INDEX
...
Summary
5
...
Chapter 1 - Biology and Structure of RNA Viruses
7
...
1.1. The order Mononegavirales 7
...
1.1.1. Measles Virus biology 9
1.1.1.1. The N protein
1.1.1.2. NTAIL and Intrinsically Unstructured Proteins (IUPs) 1.1.1.3. The P protein and P-N complexes
1.1.1.4. Replicative cycle control of MeV
1.1.1.5. Nucleocapsid electron microscopy structures
.. 1.1.2. Known N Protein structures in the Mononegavirales order 17
1.1.2.1 The RSV crystal structure
...
1.1.3. Solution state NMR studies on MeV 19
1.1.3.1. Spectrum of free NTAIL
1.1.3.2. Spectrum of NTAIL in the capsid 1.1.3.3. Model of the NTAIL in the capsid
...
1.2. Leviviridae biology and structure 22
...
1.2.1. Acinetobacter phage 205 (AP205) 23
...
1.3. The Rice Yellow Mottle Virus 25
... 1.4. RNA-protein interactions in icosahedral viruses 26
Chapter 2 - Biophysical methods for structural
...
investigations
28
... 2.1. Solid state Nuclear Magnetic Resonance (ssNMR) 28
...
2.1.1. The NMR Hamiltonian 29
...
... 2.1.3. Cross Polarisation (CP) 33 ... 2.1.4. Heteronuclear decoupling 34 ... 2.1.5. Homonuclear recoupling 34 ... 2.1.6. Spin diffusion 35 ... 2.1.7. Structural characterisation by ssNMR 36 ... 2.1.8. Recent progress in ssNMR 38 ... 2.1.8. Structure determination by ssNMR 39 ... 2.2. Circular Dichroism (CD) 40 ...
2.2.1. DeVoe polarisability theory 42
...
2.2.2. CD and proteins 42
2.2.2.1. Protein CD spectra analysis
...
2.2.3. CD and nucleic acids 45
Chapter 3 - Solid state NMR studies of viral
...
nucleocapsids
47
... 3.1. Micro-crystalline viral particles: the case of AP205 47
... 3.2. Ultra-centrifuged viral particles: the case of MeV 49
. 3.2.1. 1H-15N INEPT: NMR of the flexible portion of the capsids 50
... 3.2.2. 1H-13C CP: NMR of the rigid portion of the capsids 51
...
3.2.3. Water accessibility 55
Chapter 4 - Homology model of cleaved and uncleaved MeV
...
capsids
58
... 4.1. Sequence alignment of MEV N protein and RSV 59
...
4.2. Homology model of the MeV N protein 60
...
4.3 Structural model of the nucleocapsids 63
...
...
5.1. Why a RNA 31P-1H correlation? 65
... 5.1.1. 31P-1H of ATP 67 ... 5.1.2. 31P-1H of a 14-mer hairpin 68 ... 5.1.3. 31P-1H of AP205 69
Chapter 6 - Circular dichroism studies on viral particles 71
... 6.1. CD of viral capsid protein: the case of MeV 71
...
6.1.1. Previous CD studies on MeV N protein 71
...
6.2. CD Spectra of viral RNA 76
...
6.3.1. DeVoe calculation of nucleic acid CD 77
...
Materials and Methods
80
...
I. Sample preparation 80
.... I.1. Cloning, Expression, and Purification of MeV nucleocapsids 80
... I.2. Trypsin treatment of recombinant measles virus N-RNA 81 I.3. Cloning, Expression, Purification and Crystallisation of AP205
...
capsids 81
.. I.4. Cloning, Expression, Purification of modified RYMV capsids 82
.. II. Protein Sequences and other physico-chemical parameters 83
...
II.2. MeV N protein Trypsin digested (392aa) 84
...
II.3. AP205 coat protein sequence (130aa) 84
... II.4. Modified RYMV coat protein sequence (190aa) 85
. III. RNA hairpin crystallisation and ATP for ssNMR analysis 85
...
IV. NMR spectroscopy 86
...
IV.1. Magic Angle setting 86
...
...
IV.4. Temperature calibration 87
...
IV.5. INEPT 87
... IV.6. 13C,13C correlations on MeV nucleocapsids 88
... IV.7. 15N,13C correlations on MeV nucleocapsids 89
...
IV.8. Water-edited CP pulse sequence 90
...
IV.9. 15N,13C correlations on AP205 capsids 90
...
IV.10. 1H,15N correlation on AP205 91
... IV.11. 1H,31P correlation on ATP and the RNA-hairpin 92
...
IV.12. 1H,31P correlation on AP205 capsid 93
...
V. Homology model 94
...
VI. Circular Dichroism Spectroscopy 94
...
VI.1. Protein CD 94
...
VI.2. Nucleic acids CD 94
... VI.3. Determination of protein and nucleic acid concentration 95
...
VII. DeVoe ECD calculation 97
... VII.1. Adenine 97 ... VII.2. Guanine 98 ... VII.3. Cytosine 98 ... VII.4. Thymine 99 ... VII.5. Uracyl 99
...
Conclusions
100
...
References
103
Summary
High-resolution solid-state Nuclear Magnetic Resonance (ssNMR) is rapidly emerging as a powerful structural tool in chemistry and biology and is applicable to a wide range of problems that cannot be addressed by solution state NMR or X-ray crystallography. Steady ongoing methodological developments combined with tremendous advances in probe and spectrometer hardware have led to a variety of strategies for resonance assignment, paving the way to the first 3D structure determinations of a wide range of samples at atomic resolution, ranging from inorganic frameworks and catalysts to membrane proteins and fibrils. Solid-state NMR does not suffer from molecular weight limitations (unlike its solution counterpart) and can be applied to non-crystalline samples. Therefore, this method is uniquely positioned to answer key questions about the chemistry and the structure of macromolecular assemblies which, because of their size and structural flexibility, are often difficult to characterize.
However several important problems remain to be solved before ssNMR is ready to cope with challenging solid biochemical assemblies, and many methodological developments are still expected in this fast evolving field.
The principal goal of this thesis has been to test a panel of experimental ssNMR methodologies for the structural investigation of large non-crystalline RNA-protein complexes such as determining the architecture and function of viral capsids.
In detail, we have focused on three highly relevant biological systems, the nucleocapsids of Measles virus (MeV), of Acinetobacter phage 205 (AP205), and
of Rice Yellow Mottle Virus (RYMV). These molecules constitute a very challenging target whose molecular weight is 10 to 100 times larger than any structural study previously performed on globular proteins by ssNMR.
In these viral particles, genomic RNA is protected by multiple copies of a coat protein (the so-called N-protein, of about 400 amino acids for MeV), which are organized into complex superstructures (helical for MeV, and icosahedral for AP205 and RYMV, respectively). These systems are too large for solution state
rays analysis. However, recombinant (13C,15N)-labelled nucleocapsids can be prepared, and low resolution structures obtained by electron microscopy are available.
In the present work, solid-state NMR was used to study the nucleoproteins in the capsid superstructure. The repetitive positioning of the coat proteins provide precisely the degree of order necessary for high resolution NMR spectra. We have optimized sample conditions such that the proteins can be studied in situ, and have recorded the first (13C,13C), (15N,13C) and (1H, 31P)-correlation spectra (notably, with the aid of high magnetic fields and ultra-fast magic angle sample spinning), which constitute the first steps towards resonance assignment and structure determination, including determination of the quaternary interaction between neighboring proteins on the RNA.
The analysis of the NMR data was enriched by a series of circular dichroism (CD) spectra, which probe the relative arrangements of aromatic bases in the RNA, and of peptide groups along the capsid protein backbone.
The work is highly interdisciplinary, going from research in virology and molecular biology to quantum chemistry, via structural biology and bioinformatics.
Chapter 1 - Biology and
Structure of RNA Viruses
1.1. The order Mononegavirales
Mononegavirales is an order of negative-strand non fragmented ssRNA viruses.
Their genomes are always encapsidated by a viral coded nucleoprotein (N protein) to form a ribonucleoprotein (RNP) complex. The RNP is formed concurrently with transcription and replication by the viral RNA-dependent RNA polymerase (RdRp). The RdRp complex is composed of negative sense RNA genome, N protein (nucleoprotein), P protein (phosphoprotein), and the L protein (large subunit polymerase). The nucleoprotein polymerizes to cover the entire length of the genome. RNA genome is always found associated with N protein as RNP particle. During transcription and replication a portion of the nucleoprotein temporarily dissociates from the RNA allowing the polymerase access to the genome.
