farmaci o per coloro che sono intolleranti al trattamento farmacologico.
Figura 10. Sezione sagittale di 12 millimetri lateralmente dalla linea mediana, che dimostra il nucleo
subtalamico (STN) (colore lavanda). L’STN è uno degli obiettivi chirurgici preferiti della stimolazione cerebrale profonda (Deep Brain Stimulation) per il trattamento dei sintomi del morbo di Parkinson avanzato [2].
Gli approcci terapeutici dei sintomi non motori del Parkinson acquistano una maggiore importanza oltre al controllo di sintomi motori per migliorare la
23
qualità di vita nei pazienti del Parkinson. Il trattamento farmacologico a lungo termine dei sintomi non motori provoca l’insorgenza di vari effetti collaterali a breve e a lungo termine. Il corso della malattia, l'espressione dei sintomi motori e non motori, l'efficacia e la tollerabilità degli interventi terapeutici variano da paziente a paziente nella pratica clinica. Di conseguenza, diventa necessario seguire un regime terapeutico individualizzato con controllo ripetuto dal medico in stretta collaborazione con il paziente ed il suo badante.
Gli obiettivi primari delle terapie sopraccitate sono di alleviare i sintomi motori e non motori del Parkinson, piuttosto che curare o modificare la progressione della malattia. Pertanto, lo sviluppo di nuovi farmaci per invertire, impedire, o rallentare il progresso del Parkinson è uno degli scopi principali, se non dell'obiettivo primario, per la ricerca sul Parkinson [13].
La ricerca di terapie neuroprotettive o modificative per il Parkinson non ha dato risultati, in quanto non esistono terapie neuroprotettive o modificative dimostrate o approvate. Alcune motivazioni di questo insuccesso possono includere l'uso dei modelli di intossicazione acuta quale il 1-metil-4-fenil-1,2,3,6-tetraidropiridina (MPTP) e la 6-idrossidopamina (6-OHDA) per scoprire e verificare le nuove terapie neuroprotettive. Sebbene questi modelli suscitino la perdita selettiva dei neuroni dopaminergici, non possono riassumere esattamente il processo degenerativo del Parkinson. Altre motivazioni possono comprendere la limitata progettazione di trials clinici, l’inizio della terapia troppo tardi nel corso della malattia, la mancanza del target a livello cerebrale come pure la scelta dei target che possono non essere cruciali nella patogenesi del Parkinson.
Tutte queste problematiche rimangono delle barriere cruciali per lo sviluppo definitivo della terapia neuroprotettiva o modificativa per il Parkinson. Sebbene l'eziologia del Parkinson sporadico sia poco chiara, parecchi geni connessi con il Parkinson ereditario forniscono degli obiettivi che possono essere utilizzati per la scoperta di nuovi farmaci. L’identificazione dei difetti geneticispecifici nei casi di Parkinson familiare e
24
le vie di segnalazione governate da questi geni hanno fornito una grande comprensione nella patogenesi del Parkinson ed offrono delle possibilità nella progettazione di nuove strategie terapeutiche.
La proteina leucine rich-repeat kinase 2 (LRRK2) è un target importante del Parkinson per lo sviluppo di nuovi farmaci per la terapia del Parkinson. Sebbene le conoscenze attuali per quanto riguarda i meccanismi regolatori dell'attivazione della LRRK2 siano limitate, sta diventando sempre più evidente che l'attività chinasica dei mutanti patologici della LRRK2 è associata con la neurodegenerazione e pertanto l’attività chinasica di questa proteina è un potenziale obiettivo terapeutico. Gli inibitori della LRRK2 hanno potuto fornire degli strumenti importanti per capire i ruoli patofisiologici e fisiologici della LRRK2 come pure l'eziologia del Parkinson.
25
CAPITOLO 1
La LRRK2 e la sua inibizione come strategia
terapeutica
1.1 La chinasi LRRK2
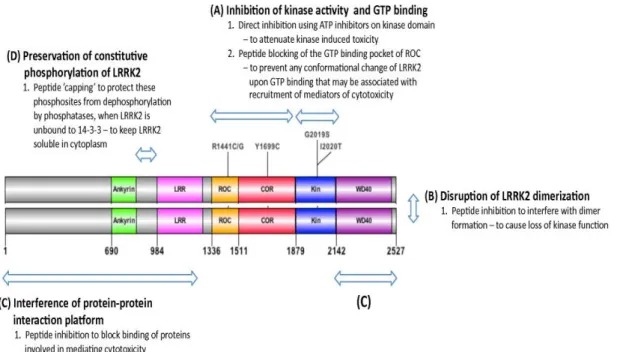
La LRRK2 è una grande proteina chinasi multidominio costituita da 2527 amminoacidi ed è un membro della famiglia delle proteine ROCO. Questa classe di proteine multidominio è caratterizzata dalla presenza di 200-250 aminoacidi del dominio Roc (per Ras of complex proteins), immediatamente seguito da un dominio di 300-400 aminoacidi definito COR. I dominii ROC e COR sono seguiti solitamente da un dominio chinasico e da vari motivi di interazione proteina-proteina che possono esistere all'interno di queste proteine. Ci sono almeno 40 membri nella superfamiglia ROCO e queste proteine sono state trovate in varie specie, compresi i mammiferi, i metazoi, i dictyostelium, le piante ed i procarioti [14].
