SAPIENZA Università di Roma
Facoltà di Scienze Matematiche Fisiche e Naturali
DOTTORATO DI RICERCA
IN BIOLOGIA CELLULARE E DELLO SVILUPPO XXXII Ciclo
(A.A. 2018/2019)
Titolo
Study of cell cycle in non-proliferating cells Dottorando
Emilia Giannella
Docente guida Prof. Milena Grossi
Tutore Coordinatore
Index
1. Glossary
2. Summary-Sommario 3. Introduction
3.1. The cell cycle
3.1.1. Cyclins and cyclin-dependent kinases 3.1.2. Inhibitors of cyclin-dependent kinases 3.1.3. Checkpoints 3.2. No proliferative states 3.2.1. Quiescence 3.2.2. Replicative senescence 3.2.3. Terminally differentiation 3.2.3.1. Muscle differentiation
3.2.3.2. Cell cycle reactivation of terminally differentiated cells
3.3. Satellite cells in muscle regeneration 3.4. Xenopus egg extracts (XEE)
4. Aims 5. Results 6. Discussion
1. GLOSSARY
CDK: Cyclin dependent kinase
Cip/Kip: CDK interacting protein/Kinase inhibitory protein Cy3-dCTP: Deoxycytidine triphosphate conjugated with Cy3 fluorescent dye
DPI: day post injection G1: Gap 1
G2: Gap 2
GF: Growth factors HSE: High speed extract LSE: Low speed extract M: Mitosis
MSC: Muscle satellite cells Mt: myotubes
P: proliferating myoblasts pRB: retinoblastoma protein
p21KO: p21 knock out
p21 scko: satellite-cell conditional p21 knock out Q: quiescent myoblasts
R point: Restriction point TA: anterior tibialis muscle
TD: terminally differentiated XEE: Xenopus egg extract
2. SUMMARY
Non-proliferating cells can be found in three conditions: reversible quiescence, replicative senescence and terminal differentiation (TD). Cells in any of these non-proliferative states can be made to re-enter the cell cycle through the removal of appropriate CKIs.
Through a CRISPR/Cas9 strategy, we have generated conditional p21 KO mice. These animals were crossed with mice expressing a tamoxifen-activatable Cre recombinase under the control of the Pax7
promoter to knockout p21 exclusively in Pax7+ satellite cells.
The first purpose of my thesis was to study the consequences of the absence of p21 in satellite cells. We show that the deletion of p21 in satellite cells only does not lead to fiber neogenesis and, after injury, does not have impact on muscle regeneration.
Skeletal muscle myotubes (Mt), a model system of TD, can be forced to reenter the cell cycle but they are not able to fully duplicate their DNA. Understanding the molecular bases that prevent Mt to a regular and complete replication is the aim of the second part of this work. We used Xenopus laevis egg extracts (XEE) to study DNA replication of different genetic material from myoblasts or myotubes to identify an eventual structure obstacle, that do not permit a complete replication of myotube. The results obtained suggest that the no-histones proteins are the possible candidates responsible for the inability of these cells to proliferate.
SOMMARIO
Le cellule non proliferanti si possono trovare in tre condizioni: quiescenza reversibile, senescenza replicativa e differenziamento terminale (TD). Cellule in qualsiasi stato non proliferativo possono rientrare nel ciclo cellulare attraverso la rimozione dell’appropriato CKI.
Attraverso il sistema CRISPR/Cas9, abbiamo generato topi KO condizionali per p21. Questi animali sono stati incrociati con topi che esprimono una Cre ricombinasi, attivabile dal tamoxifene, sotto il controllo del promotore Pax7 per eliminare p21 esclusivamente
nelle cellule satellite Pax7 +.
Il primo scopo della mia tesi era quello di studiare le conseguenze dell'assenza di p21 nelle cellule satellite. La rimozione di p21 nelle sole cellule satelliti non determina neogenesi delle fibre e, dopo il danno, non ha alcun impatto sulla rigenerazione muscolare.