The Mononegavirales order is composed of four viral families [1,2] (Fig. 1): •Bornaviridae contains the Borna Disease Virus (BDV) which affects the
nervous system in many animals, including cows and rats.
•Filoviridae has only two members, namely Ebolavirus and Marburgvirus that cause hemorrhagic fevers with mortality rates up to 90% in humans. The Centers for Disease Control and Prevention have included the Ebola and Marbourg viruses in the list of Biotterrorism Agents/Disease.
•Rhabdoviridae family contains Rabies Virus (RabV) and Vescicular Stomatitis Virus (VSV), which are both able to pass from their animal hosts to cause disease in humans. VSV is the model for Rhabdoviridae family.
•Paramyxoviridae is further on divided in two subfamily. Subfamily Paramyxovirinae includes Sendai Virus (SeV, genus Respirovirus) which typically affects rats and mice, and two viruses that cause childhood
epidemics, Measles Virus (MeV, genus Morbillivirus) and Mumps Virus (MuV, genus Rubulavirus). The genera Hanipavirus and Avulavirus infect bats and birds respectively. New Castle Disease Virus is a member of Avulavirus genera. Subfamily Pneumovirinae includes the Respiratory Syncytial Virus (RSV) that causes respiratory tract infection in human.
1.1.1. Measles Virus biology
Measles Virus (MeV) is a member of the Paramyxoviridae family of the Mononegavirales. With approximately 800,000 deaths worldwide, measles ranks
eighth as the cause of worldwide mortality and represents the main cause of childhood mortality in developing countries. Despite extensive vaccination campaigns, the disease has not been eradicated yet and to date, no effective antiviral treatment exists [3]. The genome of MeV is an RNA molecule of 15894 nucleotides in length, whose sequence is known. It contains contains six genes in tandem:
•P/V gene codes for a subunit of RNA dependent RNA polymerase (RdRp); it has chaperone role for Measles virus nucleocapsid protein.
•N gene codes for nucleocapsid protein (N protein).
•M codes for matrix protein, a hydrophobic protein on inner leaflet of membrane. Moreover it inhibits transcription process.
•L codes for large subunit of RNA dependent RNA polymerase (RdRp); it also catalyse RNA synthesis, capping, and polyadenylation.
•H codes for a type II transmembrane glycoprotein; it interacts directly with entry receptors.
•F codes for a type I transmembrane acylated glycoprotein; the fusion protein is synthesised as the precursor peptide F0 and subsequently cleaved to a
disulphide linked F1-F2 complex by a protease.
MeV is an enveloped virus. Its envelope is composed of a lipid bilayer, derived from the plasma membrane of the host cell, which contains the attachment (H) and fusion (F) proteins. Beneath the envelope, the viral matrix protein (M) associates with the cytoplasmic tails of the H and F proteins as well as the viral core particle or nucleocapsid. The viral genome is encapsidated by the nucleoprotein (N) to form a helical nucleocapsid that is the substrate for both transcription and replication. Each N subunit binds precisely six nucleotides [4]. The nucleocapsid has a left-handed helical structure which is approximately 1 Å long and has a diameter of 18 nm, with a central channel that is 4-5 nm in diameter. The nucleocapsid is rather flexible and can adopt different helical pitches resulting in conformations differing in their extent of compactness [5]. At the present moment we don’t have detailed information about the structure of the replicative complex. The lack of structural data stems from several facts:
analysis, the low abundance of L in virions and its very large size that renders its heterologous expression difficult and the structural flexibility of N and P [6].
1.1.1.1. The N protein
The primary function of the nucleoprotein is to encapsidate the viral genome. Within Mononegavirales, each N monomer interacts with a precise number of nucleotides. The number of nucleotides varies among Mononegavirales, being specific to each family: 6 nucleotides for Paramyxoviridae [4], 9 for
Rhabdoviridae [7] and 12-15 for Filoviridae [8]. Beyond its structural role, N
plays several functions. It protects RNA from nuclease degradation, it has a regulative role in the control of replicative cycle and it interact with many of host cellular partner.
In infected cells, two forms of N exist: a monomeric form (referred to as N°) and an assembled form (referred to as NNUC). N° is chaperoned by oligomeric P
protein and assembles on nascent genome/antigenome RNA. Like in VSV, RabV and SeV, MeV N° expressed by itself tends to aggregate nonspecifically often with cellular RNA [9,10,11].
From a functional point of view MeV nucleoprotein consists of two modules (Fig. 2): a N-terminal module (NCORE, aa 1-400) and a C-terminal module
(NTAIL, aa 401-525). NCOREcontains all the regions required for the self-assembly
and RNA binding, whereas NTAILis responsible for the interaction with P and
the polymerase (L-P) complex [6].
Fig. 2. Organisation of MeV N. The approximate locations of N-N, N-P and RNA-binding sites are
indicated [3].
1.1.1.2. N
TAILand Intrinsically Unstructured Proteins
(IUPs)
NTAIL has a hyper-variable sequence within Morbillivirus members. It has a
peculiar sequence composition. It is depleted in order promoting residues (W, C, F, Y, I, L) and enriched in disorder promoting residues (R, Q, S, E) (Fig.3) [12]. NTAIL is predicted to be a intrinsically disordered domain [2,6].
Intrinsically disordered or intrinsically unstructured proteins (IUPs) are functional proteins that fulfil essential biological functions while lacking any
constant secondary and tertiary structure under physiological conditions. The majority of IUPs undergo an unstructured-to-structured transition upon binding to their physiological partner, a process termed induced folding. The functional importance of disorder is inherent in their flexibility. In particular, an increased plasticity would[12]:
•allow protein interactions to occur with both high specificity and low affinity •enable binding of numerous structurally distinct targets
•provide the ability to overcome steric restrictions, enabling larger interaction surfaces.
Fig. 3. PONDR disorder prediction of N. Disorder prediction values are plotted against the residue
number [6].
Most proteins containing disordered regions are involved in signalling and regulation events that generally imply multiple partner interactions. IUPs possess peculiar sequence properties that allow them to be distinguished from globular proteins. In particular, they are generally enriched in amino acids preferred at the surface of globular proteins such as charged and polar residues (i.e., A, R, G, Q, S, P, E and K) and depleted in hydrophobic residues such as W, C, F, I, Y, V, L and N. They tend to have a low sequence complexity and a high sequence variability [13].
While conserving all these IUPs characteristic, NTAIL has some residual structure
that typifies the premolten globule proteins which are characterised by a conformational state intermediate between a random coil and a molten globule [14]. Using computational approaches, an α-helical Molecular Recognition
Element (MoRE, aa 488–499) has been identified within NTAIL. MoREs are regions within IUPs that have a certain propensity to bind to a partner and thereby to undergo induced folding [15]. In the case of MoRE, an α-helical
isolated NTAIL and in recombinant nucleocapsid, has been recently characterised
by solution state NMR [17]. NTAIL interacts with a lot host cellular proteins:
•Hsp72 which neutralise the contribution of C-terminus NTAIL to the formation
of stable P-NTAIL complex [18].
•IRF-3 (Interferon Regulator Factor 3) stimulating interferon production [19]. •An uncharacterised Nuclear Receptor; this interaction leads to the arrest of
cell cycle in G0/G1 phase. On the other hand NCORE-FγgRII interaction leads
to apoptosis. Both mechanism could contribute to immunosuppression that is hallmark of MeV infections [20].
1.1.1.3. The P protein and P-N complexes
The P protein is an essential subunit of the viral polymerase and plays a central role in transcription and replication. As the nucleoprotein, it provides several functions. It binds to the nucleocapsid through the formation of a P-NNUC
complex, thus tethering the polymerase (P-L complex) onto the nucleocapsid template. It also plays a chaperone role for the nucleoprotein in that it binds to N° thus preventing the illegitimate assembly of the latter in the absence of ongoing viral RNA synthesis. This soluble N°-P complex is used as a substrate for the encapsidation of nascent RNA [3,21]. In MeV, the P gene encodes multiple proteins, including P and V. The V protein is translated from a P messenger obtained upon co-transcriptional insertion of a G at position 753 of the P mRNA [21].