Parecchi dominii indipendenti sono stati stabiliti per la proteina LRRK2 [15]: un dominio del tipo della anchirina (ANK), un dominio ricco di leucina (LRR), un dominio Ras (sistema renina-angiotensina) che appartiene alla famiglia di Ras GTPasi (Roc), seguito dalla regione C-terminale del dominio Roc (COR), un dominio chinasico (MAPKKK) e di un dominio WD40 nella regione C-terminale (Figura 1.1).
Le variazioni genetiche sono state trovate in tutti i dominii funzionali della LRRK2, indicando che questa proteina possa servire da integratore centrale delle vie multiple di segnalazione che sono cruciali per il perfetto funzionamento di un neurone. Inoltre, la presenza sia di dominii di interazione proteina-proteina (LRR e WD40) come pure di dominii enzimatici (Roc e MAPKKK) all'interno della LRRK2, suggerisce che questa proteina possa servire da impalcatura per l’assemblaggio di un complesso multiproteico di
26
Figura 1.1 Dominii e mutazioni della LRRK2 [16].
segnale.
1.1.1 Struttura e i dominii
Dominio arricchito in ripetizioni di leucina (LRR)
Questo motivo è presente in più di 60 proteine e la sua funzione primaria sembra essere il rifornimento di una struttura versatile per la formazione di interazioni proteina-proteina. Le proteine che contengono LRR partecipano a molti processi biologici importanti, quali le interazioni recettore-ormone, l’inibizione degli enzimi, l’adesione delle cellule e il traffico cellulare, lo sviluppo iniziale dei mammiferi, il neurosviluppo, la polarizzazione delle cellule, la regolazione genica, l’apoptosi e il regolamento della dinamica citoscheletrica. Sulla base di modelling per omologia, nel dominio di LRR molte sostituzioni amminoacidiche possono essere previste sulla superficie di questo motivo strutturale e, pertanto, possono intervenire alla formazione di interazioni proteina-proteina.
Dominii Roc e COR
Il dominio Roc dei membri della famiglia ROCO rimane distaccato come un gruppo monofiletico separato della superfamiglia di piccole GTPasi associate alla Ras. Le tre sostituzioni aminoacidiche patogene nel dominio Roc della LRRK2 si presentano alla posizione R1441 (R1441C, R1441G e
27
R1441H). Il dominio COR è comune a tutte le proteine ROCO ed è sempre come C-terminale del dominio Roc. Questo schema Roc-COR è conservato durante l’evoluzione, indicando un'interdipendenza funzionale di questi due motivi. Le determinazioni strutturali cristallografiche recenti di Roc e Roc-COR suggeriscono due meccanismi differenti di dimerizzazione all'interno di queste regioni di LRRK2. Alcuni ricercatori hanno riferito che il dominio Roc da solo esiste come dimero con scambio di dominii (domain swapping), mentre altri hanno identificato il dominio COR come il motivo della dimerizzazione. Due mutazioni associate al Parkinson, Y1699C e R1628P, sono situate nel dominio COR.
Dominio di MAPKKK
Sulla base di similarità di sequenza, il dominio MAPKKK di LRRK2 appartiene alla sottofamiglia tirosin-chinasi-like (TKL) delle protein-chinasi umane, i cui membri mostrano una similarità di sequenza sia con le chinasi serina/treonina che con le tirosin chinasi. Il dominio MAPKKK di LRRK2 assomiglia di più ai recettori che interagiscono con le protein-chinasi (RIPKs), che sono sensori cruciali dello stress cellulare e possono attivare le vie delle MAP chinasi (Mitogen activated protein kinase). Le mutazioni G2019S e I2020T collegate al Parkinson si presentano ai siti catalitici del dominio MAPKKK e possono alterare l'attività della chinasi.
Dominio WD40
I dominii WD40 probabilmente mediano le interazioni proteina-proteina. Ci sono parecchi esempi in cui i dominii WD40 sono fusi ai dominii con attività chinasica prevista, simili alla disposizione del dominio C-terminale della LRRK2. La mutazione di G2385R si trova all'interno del dominio WD40.