Fig. 4. General organisation of MeV P protein. Globular and disordered regions are represented by large
and narrows boxes respectively. The region overlapping the V ORF is represented by dotted lines [6].
P has a modular organisation consisting of six distinct regions (Fig.4) [6]:
•The N-terminal (PNT) domain, common to both P and V proteins, contains a hydrophobic region and a N°-binding region with α-helical potential.
•The C-terminal (PCT) domain, unique to the P protein, which contains a disordered region of greatly variable length, a central disordered region (overlapping the V ORF), a coiled-coil region, a disordered linker and the XD domain.
PNT domain, which is poorly conserved, provides several additional functions required for replication, whereas PCT domain is the most conserved in sequence and contains all regions required for virus transcription. Moreover through the coiled-coil region of PCT, P protein forms functional tetramers. CD, NMR and stokes radius studies and computational predictions, converge to show that PNT belongs to the class of IUPs [22,23].
Fig. 5. (A) Schematic representation of N°-P and NNUC-P complexes of MeV. Disordered regions are
represented by lines. The N°-P complex has been represented with a 1:4 stoichiometry (B) Model of the polymerase complex actively replicating genomic RNA. The P molecule delivering N° has been represented as distinct from that within the L-P complex. The encapsidation complex, N°-P, binds to the nucleocapsid template through three of its four PX arms. The extended conformation of NTAIL and PNT would allow the
The P protein interaction with N leads to the formation of the encapsidation complex (N°-P) in which P to N° binding is mediated by the dual PNT-NCORE
and PCT-NTAIL interaction. On the contrary, in the P-NNUC complex, binding is
mediated only by PCT-NTAIL interaction and PNT is free to interact with L. In
this way P acts as a bridge between the nucleocapsid template (NNUC) and the
polymerase (P-L) complex [3] (Fig. 5).
The P region responsible for the interaction with NTAIL and the induced folding
of this latter has been mapped to the C-terminal module (XD, aa 459–507) of P. Kingston and coworkers in 2004 solved the crystal structure of a chimeric form mimicking this complex XD-MoRE [24]. The structure of XD consists of a triple α-helical bundle. The surface of XD contains a large hydrophobic cleft that could accommodate the MoRE of NTAIL, promoting the induced folding of the
latter. The model showed that only 18 NTAIL residues (out of 125) participate in
the interaction with XD. Moreover in 2005 SAXS studies allowed to derive a low resolution model of the NTAIL-XD complex [25]. This model shows that most of
NTAIL (aa 401-488) remains disordered within the complex (Fig. 6).
Fig. 6. Low resolution structure of the NTAIL-XD complex derived by SAXS seen in two orientations
1.1.1.4. Replicative cycle control of MeV
The self-regulatory model for replication, which was proposed more than twenty years ago for all Paramyxoviridae, is still accepted in broad outline [26]. The genomic RNA functions as a template for mRNA synthesis and at the same time as a template for full-length antigenome synthesis. Transcription and replication activities are carried out by the same RNA-dependent RNA polymerase (RdRp) that is composed of the large (L) protein and of the phosphoprotein (P). Both types of RNA synthesis on the same N:RNA templates. During transcription RdRp responds to cis-acting signals to polyadenylate and terminate each mRNA. As a consequence, RdRp restarts at successive mRNA start sites to synthesise each mRNA in turn. During RNA replication, in contrast, RNA synthesis and assembly with N occur concomitantly, and vRdRp ignores all polyadenylation and termination signal to form a full-length antigenome nucleocapsid.
Fig. 7. Schematic representation of MeV RNA synthesis. The genome and antigenome nucleocapsids
(N:RNAs) are represented as long boxes with two widths; the central wider region indicates the entire coding region (from the start codon of N to the stop codon of L). Only the N/P junction is indicated, and the drawing is not to scale. The narrow regions at the ends represent the noncoding regions where the two bipartite replication promoters are found. The bent arrows show the sites and direction of RNA synthesis.
le, tr, and mRNA synthesis are independent of N° availability, whereas genome/antigenome synthesis and
assembly with N takes place concurrently [27].
viral genomes and antigenomes, respectively [26]. The first mRNA (that of N protein) starts around 50 nt from the genome 3’end, and the 50 nt upstream are called the leader region le. In a similar way the last mRNA (that of L protein) is followed by the trailer region tr at the 5’ end. The specific encapsidation signal is thought to lay within the leader and trailer extremities of the antigenome and genome RNA strands.
N plays a central role in the regulation of replicative cycle. Because each mature genome is associated with many N subunits, on-going protein synthesis is required for genome synthesis at any time of infection. Hence the component limiting for replication was assumed to be unassembled N protein (N°). The nucleocapsid assembly was proposed to initiate on nascent le and tr RNAs before RdRp had released these chains at or near the le/tr-gene junctions. If N° levels exceed a critical concentration antigenome RNA synthesis and nucleocapsid assembly become coupled, and the RdRp is a replicase that synthesises complementary N:RNAs. In the absence of sufficient levels of N° to permit the assembly on nascent le, RdRp would release the le chain near the N mRNA start site, RdRp becomes a ‘‘transcriptase’’ that responds to transcriptional signals (in particular to polyadenilation and termination signals) and it is free to initiate N mRNA synthesis (Fig. 7) [27]. In this way relative levels of transcription and replication are controlled by the availability of N°. At the present moment, because of the lack of structural data, we don’t know exactly how RdRp switches between its dual function as transcriptase and replicase remains unclear.
1.1.1.5. Nucleocapsid electron microscopy structures
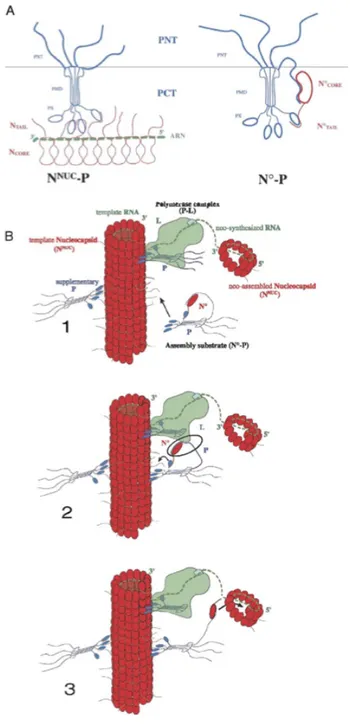
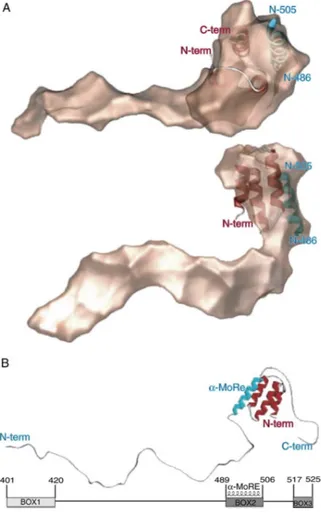
The helical parameters have been studied for the first time by negative stain and metal shadowing electron microscopy (EM). These first investigations showed that in the measles virus native nucleocapsid there are about 13 subunits of N per helical turn but the helical pitch seemed to be variable.The first cryelectron microscopy (cryo-EM) structure of a MeV nucleocapsid-like structure was published in 2004 by Schoehn and coworker [28]. Recombinant measles virus native nucleoprotein (N) were too flexible for high-resolution Fourier-Bessel image analysis or even by single-particle-based approaches. Removal of the C-terminal tail of N by trypsin treatment resulted in structures that were much more rigid and seemed more regular. The treatment with trypsin removes the first 13 amino acid residues and the last 120 amino acid residues, living a fragment (14-405) of 43.5kDa. The iterative helical real space reconstruction method allowed the identification of two left handed helical states for which a reconstruction could be calculated. The model with the highest resolution (11.8 Å) show that N monomers were connected to their neighbours
by two narrow connections, one close to the helical axis and another toward the middle of the monomers. There were no connections between N molecules in subsequent helical turns. The resulting model showed a helix pitch of 50.8 Å with a 12.35 subunits per helical turn (Fig 8a) [28].
Fig. 8. a) cryo-EM reconstruction of trypsin-digested N-RNA from [28]. The black broken line shows the
orientation of the line that connects subunits in subsequent turns. b) and c) uncleaved or digested nucleocapsids with NanoW in a double carbon layer [29].
More recently Recombinant measles virus nucleoprotein-RNA (N-RNA) helices were analysed by negative-stain electron microscopy. Three-dimensional reconstructions of both trypsin-digested and intact nucleocapsids were done by negative-stain electron microscopy by trapping the sample between two layers of carbon film and by using NanoW stain instead of the more traditional uranyl acetate. This preparation technique enabled to image intact measles virus nucleocapsids had the advantage of maintaining the helix in a more rigid state. Final three-dimensional reconstructions of trypsin-digested and intact measles nucleocapsids at a resolution of 25 Å were obtained [29]. Removal of the NTAIL
leads to a compaction of the helix, with the pitch shortening from 57.2 Å to 48.7 Å and a diameter constriction from 200 Å to 190 Å, in line with the previous cryo-negative-stain EM work. Even with this new approach the number of subunits per turn in the digested nucleocapsid was found to be 12.33. On the other hand the intact nucleocapsid helix accommodates a nearly integer number of 12.92 subunits per turn, which agrees with the 5% increase in diameter. NTAIL
is not visible in the reconstruction (Fig 8b,c and Fig Xa,b).
1.1.2. Known N Protein structures in the
Mononegavirales order
Virus (RabV) and last Respiratory Sincitial Virus (RSV) have been reported
[30,31,32,33]. Only VSV, RabV and RSV N protein structures were resolved in presence of a random RNA molecule. All these proteins are organised in a core region containing two domains, namely N- and C-terminal (NTD and CTD), that are connected through a hinge region. RNA is accommodated in a cavity between the two domains. The core region has N- and C-terminal extensions, termed N- and C-arm, that fold only in the context of quaternary interactions in the nucleocapsid particle. A similar organisation is also found in the Flu A
Virus N protein (Family Orthomyxoviridae) [34].
It appears that even the general organisation of the RNA binding motif is conserved in all Mononegavirales known N proteins. The N/RNA contacts in the groove are not base-specific and all Mononegavirales N proteins have a cavity that is tailored to bind three stacked bases. Anyway the number of nucleotide per monomer is specific for each family or subfamily in Order and this is due to the fact that NTD and CTD may change their relative orientations in the RNA binding region, resulting in different lateral N contacts and hence in different RNA binding mode [34].
1.1.2.1 The RSV crystal structure
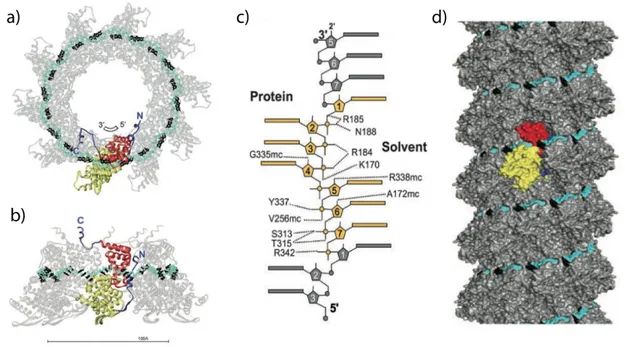
RSV is classified into the Pneumovirus genus with the Pneumovirinae Subfamily of the Paramyxoviridae. In 2009 Tawar and coworker obtained crystals of recombinant decameric RSV N-RNA rings diffracting to 3.3 Å resolution [33]. This complex mimics one turn of the helical nucleocapsid. Both core domains are α-helical bundles, with 10 helices in the NTD and 4 in the CTD. The RNA chain run within a surface groove surrounding the N-protein ring. Each N subunit interacts with seven nucleotides, resulting in a RNA chain of 70 nucleotides. Because the RNA has a random sequence, the bases were all modelled as cytosines. The ring is stabilised by the insertion of the N arm of one subunit into the compact fold of the adjacent subunit (Fig 9a,b,c).
A cryo-EM electron density of the nucleocapsid-like particles was obtained with a resolution of 26 Å (Fig 9d). The atomic resolution ring structure was fitted inside the EM map using the least possible distortion of the ring contact. This resulted in detailed model of helical RSV nucleocapsid. Notably the region between helical turns is occupied by the mobile C-arm, which may play a functional role by providing flexibility to the nucleocapsid [33]. Like NTAIL, the
Fig. 9. Crystal structure of RSV the decameric ribonucleoprotein ring complex. Yellow and red indicate
NTD and CTD, respectively, and the N and C arms are blue. The RNA is displayed with the backbone in cyan and the bases in black. a) View down the ring axis. b) Side view. The bar indicates 100 Å. c) Details of the interactions with RNA. d)The helical nucleocapsid modelled from the contacts in the ring, using the cryogenic negative-stain electron micrograph of recombinant RSV nucleocapsid-like helices. One subunit is highlighted [33].
1.1.3. Solution state NMR studies on MeV
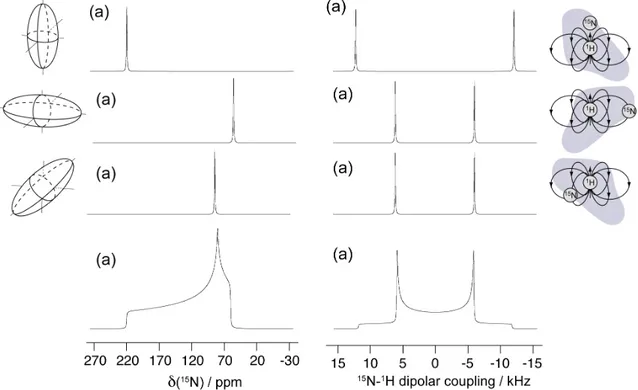
NMR of proteins in solution is a powerful structural method that, however, becomes increasingly difficult with increasing molecular weight because of the slow molecular reorientation dynamics [36]. The slow tumbling associated to the large mass of MeV nucleocapsid particles precludes detection of the signals of folded globular domains of the N protein by solution-state NMR, and only the most dynamic part of the capsid protein, the NTAIL, appear in solution studies.
1.1.3.1. Spectrum of free N
TAILChemical shifts and Residual Dipolar Coupling (RDC) were used in combination to probe the structure on isolated NTAIL [17]. The investigation reveals that the
majority NTAIL behaves like an intrinsically disordered polypeptide but for a
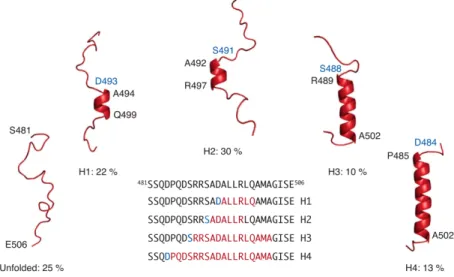
small region, the MoRE which exists in a rapidly interconverting conformational equilibrium between unfolded form and conformers containing one of four discrete α-helical elements. All of these forms are stabilised by N-capping by aspartic acids (Asp493, Asp 484) or serines (Ser 491, Ser 488) residues (Fig. 10). Previously a crystal structure of the chimeric complex between a short construct of NTAIL and the XD domain of P protein show that NTAIL assumes in the MoRE
region a helical structure that is similar to the longest of the four helical elements present in isolated NTAIL [24].
Fig. 10. NTAIL preferentially adopts a dynamic equilibrium between a completely unfolded state and
different partially helical conformations each represented by a single cartoon structure for clarity. All helices are stabilized by N-capping interactions through aspartic acids or serines (blue residues). The location of the helices within the MoRE is shown in the primary sequence [17].
1.1.3.2. Spectrum of N
TAILin the capsid
The authors have therefore used solution state NMR to characterise the conformational behaviour of NTAIL in 15N,13C labelled nucleocapsids. From EM it
is possible to estimate the molecular mass distribution of the objects in the sample to fall in a range between 2 to 50 MDa that would normally preclude detection of solution state NMR signal of a folded globular protein [17].