1.1.2 Attività chinasica
28
dominio MAPKKK della LRRK2 e l’effetto che le mutazioni associate al Parkinson potrebbero avere su questa proprietà enzimatica. La LRRK2 è una chinasi funzionale, capace di subire sia l’autofosforilazione che la fosforilazione del substrato generico della proteina basica della mielina (MBP). Uno studio, facendo uso di proteina ricombinante espressa nei batteri, ha dimostrato che l’autofosforilazione della LRRK2 è una reazione intermolecolare che mira a due residui all'interno del segmento di attivazione, il T2031 ed il S2032, con il residuo della serina della mutazione G2019S che genera un sito supplementare di autofosforilazione. Al contrario, un altro studio facendo uso di proteina espressa nei mammiferi, ha dimostrato che la LRRK2 dimerica subisce l’autofosforilazione intramolecolare e per questa attività è richiesto un C-terminale intatto, ma non l’N-terminale.
C'è un contrasto nella letteratura per quanto riguarda l'effetto che hanno le mutazioni associate al Parkinson sull’attività chinasica della LRRK2. Per esempio, parecchi studi hanno dimostrato che le mutazioni della LRRK2 collegate al Parkinson familiare (I1122V, R1441C, R1441G, R1514Q, Y1699C, G2019S, I2020T) aumentano l'attività chinasica come misurato dall’autofosforilazione, indicando un meccanismo di guadagno di funzione (gain-of-function) per la malattia associata alla LRRK2. Tuttavia, altri studi hanno trovato che soltanto la mutazione di G2019S ha aumentato l'attività chinasica della LRRK2, mentre tutte le altre mutazioni studiate non influenzanooppure inibiscono l'attività chinasica.
1.1.3 Attività GTPasica
La LRRK2 è una proteina GTP/GDP-binding, come misurato dal legame specifico al GTP-agarosio e al GTP radiomarcato. Sia il wild type (WT) che la forma mutata della LRRK2 collegata al Parkinson si legano al GTP ed al GDP. La LRRK2-K1347A, che porta una mutazione che altera la sede di legame al GTP, non si lega sensibilmente a GTP e riduce l'attività chinasica. Questo risultato è stato ulteriormente confermato dalla recente struttura
29
cristallina del dominio Roc della LRRK2 nel complesso con GDP-Mg(2+) a risoluzione 2.0A. Il sistema cristallino mostra un dimero del dominio Roc. Due residui patogeni associati al Parkinson, i R1441 e I1371, situati all'interfaccia dei due monomeri possono alterare la dimerizzazione del Roc e regolare l’attività GTPasica e/o l'attività chinasica della LRRK2. Uno studio recente ha suggerito che la dimerizzazione di LRRK2 sia associata con il legame alla membrana e l'attività incrementata della GTPasi. Le mutazioni in LRRK2 all'interno dei dominii Roc e COR, associate al Parkinson familiare (I1371V, R1441C, R1441G e Y1699C), sembrano aumentare il GTP-binding come misurato dal legame al GTP-agarosio, mentre le mutazioni al di fuori di questi dominii non influenzano il GTP-binding se confrontati con la WT-LRRK2. Tuttavia, altri studi hanno indicato che la mutazione R1441C non aumenta il GTP-binding [17].
Il dominio Roc della LRRK2 divide l'omologia di sequenza con tutte e cinque le sottofamiglie della superfamiglia Ras delle piccole GTPasi (Ras, Rho, Rab, Sar/Arf e Ran) e contiene dei motivi conservati per l’attività GTPasica. Tre gruppi indipendenti hanno dimostrato che la LRRK2 ha attività intrinseca di GTPasi e subisce l'idrolisi intrinseca di GTP. La LRRK2 purificata full-length ha soltanto deboli attività GTPasiche, suggerendo che se è attiva nella cellula può richiedere delle proteine accessorie. Considerevolmente, il dominio Roc della LRRK2 è sufficiente per la sua attività intrinseca di GTPasi. La LRRK2 lega ed idrolizza il GTP in modo simile alle altre piccole GTPasi relative alla Ras. Sulla base delle analisi in vitro, le mutazioni R1441C/G e Y1699C associate al Parkinson sembrano diminuire il tasso di idrolisi di GTP in confronto alla WT-LRRK2, indicando che questi mutanti passano più tempo nello stato attivo legati al GTP.