The HSQC (Heteronuclear Single Quantum Correlation) spectrum of the intact nucleocapsid shows signals deriving only from the NTAIL, indicating that it
remains flexible and dynamic even if attached to the nucleocapsid particle. In such spectrum signals for the first 50 amino acids (residue 401-405) are absent, indicating that the first stretch of 50 amino acids on N-tail is conformationally restricted. However the spectrum is superimposable to that of isolated NTAIL,
demonstrating that the local conformation behaviour of residues 450-525 of NTAIL is conserved in the viral particle. The large variation of the peak
intensities indicate differential flexibility along the chain, with MoRE having particularly low intensities. These results suggested that the MoRE of NTAIL
slowly exchanges on and off the surface of the nucleocapsid. Analysis of the intensity of the peaks showed that more than 95% of the MoRE population is bound. The R2 values increase around residue 460, which, combined with the absence of signals of the first 50 residues of NTAIL, indicates that the first stretch
1.1.3.3. Model of the N
TAILin the capsid
In 2010 Desfosses and coworkers dock into the EM density map of both cleaved and uncleaved MeV nucleocapsid particles the RSV protomer with 6 RNA nucleotide bound, on the basis of secondary structure homology [29]. RSV N-RNA fit nicely into the electron density map and even the distal β-hairpin of the NTD. The helical model would place NTAIL toward the helix interior, which
would have consequences for contacts between subsequent helical turns. Moreover the docking of the atomic structure of RSV N-RNA subunit into the electron microscopy density map supported a model that places the RNA at the exterior of the helix. To obtain a continuous RNA molecule in the built viral particle, an energy optimisation on RNA was performed. In particular the three solvent facing nucleotides (1,5,6) were optimised.
Fig. 11. Three-dimensional reconstructions of MeV nucleocapsids of digested nucleocapsid a) and
uncleaved b) nucleocapsid. The fit of the RSV N-RNA atomic structure is shown as an overlay[29]. c) and d) Proposed model of the location of NTAIL in intact nucleocapsids. The three-dimensional coordinates of
the RSV N-RNA subunit docked into the EM density map of MeV N-RNA were used. The conformational sampling algorithm flexible-meccano was used to build chains from the C terminus of the folded domain of NCORE (successive NCORE monomers are coloured green and yellow) [17].
In 2011 the behaviour of NTAIL was further on investigated by building explicitly
the NTAIL on Desfosses model of 2010. In such model NTAIL obey random coil
statistic and can indeed exfiltrate from the interior of the capsid via the interstitial space between two adjacent helix turns, providing an explanation for the decrease in pitch between intact and cleaved capsids. The maximal angular freedom is only achieved after approximately 50 amino acids, providing an explanation for the lack of the signals of the first 50 residues of NTAIL. The
surface. Because the RNA is sequestered on the outer surface of the capsid, this would place the RNA in the vicinity of NTAIL [17].
1.2. Leviviridae biology and structure
Leviviridae is a family of ssRNA bacteriophages that infect a wide variety ofGram-negative bacteria causing lytic infection. Based on different physical and serological properties and on their genetic map, ssRNA coliphages have been divided in two genera: Levivirus (examples MS2 and GA phages) and
Allolevivirus (examples Qβ and SP phages) [37]. Positive-sense genome of the
leviviruses encode for four proteins: •MP (maturation protein)
•CP (coat protein)
•R (replicase). It is a RNA-dependent RNA polymerase. •L (lysis protein)
Even the genome of alloleviviruses codes for four proteins: •MP (maturation protein)
•CP (coat protein)
•R (replicase). It is a RNA-dependent RNA polymerase.
•RT (read through protein)which is a C-terminal extension of CP.
Hence the major difference between the two genera is the presence of the read-through protein, a minor capsid constituent, that is absent in members of the genus Levivirus. On the other hand, the lysis gene is absent from Allolevivirus members. In such genus the maturation protein has also a role in host lysis [38]. Phage RNA contains not only genetic information. In fact it assumes a specific higher order structure that is responsible for regulation of replication and translation and many other function. The most important of this secondary RNA structure is the operator hairpin [39].
All ssRNA Leviviridae share the same special way to control translation of their replicase gene [40]. The start region of this gene can fold into a phage-specific hairpin structure that has affinity for its own coat protein (Fig. 12). When the concentration of the coat protein during infection becomes high enough, a dimer bind the stem-loop structure of the R operon and precludes further ribosome binding to RNA molecule. This complex is also consider to be the nucleation point for capsid assembly.
Fig. 12. Replicase-operator hairpins in several RNA phages. Bases shown to have sequence-specific interaction with the coat protein are encircled. Replicase start codon are boxed and nucleotides that interact in a base-specific way with the coat protein are encircled [39].
Interestingly it has been shown that non-specific RNA sequences are able to interact with CP dimers and contribute substantially to the assembly pathway [40]. The secondary binding sites probably consist of hairpins resembling the operator, and they are likely to bind CP dimers in a similar manner.
1.2.1. Acinetobacter phage 205 (AP205)
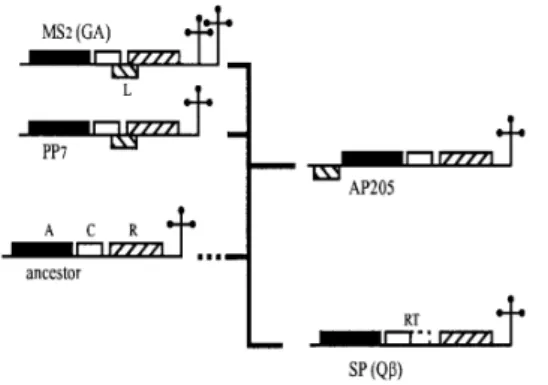
AP205 is a member of the genus Levivirus with a particularly long genome (4268 nt). Its genome is only 9 nucleotides shorter than that of SP phage which has the longest ssRNA phage known today [39]. AP205 is the first example of a ssRNA phage with a different genetic map. Although the three major genes occupy positions identical those of MS2 phage, the lysis gene occupies an unusual position (ORF1). It is likely that the lysis gene has arisen more than once in the ssRNA phages and that L protein in MS2 and AP205 are functional analogues rather than homologues. The operator of AP205 has features from both leviviruses and allolevivirus [39]. Based on these findings and on other structural and genetic analysis (for example on 5’ terminal hairpin and 3’ UTR structure) a tentative of phylogenetic tree of principal ssRNA bacteriophages was made [39]. This three put AP205 in an intermediate position between
Levivirus (MS2 and PP7) and Allolevivirus (Qβ and SP phages) (Fig. 13).
The coat protein of AP205 is 130 amino acids long. The length of coat protein is remarkably constant in all RNA phages. 180 monomers compose the AP205 viral particle. At the present moment high resolution crystal structures are reported for phage MS2, fr, Qβ, GA and PP7 [41].
MS2, Qβ, PP7 and AP205 nucleocapsid structures were determined by cryo-EM with a resolution estimated to be between 17Å and 24Å [42]. The general RNA organisation of the RNA inside the capsids is similar in all four phages. However the details of RNA densities inside capsids of PP7 and AP205 differ significantly from the other two phages studied.
Fig. 14 a)-d) Exterior of the virions and e)-h) interior of a hemisphere of the cryo-EM reconstructions of
bacteriophages a) MS2, b) Qβ, c) PP7 and d) AP205 viewed along the 5-fold axis (red, RNA) [42].
AP205 has a diameter of about 29 nm. Holes were found in the capsid at the 3-fold and 5-3-fold axes except for AP205, in which the holes at the 5-3-fold axes are not visible, probably due to the different packing of the RNA inside the capsid (Fig. 14). AP205 showed three spheres of RNA density below the capsid shell at about 30Å, 70Å and 100Å. The observed dispersed RNA density is in concordance with RNA binding that would occur mainly through non-specific interactions as well as specific operator-CP interactions[42].
Differently from MS2 and Qβ phages in which RNA density appear like a series of disconnected bulges embedded partly in the capsid density, in AP205 the density distribution is dispersed and form continuous networks (Fig. 15). Apart from a few positions, such RNA network seems to be not in contact with the capsid. This implies that the genome interact only loosely with the capsid, probably only through non-specific RNA backbone interactions.
Fig. 15 The RNA density (red) is spheroidal and discontinuous in a) MS2 and b) Qβ. The reconstructions of c) PP7
and d) AP205 show continuous networks of RNA. e)-h) At lower σ values, the RNA network of PP7 is broken and resembles the MS2 and Qβ structures, whereas in the AP205 reconstruction only a ring around the 5-fold axis is left. [42].