Esiste un collegamento fra l’attività intrinseca di GTPasi e l’attività chinasica della LRRK2. Parecchi studi hanno dimostrato che l'attività intatta della GTPasi è richiesta per l’attività chinasica della LRRK2, mentre l'attività di GTPasi funziona indipendentemente dall’attività chinasica. Inoltre, il
30
legame al GTP stimola l'attività della chinasi, dimostrando il regolamento intramolecolare fra il dominio Roc ed il dominio MAPKKK. Basandosi sulla struttura dimerica recentemente apprezzata della LRRK2, è stato previsto che i dominii dimerici del Roc o del Roc-COR fungano da interruttori binari che regolano l'attivazione della chinasi. È stato ipotizzato che, nella conformazione legata al GTP, la dimerizzazione del Roc o dei dominii Roc-COR induce ulteriormente l'autoassociazione dei dominii della chinasi, permettendo l’autofosforilazione e l'attivazione successiva dell’attività chinasica.
1.1.4 Ipotetici substrati
Due substrati eterologhi sono stati proposti per la LRRK2: la moesina e la 4E-BP [eIF4E (fattore di inizio eucariotico 4E)-proteina legante].
La moesina è una delle tre proteine ERM (Ezrina/Radixina/Moesina). Il ruolo principale delle proteine ERM è l’ancoraggio del citoscheletro alla membrana plasmatica e quindi il condizionamento dei processi cellulari relativi alla dinamica citoscheletrica alla superficie delle cellule, quale il mantenimento dei coni di crescita di un neurone. Il sito sulla moesina che è fosforilato dalla LRRK2 (Thr558, con un sito secondario a Thr526) si trova nel dominio C-terminale. Tuttavia, non è stato ancora dimostrato che la LRRK2 sia una chinasi autentica per la moesina in vivo [16].
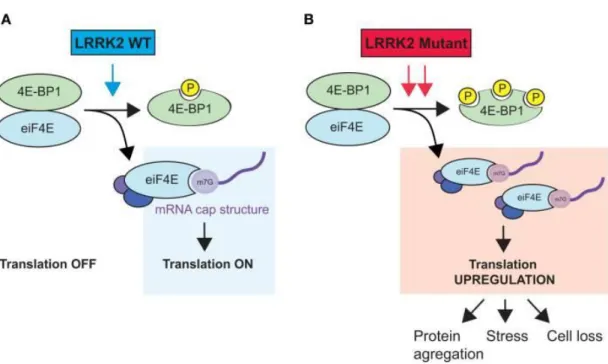
La proteina 4E-BP è un regolatore negativo coinvolto nella traduzione delle proteine tramite il suo legame con il fattore eIF4E (Figura 1.2). Lo stress ossidativo ed altri stimoli che hanno un impatto sulla traduzione delle proteine influenzano la fosforilazione della 4E-BP. Alcuni ricercatori hanno proposto che la LRRK2 modulasse questo sistema tramite la fosforilazione della 4E-BP. La fosforilazione della 4E-BP da parte della LRRK2 provoca il rilascio del fattore eIF4E dal suo legame con la 4E-BP. Nelle colture cellulari di mammiferi, la sovraespressione della LRRK2 aumenta la fosforilazione della 4E-BP ad una serie di siti; gli stessi ricercatori hanno proposto che la LRRK2
31
per prima cosa fosforila la 4E-BP su Thr37/Thr46, che funge da stimolo per un’ulteriore fosforilazione da parte di altre chinasi ai siti secondari compreso il sito Ser65/Thr70. Inoltre, è stato osservato che la sovraespressione di 4E-BP salvaguarda gli effetti dei mutanti di LRRK in vivo, usando dei modelli della Drosofila che mostrano una sensibilità aumentata allo stress ossidativo. Tuttavia, l’effetto della LRRK2 sulla 4E-BP deve ancora essere dimostrato su modelli di LRRK2 nei mammiferi [18].
Figura 1.2 Modello ipotetico del regolamento di LRRK2 nella traduzione delle proteine. (A)
WT-LRRK2 lega e fosforila la 4E-BP. Il eIF4E libero interagisce con altri fattori di inizio (eIF4A e eIF4G) e la traduzione abbia inizio. (B) Mutanti di LRRK2 con aumentata attività chinasica causano la iperfosforilazione di 4E-BP1, che stimola ulteriormente la sintesi delle proteine. La upregulation della traduzione proteica può condurre a varie conseguenze, quale l'aggregazione della proteina, lo stress delle cellule ed infine, la morte cellulare. Il legame di LRRK2 a 4E-BP1 non è descritto qui. [Da Véronique Dorval and Sébastien S. Hébert. (2012 Feb) LRRK2 in transcription and translation regulation: relevance for Parkinson’s disease. Frontiers in Neurology, Volume 3, Article 12, 1-6.]