1.3. The Rice Yel low Mottle Virus
The species Rice Yellow Mottle Virus (RYMV) belongs to the genus
Sobemovirus which is not assigned to any family. The genus contains 13
definitive species including Southern Bean Mosaic Virus (SBMV), the type species [43]. RYMV causes an emergent disease that was first observed in 1966 in Kenya. Since then, it has been reported in nearly all rice-growing countries of sub-Saharan Africa. RYMV is transmitted by coleopterous insects and is also disseminated abiotically. It has a narrow host range limited to wild and cultivated rices and a few related grasses
RYMV has icosahedral particles of 25 nm in diameter. The virions contain a single coat protein (CP) of 29kDa, a genomic RNA (gRNA) and a subgenomic RNA (sgRNA) [44].
The genomic RNA is one single-strended positive sense molecule of 4450 nucleotides in size. The 5’ terminus of the RNAs has a genome-linked protein (VPg). RYMV often encapsidates, in addition to its genomic RNA, a viroid-like satellite RNA (satRNA). satRNA has 220 nt and is the smallest naturally occurring viroid-like RNA known today [43].
RYMV genome is organised in four overlapping open reading frames (ORF). •ORF1 code for P1 protein that is required for infectivity in plants and in
•ORF2b is translated as a polyprotein fused with ORF2a after a programmed ribosomal frameshifting mechanism. It contains RNA-dependent RNA polymerase
•ORF4 is traslated from the subgenomic RNA and encodes the coat protein. The capsid contain 180 copies of the CP subunit arranged with a T=3 symmetry. The monomers exist in 3 different conformations. The structure of RYMV was determined by X-ray crystallography at 2.8Å resolution [44]. Each subunit adopts a eight stranded jelly-roll β-sandwich fold. The N-terminal 49 residues are disordered in the crystal. Moreover the N-terminal sequence of the coat protein contains a putative bipartite nuclear localisation signal. This highly basic region is thought to interact with viral RNA and to stabilise the virion. Like other Sobemovirus particles RYMV capsid is stabilised by divalent cations (Ca2+ and Mg2+), pH-dependent protein-protein interactions, and salt bridges
between protein and RNA. Interestingly the viral particles swell in vitro if divalent cations are removed or if the pH is shifted from slightly acidic to basic and the swelling process is reversible [45].
There are not information about RNA conformation in RYMV capsids, whereas in SBMV crystal structure the RNA was found to be in a rigid, compact form [46]. The localisation of a considerable amount of RNA was possible. Diffraction datas did not yield information on the RNA conformation although a dsRNA fragment, comprising 9 base pairs, could be successfully modelled, involving interactions between several arginines and lysines and RNA phosphate groups [47].
Our RYMV sample contains non native viral particles composed by 60 copies of CP instead of 180. This is achieved by truncating its N-terminal part. All 60 subunits are supposed to be identical, hence NMR peak assignment might be much easier than in case of the native capsids, where monomers exist in three different conformations.
1.4. RNA-protein interactions in
icosahedral viruses
Non-enveloped icosahedral RNA viruses are excellent model for studying protein-protein and RNA-protein interactions. Of particular interest is the study of the organisation of the genetic material inside the virion. Information about the positioning of the viral RNA within the virion is steadily increasing. In general there is a little understanding of how the genomic RNA as a whole
interacts with the virus capsid. It is likely that the folding of the RNA during encapsidation is guided by coat protein, which can cause a different folding of the RNA molecule. However the identification of a protein binding motif in the genomic RNA seems to have a central role.
Crystallography and electron-microscopy studies showed that a major feature seems to be the tight association of a significant part of the viral genomic RNA with the capsid, often manifesting itself as a density corresponding to A-type RNA double helical segments [41]. To a certain extent, these structures elements follow the symmetrical order of the capsid protein shell, while the remainder of the RNA is then tucked away in the interior of the virion [42]. Detailed information on the specific interaction between these RNA helices and the capsid, however, is lacking. The most characterised system is the operator-CP interaction in MS2 phage, but nothing is known about the structural organisation of the rest of the MS2 phage genome [48]. A possibility is that the additional interactions between the entire genome and the capsid are not identical to that of the operator hairpin, but they are characterised by more aspecific interactions.
It is clear that for a detailed description of the RNA-protein interactions in icosahedral viruses crystallography needs to be complemented by other biochemical and biophysical techniques. In fact the process of crystal formation of virus material depends on the capsid structure only. Although, in the crystal the capsids are aligned, the RNA may not and therefore appears as disordered. Through the procedure of signal accumulation and averaging only general features such as repetitive structures can be visualised, whereas all unique features are lost [41].
We propose Circular Dichroism and solid state NMR as complementary technics for the study of RNA-protein interaction in nucleocapsids.
Chapter 2 - Biophysical
methods for structural
investigations
2.1. Solid state Nuclear Magnetic
Resonance (ssNMR)
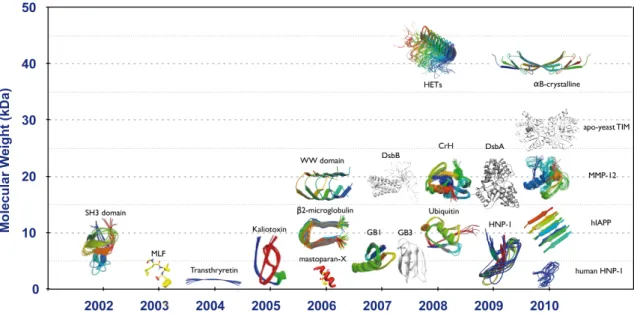
Since the first structure determination of a microcrystalline model protein domain in 2002 [49], solid-state NMR (ssNMR) spectroscopy has significantly grown thanks to combined progress in sample preparation, probe and magnet technology and radio-frequencies irradiation schemes. In last few years an increasing number of structures of micro-crystalline globular proteins or protein domains has been resolved by ssNMR (Fig. 16). ssNMR is now open to structural characterisation of complex biological systems such as membrane proteins, big protein-protein complexes and protein fibrils [50].
Fig. 16. Summary of the structures of proteins, protein domains and fibres determined to date by
In this section, I will expose first the basics of magic-angle spinning (MAS) solid-state NMR experiments available today, namely cross-polarisation (CP), heteronuclear decoupling and homonuclear recoupling. Then, I will summarize the recent progress in the field of protein structure investigation by ssNMR.
2.1.1. The NMR Hamiltonian
The physical interaction of nuclei with non-zero magnetic moments are quantum-mechanically described by the Hamiltonian operator, Ĥ. Each term of the operator describes a possible interaction involving the nuclear spins, namely:
HZ describes the Zeeman interaction with the external magnetic field B0:
where γI is the gyromagnetic ration of spin I, is Plank’s constant divided by
2π, and Îz is z component of the nuclear spin operator. The product -γB0 is
called the Larmor frequency of spin I.
HCS describes the chemical shift Hamiltonian. The electron distribution
surrounding each nucleus induces small local fields that can interfere with the external magnetic field B0, shielding or un-shielding its effects:
The term σzz is the component along the magnetic field of the chemical shift
anisotropy tensor. Due to the fact that the electron distribution is not spherically symmetric, σzz depends on the molecular orientation Ω of the
molecule containing the spin I with respect to B0. In an isotropic liquid, the
molecules tumble rapidly through every conceivable molecular orientation, with equal probability for each orientation. The observed chemical shift, is therefore the isotropic average of the product
In powdered solids, all possible orientations are present, and the obtained spectrum is a sum of the contribution derived from each specific molecular orientation with respect to B0.
ĤJ is the Hamiltonian term which describes the J-couplings, the indirect magnetic interactions among nuclear spins, through the involvement of the electrons. This term has the form:
where Îj and Îk are the spin operator of the two nuclei involved in the
interaction and Jjk represents the isotropic coupling constant.
ĤQ is the quadrupolar interaction Hamiltonian, and it is non-zero only for nuclei
with spins greater than 1/2.
ĤD describes the dipolar interactions. These represent the direct, through space
magnetic interactions among nuclear spins. They can be intramolecular or intermolecular. The general form of ĤD between two spins j and k is:
where ejk is a unit vector parallel to the line joining the centers of the two
nuclei, μ0 is the value of magnetic permeability in a classical vacuum and rjk is
the distance between the two nuclei.
If the spins are of the same isotopic species (homonuclear case), the Hamiltonian in polar coordinates has the form:
where θjk is the angle between the vector joining the spins and the external
magnetic field. Whereas for the heteronuclear dipolar interaction it is:
In an isotropic liquid, the term (3cos2θ
jk -1) is averaged to zero due to the
tumbling and hence ĤD in a liquid is 0. In a solid, the dipole-dipole part of the
Halmiltonian is given by the sum of ĤDjk terms for every spin pair in the sample.
The observation of anisotropic interactions is the major difference between solution and solid-state NMR. In the case of a solid sample (such as a powder), all possible orientations are present simultaneously, and the NMR spectrum is the sum of the contributions from the uniform distribution of all these molecular
orientation. The typical narrow isotropic resonances known from liquid-state NMR are turned into the so-called “powder patterns” in the solid phase (Fig. 17). The major task in solid-state NMR is to manipulate the Hamiltonian with the aim to recover the typical narrow isotropic resonances of liquid-state NMR and to maintain (or reintroduce) sufficient spectral information for structure determination.
Fig. 17. Numerical simulation of the NMR spectrum for three particular sample orientations (a-c) and
for a static powder patterns (d) of a 13C carbonyl chemical shielding tensor (left) and of a amide 1H-15N
dipolar coupling tensor (right).
The observation of anisotropic interactions is the major difference between solution and solid-state NMR. In solution CS anisotropy and dipolar coupling are averaged to zero due to molecular tumbling, and hence only the isotropic parts in the Hamiltonian remain.
I the case of a solid sample (such as a powder), all possible orientations are present simultaneously, and the NMR spectrum is the result the uniform distribution of al these molecular orientation. The typical narrow isotropic resonances known from liquid-state NMR are turned into the so-called “powder patterns” in the solid phase.
The major task in solid-state NMR is to manipulate the Hamiltonian with the aim to recover the typical narrow isotropic resonances of liquid-state NMR and to maintain (or reintroduce) sufficient spectral information for structure determination.
2.1.2. Magic Angle Spinning (MAS)
MAS is a widespread method for increasing resolution in ssNMR and consists of rotating the sample rapidly about an axis tilted by an angle of 54.7° (magic angle) with respect to the external magnetic field B0 [52]. Depending on the
spinning speed used and the magnitude of the anisotropic interactions, it is possible to increase resolution by averaging out chemical shift anisotropies and through space dipolar couplings.
Fig. 18. Simulated static powder 13C spectrum of a glycine powder sample (a), along with spectra
recorded using MAS with spinning frequencies of 1 to 10 kHz (1, 2, 5 and 10 for spectra (b) to (e), respectively).
As seen in the previous section, the chemical shift produced by ĤCS and ĤD is a
combination of an isotropic term, that is orientation independent, and an anisotropic term with a (3cos2θ - 1) dependence on the orientation with the
external magnetic field. Under MAS, the orientation dependence becomes time-dependent, and it is average to zero (at 54.7° the term 3cos2θ - 1 becomes zero)
if the MAS frequency is larger than the breath of the anisotropic interaction (the powder linewidth). At slower MAS rates, the time dependent terms give
rise to a series of sharp bands, spaced by the spinning frequency (“sideband pattern”).
Averaging of dipolar interactions by MAS is only possible for the chemical shift and heteronuclear dipolar couplings (the so-called inhomogeneous interactions). For proton homonuclear dipolar couplings (a so-called homogeneous interaction), noncommuting parts of the Hamiltonian prevent this averaging unless the spinning speed is far in excess of the width of the powder spectrum. Due to the large dipolar couplings amongst protons, even at high spinning speeds it is thus difficult to resolve individual 1H resonances. For these reason ssNMR has
focused on the detection of rare nuclei such as 13C and 15N. Due to their low
gyromagnetic ratio and low natural abundance, they have relatively weak dipolar couplings that can be averaged out with moderate spinning speeds and high power decoupling. However, these two factors also lead to low signal intensities as compared to protons.
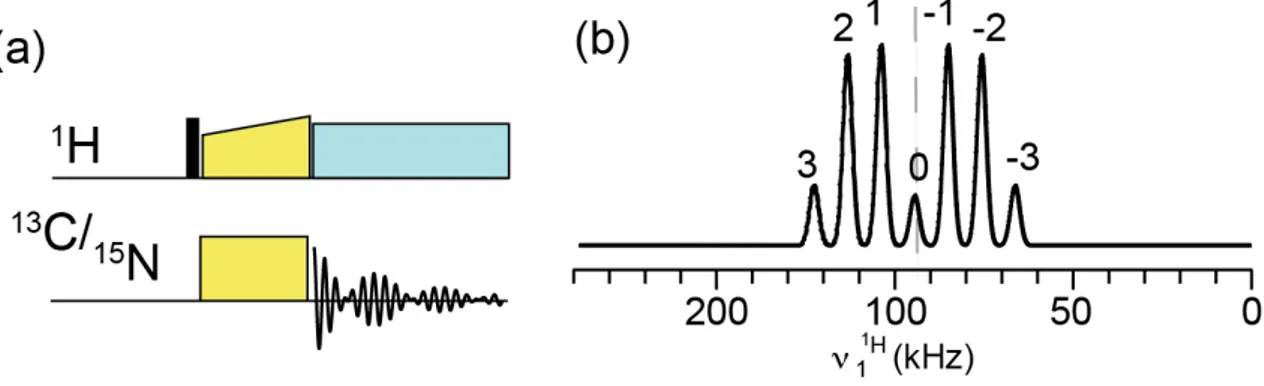
2.1.3. Cross Polarisation (CP)
Cross Polarisation is an experimental method for increasing the signal sensitivity of rare nuclei, developed by Hartmann and Hahn [53]. It employs double resonance irradiation of the system such that a dilute spin S (typically
13C or 15N) derives an initial magnetisation state from a network of abundant
spins I (typically 1H) that are in close proximity via the heteronuclear dipolar
coupling interaction. The larger polarisation of 1H is due to large γ which results
in large Boltzmann population differences.
Fig. 19. Pulse sequence elements for CP and schematic Hartmann-Hahn matching profiles.
In a CP experiment protons are first excited by a 90° pulse. In the second step both high and low-γ nuclei are “spin-locked” with rf-fields that result in nutation frequencies νI and νS satisfying one of the Hartmann-Hahn matching
where νR is MAS frequency and B1I and B1S are the amplitude of the rf-fields
applied to the I and S spins, respectively (Fig. 19).
The amount of bulk magnetisation of the dilute spin S is increased by a factor γI/γS, thus directly improving the signal intensity.
2.1.4. Heteronuclear decoupling
As we have seen, the large γ of 1H spins results on the one hand in a good
sensitivity due to the large population differences, but on the other hand, it leads to significant line broadening. Because of their large gyromagnetic ratio, protons build a network of large dipolar couplings which is not possible to fully average with spinning alone [55].
As a result, it is also necessary to apply decoupling irradiation as part of the pulse sequence, so to average the spin component of the dipolar coupling Hamiltonian. In the CW scheme, this entails applying a high-level rf field at the Larmor frequency of the protons [56]. In this way protons precess around the applied field so that, over an integer number of cycles, the average dipolar coupling reduces to zero. In practice, the strength of the rf field must exceed both the heteronuclear carbon-proton coupling, but must also exceed the homonuclear proton-proton coupling.
Many decoupling schemes have been developed to increase the efficiency of line-narrowing across a range of spinning frequencies, generally, where the irradiation is divided into blocks of pulses of varying phase. TPPM (“two-pulse phase-modulated decoupling”), swept-TPPM and SPINAL-64 (“small phase incremental alteration), are heteronuclear decoupling schemes widely used in biomolecular studies.