32
1.2 Strategie terapeutiche per la malattia del Parkinson
associata alla LRRK2
Il fatto che la G2019S, la mutazione più frequente nel Parkinson associato a LRRK2, causi l'aumento dell’attività chinasica della LRRK2 e che tale attività chinasica porti alla citotossicità, suggerisce che l'inibizione dell’attività chinasica potrebbe essere utile dal punto di vista terapeutico. Tuttavia, data la complessità della molecola della LRRK2, è possibile che ci siano anche altri aspetti della funzione che potrebbero essere buoni target. Infatti sono state analizzare le varie possibilità degli agenti terapeutici verso la LRRK2 quali l’inibizione della chinasi, l’inibizione del GTP-binding ed altre alternative possibili per il trattamento del Parkinson associato alla LRRK2 (Figura 1.3) [18].
Figura 1.3 Rappresentazione schematica delle potenziali strategie terapeutiche per la malattia del
Parkinson associata alla LRRK2. I punti terapeutici possibili di intervento per LRRK2 sono: (A) l’inibizione dell’attività chinasica e il GTP-binding, (B) la rottura della dimerizzazione di LRRK2, (C) interferenza con l’ interazione proteina-proteina e (D) conservazione di fosforilazione costitutiva di LRRK2.
33
1.2.1 Attività chinasica e GTPasica come target
Un approccio per la manipolazione della LRRK2 è lo sviluppo di inibitori della chinasi; e questo viene solitamente effettuato preparando composti che competono per l’ATP, nella tasca di legame per l’ATP della LRRK2. La selettività della maggior parte degli inibitori della chinasi è pertanto incompleta, dato che la maggior parte inibisce anche altre chinasi in base alla similarità strutturale in questa regione. Come si poteva prevedere, parecchi inibitori noti delle chinasi inibiscono anche la LRRK2, ma tutti hanno alcuni effetti non specifici [19-21]. Poiché l'inibizione non specifica di altre chinasi può condurre a risultati ambigui, il composto ideale con cui esaminare la funzione e la disfunzione della LRRK2 dovrebbe essere altamente selettivo.
Recentemente sono stati descritti due composti, il LRRK2-IN1 e il CZC-25146, che presentano migliore selettività verso la LRRK2 rispetto agli altri inibitori chinasici [22,23]. Questi composti sono stati utilizzati in laboratorio per esplorare la funzione della LRRK2 perché possono penetrare nelle cellule, ma il loro studio probabilmente non procederà fino ai test clinici perché non si accumulano a livello cerebrale. Sperimentalmente, si può infondere gli inibitori attraverso i ventricoli cerebrali, ma questo non può essere utilizzato negli esseri umani. Di conseguenza, per provare se l'inibizione della chinasi potrebbe funzionare nei pazienti, sarebbe necessario sviluppare degli inibitori specifici e potenti, che preferibilmente possano essere somministrati oralmente.
L’applicazione degli inibitori della chinasi alle cellule che esprimono la LRRK2 causa la perdita di fosforilazione su Ser910 ed al Ser935, situati all'estremità amminica del dominio LRR, che viene ulteriormente associata con la perdita di legame con la proteina 14-3-3 ed alla riorganizzazione della LRRK2 in strutture filamentose. Al contrario, la kinase-dead LRRK2 conserva la fosforilazione ed il 14-3-3-binding e rimane citoplasmatica [24]. Pertanto, almeno in queste analisi cellulari, la kinase-dead non dà gli stessi risultati della chinasi inibita.
34
In alcuni mutanti patogeni della LRRK2 (R1441C/G, Y1699C e I2020T, ma non il G2019S) è abolito il legame con la 14-3-3 e non preservano la fosforilazione della LRRK2 costitutiva. Inoltre, questi mutanti hanno più inclusioni di LRRK2 nel citoplasma rispetto alla WT-LRRK2 legata alla 14-3-3. Di conseguenza ci si deve chiedere se gli inibitori della chinasi possano spingere LRRK2 verso piuttosto che lontano da una forma patogena. Se questo è vero, non si possono ancora prevedere gli effetti degli inibitori della chinasi né si può prevedere se questi effetti potrebbero essere gli stessi di quelli della
kinase-dead. Sebbene ci siano dei dati che sostengono l’effetto preventivo
degli inibitori della chinasi verso la tossicità neuronale, gli esperimenti eseguiti fin ad oggi sono stati effettuati con gli agenti meno selettivi, quindi i dati devono essere rivalutati utilizzando inibitori della chinasi più potenti e più selettivi.
Pertanto, sono stati valutati altri approcci per la LRRK2 usando degli inibitori competitivi dell’ATP. Le chinasi partecipano spesso a cascate di segnale ordinate e così gli effettori a monte e a valle della LRRK2 possono essere target validi per la terapia farmacologica di pazienti del Parkinson. La modulazione dell'attività delle proteine coinvolte in queste cascate potrebbe compensare il danno della funzione della LRRK2 causato dalle mutazioni. Per esempio, secondo le indicazioni nei modelli di Drosofila, la co-sovraespressione di 4E-BP nel mutante G2019S previene la neurotossicità [25]. Poiché la 4E-BP è anche implicata nel bersaglio della via della rapamicina nei mammiferi, dovrebbe essere possibile regolare gli effetti della 4E-BP indotti dalla LRRK2 usando la rapamicina. Un gruppo di ricercatori ha mostrato che può essere un approccio plausibile nei modelli di Drosofila, in cui hanno indicato che il trattamento con la rapamicina ha impedito la perdita di neuroni dopaminergici [26].
Un altro bersaglio possibile è la modulazione dell’attività GTPasica della LRRK. Il legame di GTP alla LRRK2 potrebbe cambiare la sua conformazione, e di volta in volta, mediare l'interazione con differenti
35
proteine. Le GTPasi passano ciclicamente dallo stato di legame al GTP a quello con il GDP con differenti affinità per effettori eterologhi. Per la LRRK2, i dati sui mutanti R1441C e Y1699C che hanno attività GTPasica più bassa suggeriscono che la forma legata al GTP sia associata alla malattia. Così un potenziale bersaglio terapeutico per il Parkinson associato alla LRRK2 potrebbe essere il blocco della tasca GTP-binding del ROC o dello stimolo dell’attività GTPasica per limitare un'interazione patogena [27]. La modulazione dell’attività GTPasica potrebbe essere particolarmente utile per i mutanti R1441- e Y1699- della LRRK2, ma per ora non ci sono strumenti disponibili per raggiungere tale scopo.
1.2.2 Strategie fuori dalle regioni enzimatiche
Poiché la LRRK2 ha molti dominii di interazione proteina-proteina senza attività catalitica, è possibile che la LRRK2 sia un'impalcatura che serve da piattaforma su cui le varie proteine si assemblano per eseguire una funzione specifica. Questi dominii possono svolgere un ruolo nella mediazione della citotossicità indotta dall’attività chinasica della LRRK2. È stato dimostrato che la delezione della porzione N-terminale di LRR o di WD40 di G2019S e/o di R1441C previene la tossicità indotta dalla LRRK2 in cellule neuronali [28,29]. Ciò può essere un punto di intervento terapeutico per mezzo di inibizione del peptide, bloccando il legame delle proteine coinvolte nella mediazione della citotossicità indotta da LRRK2 come è stato realizzato per altre proteine globulari che interagiscono [30].
Poiché l'attività chinasica di LRRK2 dipende dalla sua dimerizzazione, un modo per inibire selettivamente questa attività potrebbe essere la disgregazione del dimero LRRK2. Ciò può essere raggiunto usando peptidi inibitori per interferire con il ripiegamento proteico o la formazione dimerica. Questo approccio ha avuto successo negli studi dell’HIV in cui un peptide inibitorio è stato usato per inibire il ripiegamento della proteasi dell’HIV-1, un enzima importante nel ciclo vitale del virus, quindi inibire il ciclo vitale del
36
virus e controllare l'AIDS [31]. L’interferenza sulla dimerizzazione potrebbe essere utile come strategia terapeutica per il mutante Y1699C di LRRK2, che presenta una forza di interazione fra i dominii ROC e COR abnormalmente elevata.
Come discusso precedentemente, certe forme mutanti di LRRK2 perdono la capacità di legare le proteine 14-3-3. I meccanismi che sono alla base della perdita di fosforilazione costitutiva e del legame con le proteine 14-3-3 non sono conosciuti, ma è possibile che l'effetto funzionale di questi cambiamenti sia di rendere la LRRK2 meno solubile. Da questo punto di vista, un intervento possibile potrebbe essere la progettazione di un peptide che servisse da “cappuccio„ per la protezione di questi siti, quando non legati a 14-3-3, per mezzo della defosforilazione da parte delle fosfatasi. Ciò potrebbeimitare più esattamente quello che accade con la kinase-dead LRRK2, che sembra non essere tossica nei sistemi-modello.
1.3 Problematiche nello sviluppo di farmaci per la LRRK2
Dato che potenzialmente esistono vari modi per interferire con le disfunzioni di LRRK2, non è ancora possibile sviluppare farmaci per il trattamento del Parkinson associato alla LRRK2.