2.1.5. Homonuclear recoupling
The effect of MAS is not only the suppression of unwanted line broadening: dipolar interactions which contain desired structural information are partially cancelled out. However, if pulse trains are applied in a synchronised manner with the MAS, the dipolar interactions can be partially reintroduced in a process called “recoupling”. In this way, for example, spectra can be recorded containing correlations between spins which are in proximity through space [55]. A large number of pulse sequences has been developed over the last 20 years to provide efficient homonuclear recoupling. A first set of techniques, the so-called “first-order recoupling”, prevent MAS averaging of dipolar coupling. RFDR (Radio-Frequency DRiven dipolar Recoupling), HORROR (double-quantum
homonuclear rotary resonance), DREAM (dipolar recoupling enhanced by amplitude modulation) are some examples of first-order recoupling. These schemes are particularly useful to allow magnetisation transfer among neighbouring nuclei, but they are incapable of transferring magnetisation over long distances. Polarisation transfer mediated by weak dipolar interactions characteristics of long distances are cut off in the presence of strongly-coupled nuclei present at shorter distances. This phenomenon is commonly referred to as dipolar truncation [57]. This drawback prevents the use of these methods for the identification of a complete spin-system, or for detection of long distance contacts that are important for structural determination.
A second set of techniques, named “second-order recoupling” allow to alleviate the effect of dipolar truncation by generating an effective hamiltonian which contains homonuclear or heteronulcear dipolar coupling terms. The most popular is the PDSD scheme (proton-driven spin diffusion), which allows a coherent polarisation exchange between coupled 13C spins in the presence of
dipolar couplings to the 1H bath which provide the energy necessary for the transfer. PDSD has allowed the first determination of a 3D structure of a protein in the solid-state [43] (SH3 selectively labelled). Other schemes are DARR (Dipolar Assisted Rotational Resonance), MIRROR (mixed rotational and rotary resonance), PAR (proton assisted recoupling) and PARIS-xy (phase-alternated recoupling irradiation scheme).
Finally 2D proton-mediated correlation data (CHHC and NHHC spectra) probe close proximities between the protons directly bonded to the correlated (13C,13C)
or (15N,13C) nuclei, and provide important additional information about
intraresidue connectivities and long range inter-residue contacts [57]. Even if these schemes take advantage from the high number of proton-proton contacts which are present in the molecule, they are not very sensitive. However such methods have allowed the structural determination of different proteins (kaliotoxin 4 kDa, GB1 6 kDa, MMP12 17.6kDa).
2.1.6. Spin diffusion
Spin diffusion is a magnetisation spatial diffusion process, that takes place without diffusion of matter. The process is driven by flip-flop transitions between dipolar-coupled spins, where both spins change their magnetic quantum number in an energy conserving process. The time evolution of the spatial distribution of the magnetisation can be described by a diffusion equation. This process is particularly important in extended systems of dipolar coupled nuclei, like those formed by 1H in a proton rich sample (“proton bath”). Some long
range recoupling schemes are driven by the “proton bath” which mediate the energy conserving transfer.
2.1.7. Structural characterisation by ssNMR
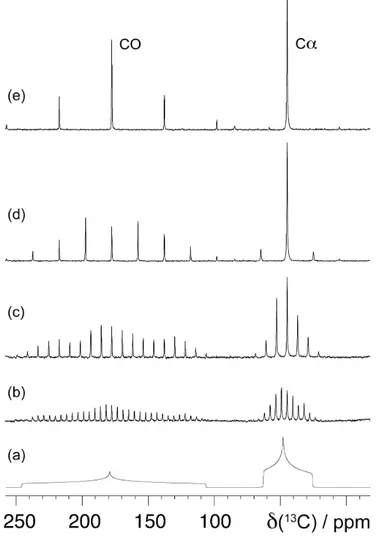
Before a structural characterisation in a multiply labeled sample can proceed, the observed signals must be identified and assigned to the NMR-detectable nuclei [36].
Intra-residue resonance assignment in polypeptides usually begins with homonuclear (13C,13C) correlation experiments that allow identification of
different amino acid types based on their characteristic chemical shift correlation pattern (Fig. 20). Carbon-carbon correlations can also be related to nearest neighbour 15N spins.
The connectivity between adjacent polypeptide residues (the inter-residue assignment) can be established through a set of triple resonance experiments to transfer magnetisation from 1H to 15N by a first 1H-15N CP step, and the to 13C
by a second 15N-13C CP step (Fig. 20).
Fig. 20. ssNMR experiments for inter-residue assignment (left) and intra-residue resonance assignment
(right)
The NCA spectrum contains only correlations of the backbone amide 15N of a
residue i with the Cα(i) of the same residue. NCO spectrum provides the corresponding sequential correlation by correlating the 15N(i) with the adjacent
CO(i-1). These two experiments can be extended with additional spin diffusion mixing scheme. For example RFDR and DREAM mixing schemes can be used respectively for polarisation transfer from CO(i-1) to Cα(i-1) (NCOCA experiment for inter-residue assignment) or from Cα(i) to Cβ(i) or Cγ(i) (NCACX experiment for intra-residue assignment). NCO, NCA and NCOCA/ NCACO spectra are in principle sufficient for the determination of the
!"#!$ #!$!" #!"!$ #!"!% !" !&' #! !"! !%! !(! # !&' #)!"*!%!+ !!! #!"!+ # !" !% !+ t1 15N x t2 t3 13C 1H x
SPINAL64 CW SPINAL64 SPINAL64 ~1 ms ≤ 7 ms ~6 ms ≤ 7 ms ~ 15 ms φ1 φ1 = (+y, –y) φ2 = (+x, +x, –x, –x) φ3 = (+x, +x, +x, +x, –x, –x, –x, –x) φr = (+x, –x, –x, +x, –x, +x, +x, –x) φ2 φ3 φr CA MIRROR/DARR φ3–π/2 ~50 ms t1 15N x t2 t3 13C 1H x
SPINAL64 CW SPINAL64 SPINAL64 ~1 ms ≤ 7 ms ~6 ms ≤ 7 ms ~ 15 ms φ1 φ1 = (+y, –y) φ2 = (+x, +x, –x, –x) φr = (+x, –x, –x, +x, –x, +x, +x, –x) φ2 φ3 φr C’ MIRROR/DARR ~50 ms φ3–π/2 x x φ3+π/2 x φ3+π/2 φ3 = (+x, +x, +x, +x, –x, –x, –x, –x) φ1 = (+y, –y) φ2 = (+x, +x, –x, –x) φ3 = (+y, +y, +y, +y, –y, –y, –y, –y) φr = (+x, –x, –x, +x, –x, +x, +x, –x) t1 15N x t2 t3 13C 1H x
SPINAL64 CW SPINAL64 SPINAL64 ~1 ms ≤ 7 ms ~6 ms ≤ 7 ms ~ 15 ms φ1 φ2 x φr CA CW φ3 C’ ~6 ms x x x #!"!$ #!$!" !"#!$ backbone sidechain
shift dispersion of carbonyl signals, a further homonulear magnetisation transfer to 13Cβ is usually mandatory (Fig. 20). Running these correlations as 3D
15N-13C-13C spectra helps to solve the problem of ambiguity and to improve the
process of assignment. Additional information about intra-and interresidue connectivities can be obtained from 2D proton-mediated correlation data (CHHC or NHHC).
Fig. 21. General approach for determining the structure of multiply labeled proteins in solid-state NMR
under MAS conditions [36].
The chemical shifts represent sensitive probe of local molecular conformation, so the spectral assignment allow the direct determination of backbone secondary structure. Empirical studies reveal that deviation from reference 13Cα and 13C β
chemical shift values can be interpreted in terms of the local backbone conformation. Computational methods can used to predict how conformation-dependent chemical shift change as a function of the dihedral angles (ψ,ϕ). In addition to the conformation-dependent chemical shift, distances, encoded as dipolar couplings, can be used to refine the local conformation (Fig. 21).
Similar to the solution state NMR, structure determination in the solid state involves the calculation of a series of molecular conformations that are consistent with the experimental derived distance or angle constraints. Internuclear distances and torsion angles define the three-dimensional conformation of a protein (Fig. 21). In particular, medium and long-range distance parameters that connect the different residues are important tools to
![Fig. 8. a) cryo-EM reconstruction of trypsin-digested N-RNA from [28]. The black broken line shows the orientation of the line that connects subunits in subsequent turns](https://thumb-eu.123doks.com/thumbv2/123dokorg/7537898.107807/18.892.210.727.226.487/reconstruction-trypsin-digested-broken-orientation-connects-subunits-subsequent.webp)