Durante lo sviluppo di un farmaco, il primo passo nella valutazione della sua efficacia è il test su un modello animale adatto. Esistono più di dieci modelli di topo che portano LRRK2-transgeni guidati da promotori differenti. Purtroppo, questi modelli non riassumono completamente tutte le caratteristiche cliniche osservate in un paziente con Parkinson. Sebbene una certa perdita di neuroni dopaminergici sia osservata in alcuni di questi modelli, la perdita non è progressiva ed è piuttosto limitata con leggeri deficit motori. Tuttavia, è stato riferito che l'espressione acuta di LRRK2 mutata, usando dei vettori virali, riproduce la perdita di cellule neuronali dopaminergiche incontrata nel Parkinson [32]. Questi modelli potrebbero essere utili in quanto forniscono un'opportunità di valutare l'applicazione acuta degli inibitori e
37
potrebbero essere il primo livello di agenti di test su animali transgenici usati per gli esperimenti a lungo termine, possibilmente usando dei fenotipi diversi dalla perdita neuronale dopaminergica, se si può dimostrare che sono affidabili.
Ci sono alcune limitazioni generali nello sviluppo dei farmaci per la LRRK2. In primo luogo, l'agente terapeutico potenziale dovrebbe essere in grado di attraversare la barriera ematoencefalica (BEE). Il candidato LRRK2-IN1 valutato biochimicamente e nei modelli in vitro, una volta provato in vivo, non colpisce la fosforilazione di Ser910 e di Ser935 della LRRK2 nel cervello di topo, ma abolisce la fosforilazione di Ser910/Ser935 nei reni, indicando che LRRK2-IN1 ha permeabilità limitata verso la BEE.
Una conseguenza della scarsa penetrazione degli agenti terapeutici LRRK2 attraverso la BEE sarebbe l’accumulo di questi nei tessuti periferici. Ciò potrebbe suscitare un problema di tossicità potenzialmente importante, come è stato recentemente dimostrato nei topi, nei quali l’abbattimento della LRRK2 ha causato il deterioramento renale [33,34]. Quindi sarebbe indispensabile controllare la funzione degli organi in cui la LRRK2 è altamente espressa, specialmente riguardo alla funzione renale e polmonare. La LRRK2 è altamente espressa nei linfociti B, quindi anche la funzione immunologica dovrebbe essere valutata.
Un altro problema nello sviluppo di farmaci per la LRRK2 è la loro selettività. Anche i farmaci selettivi verso la LRRK2 potrebbero avere degli effetti collaterali significativi se usati ad alte concentrazioni per un periodo abbastanza lungo. È infatti ben noto che non esiste nessun inibitore delle chinasi altamente selettivo [35]. Se la concentrazione dell'inibitore è abbastanza elevata, questo potrebbe inibire altre chinasi strutturalmente simili e causare effetti secondari multipli.
Presupponendo che i farmaci per la LRRK2 fossero a disposizione, un problema difficile da risolvere, sarebbe il periodo appropriato per iniziare il trattamento. Da un lato, sarebbe logico iniziare il trattamento profilattico in
38
portatori di mutazioni prima dello sviluppo dei sintomi classici della malattia di Parkinson. In questo caso, una terapia appropriata potrebbe potenzialmente impedire o ritardare la degenerazione neuronale. Dall’altra parte, le mutazioni nel gene LRRK2 hanno penetranza incompleta e variabile età di esordio. Di conseguenza, il trattamento profilattico comporterebbe un trattamento a lungo termine di individui che non necessariamente esprimeranno il fenotipo della malattia. Ciò potrebbe condurre all'idea alternativa che qualsiasi terapia dovrebbe limitarsi agli individui che hanno parkinsonismo. Inoltre alcuni aspetti motori del Parkinson sono trattabili, anche se imperfettamente, con levodopa e tramite gli approcci chirurgici quale la stimolazione cerebrale profonda. Quindi una strategia terapeutica basata sulla LRRK2 dovrebbe verificarsi vantaggiosa rispetto ai trattamenti attuali. L'efficacia, la valutazione della progressione dei sintomi del Parkinson e la sicurezza dei trattamenti per periodi più estesi dovranno essere considerati nello sviluppo dei regimi terapeutici LRRK2.
Un'altra domanda ragionevole è se la terapia basata sulla LRRK2 possa essere utile anche per il Parkinson sporadico. Questa domanda però non può avere risposta in assenza di un agente terapeutico. Tuttavia, con un composto appropriato a disposizione, sarebbe possibile verificare l'ipotesi secondo la quale il Parkinson associato alla LRRK2 ed il Parkinson sporadico sono collegati meccanisticamente.
A sostegno di questa ipotesi, e cioè che la LRRK2 può svolgere un ruolo nel Parkinson sporadico, c'è lo studio Genome-Wide Association GWAS secondo cui è presente un segnale intorno al locus LRRK2 nel Parkinson sporadico che non è rappresentato dalle mutazioni specifiche della LRRK2 [36]. Sebbene la base genetica di questa associazione sia poco chiara, un'interpretazione è che i cambiamenti nella regolazione della WT-LRRK2 rappresentino un rischio per il Parkinson nell’arco della vita. Per estrapolazione questo sosterrebbe l'idea che la LRRK2 sia collegata meccanisticamente alla malattia sporadica e che le terapie LRRK2 dovrebbero
39
essere provate nella malattia idiopatica.
Recentemente, la LRRK2 è stata associata con un rischio aumentato in alcuni cancri, in particolare il melanoma [37,38]. I pazienti con mutazione G2019S hanno un rischio triplo di sviluppare il melanoma prima dell'insorgenza del Parkinson. Sarebbe molto difficile da prevedere se un inibitore della chinasi LRRK2 potesse avere un impatto sul cancro, ma potrebbe essere considerato un obiettivo in più per lo sviluppo di un farmaco.
40
CAPITOLO 2
Inibitori della chinasi LRRK2 e loro
applicazione nel Parkinson
2.1 Inibitori della chinasi LRRK2 a piccola molecola
I primi inibitori LRRK2 derivati dallo screening di librerie di composti erano principalmente ATP-competitivi. Soltanto pochi dei composti sviluppati inibiscono specificamente la LRRK2; la maggior parte di essi inibisce più di una chinasi.
La staurosporina è uno degli inibitori della chinasi ampiamente utilizzati.Questo composto non selettivo inibisce in modo equipotente la WT-LRRK2 e la WT-LRRK2-G2019S (troncata e full-length) con un IC50 che varia da 0,2 a 40 nM (Figura 2.1). Il suo effetto inibitorio verso la LRRK2 è stato determinato con delle analisi in vitro differenti, per esempio, radioattività, TF-FRET e analisi AlphaScreen. Queste analisi hanno utilizzato substrati differenti quali i peptidi sintetici, come il LRRKtide e potenziali substrati fisiologici come la GST-Moesina. La staurosporina ha avuto un simile profilo inibitorio contro l’autofosforilazione di LRRK1/LRRK2 e la fosforilazione della MBP [15].
41
Anche i suoi derivati isoindolinonici K-252a/b e G hanno mostrato di inibire la WT-LRRK2 e la LRRK2-G2019S in concentrazione nanomolare, mentre gli analoghi della maleimide GF109203X e Ro31-8220 inibiscono la WT-LRRK2 e la LRRK2-G2019S soltanto in concentrazione micromolare (Figura 2.2). Ro31-8220 è un potente inibitore della fosforilazione di MBP con un IC50 di 50 nM nell'analisi di TF-FRET.
Figura 2.2 Derivati di maleimide come inibitori LRRK2 [15].
La potenza inibitoria della 5-Iodotubericidina e del Sorafenib (Figura 2.3) era quattro volte maggiore per la LRRK2-G2019S rispetto alla WT-LRRK2. Il Sorafenib è fino al 50% più selettivo per la WT-LRRK2 rispetto alla WT-LRRK1. Nelle colture primarie di neuroni corticali di ratto (analisi TUNEL) una concentrazione 5 μM di Sorafenib può completamente proteggere contro la tossicità indotta dalla LRRK2-G2019S e mostra un’azione protettiva contro la neurodegenerazione indotta dalla LRRK2-G2019S in C. elegans e nella Drosophila [20].
5-iodo-tubericidin Sorafenib
![Figura 2. Il Parkinson si sviluppa da interazioni complesse tra fattori ambientali e genetici e comprende delle vie molecolari multiple che interferiscono conducendo alla neurodegenerazione [4].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7647800.118952/10.893.176.788.126.738/parkinson-interazioni-complesse-ambientali-molecolari-interferiscono-conducendo-neurodegenerazione.webp)
![Figura 5. Strutture chimiche della levodopa e degli inibitori della dopa-decarbossilasi [11b]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7647800.118952/20.893.208.748.515.626/figura-strutture-chimiche-levodopa-inibitori-dopa-decarbossilasi-b.webp)
![Figura 7. Agonisti dopaminergici ergolinici [11d].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7647800.118952/21.893.227.782.619.1072/figura-agonisti-dopaminergici-ergolinici-d.webp)
![Figura 1.1 Dominii e mutazioni della LRRK2 [16].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7647800.118952/26.893.168.787.149.415/figura-dominii-e-mutazioni-della-lrrk.webp)