I miotubi (Mt), un sistema modello di TD, possono essere forzati a rientrare nel ciclo cellulare ma non sono in grado di duplicare completamente il loro DNA. Comprendere le basi molecolari che impediscono a Mt una replicazione regolare e completa è lo scopo della seconda parte di questo lavoro. Abbiamo usato gli estratti di uova di Xenopus laevis (XEE) per studiare la replicazione del DNA di diversi materiali genetici da mioblasti o miotubi per identificare un eventuale ostacolo strutturale, che non consenta una replicazione completa del miotubo. I risultati ottenuti suggeriscono che le proteine non istoniche sono i possibili candidati responsabili
3.INTRODUCTION
2.1. THE CELL CYCLE
The cell cycle is a coordinated series of events that leads to the division of a single cell into two daughter cells. The eukaryotic cell cycle can be divided into two periods: interphase and mitosis (M) (Morris et al., 2013). During the interphase the cell grows, accumulates nutrients needed for the next steps, and duplicates its DNA. In mitosis the cell divides into two distinct cells. Interphase is
composed of three phases: G1 (gap 1), S (synthesis) and G2 (gap 2),
while, mitosis consists of two coupled processes: karyokinesis, in which the cell’s chromosomes are divided between the two daughter cells, and cytokinesis, in which the cell’s cytoplasm divides in half, forming distinct cells. Most cells of an adult vertebrate spend much
of their time in a non-proliferative state, termed G0. Physiologically,
non-proliferating cells can be found in three conditions: reversible quiescence, replicative senescence, and postmitotic state; the last one characterizes and defines terminal differentiation. Transitions above mentioned states are regulated by different cellular proteins, such as cyclins and cyclin-dependent kinases (CDK).
2.1.1. CYCLINS AND CYCLIN/KINASE COMPLEXES
Cyclins are molecules synthesized and cyclically degraded by the cell in response to external signals. The action of the cyclins takes place following their association with specific proteins kinases, known as cyclin-dependent kinases or CDK. The formed complexes, consisting of a regulatory subunit (cyclin) and a catalytic subunit (CDK), are active in different moment of the cell cycle and cause a cascade of events that characterizes and regulates the various phases of the cell cycle (Murray, A.W., 2004). CDKs are serine/threonine kinases that phosphorylate substrates required to trigger cell cycle progression. Five of nine identified CDKs are active during the cell cycle: CDK4,
M. During the different phases of cell cycle, the expression of CDKs is relatively constant. The regulation of their activity depends essentially on the availability of the cyclins, phosphorylation and dephosphorylation events and the presence of specific inhibitors
(Ekholm, SV, and Reed, SI, 2000). During the transition from G0 to
G1 phase, mitogenic signals converge primarily on the expression,
assembly and activation of cyclin D/CDK complexes. Cyclins D (D1, D2, D3) are expressed differently in different organs and tissues (Sherr et al., 1993), are transcribed in response to mitogenic signals,
mediating progress through phase G1. Cyclins D can heterodimerize
specifically with cdk4 and 6 (Ekholm and Reed, 2000). In the late G1
phase exists a specific point, called the "restriction point" (R point), beyond which cell became independent from the presence of growth factors and completes its cell cycle, regardless of environmental conditions. The activity of the cyclin D/CDK complexes, as well as the mitogenic stimuli, is no longer required until the cell completes
the division and re-entry in the next G1 phase (Matsushime, H. et al.,
1994). The kinase activity of the cyclin D/CDK complexes phosphorylates and inactivates the retinoblastoma protein (pRb), p107 and p130. These phosphorylated proteins release the associated factors, belonging to the E2F family, which activate the transcription of a series of genes necessary for the beginning of S phase, for the metabolism and the DNA replication (Trimarchi, JM, and Lees, JA,
2002). During G1 phase, cyclin D/CDK complexes have a non-
catalytic role sequestering specific cell cycle inhibitors, releasing and activating the cyclin E/CDK2 complex (Sherr, C.J., and Roberts, J.M., 1995). The activity of the cyclin E/CDK2 reaches its peak in
the G1/S transition of the cell cycle while decreases rapidly in the
early S phase (Welcker, M. et al., 2003). This complex can phosphorylate many proteins, among which pRb, and can regulate various substrates involved in the expression of histone genes, in the duplication of centrosomes and in the DNA replication (Yu, Q., and Sicinski, P., 2004). CDK2 can, also, forms complexes with cyclin A.
Cyclin A is expressed in S phase, accumulates in the late G2 and it is
degraded during M phase where it assumes the role of "mitotic
(Fung and Poon, 2005); cyclins A and B bind the CDK1 and their degradation is necessary for the completion of mitosis (Yam et al., 2002). Cyclin B (B1, B2, B3) binds and activates the CDK1, forming the so-called "M-phase promoting factor" (MPF), which phosphorylates essential substrates for entry into phase M (Fung and Poon, 2005).
Fig.3.1 | Cell cycle and its regulation
2.1.2. INHIBITORS OF CYCLIN-DEPENDENT KINASES The cell cycle is orchestrated by the action of CDKs, whose activity is positively regulated by cyclins and negatively by CKIs. The latter belong to two families: INK4 or Cip/Kip (Sherr, C.J. et al., 1999). The first family includes four members, indicated, according to their molecular weight, as p15 (Hannon, G.J., and Beach, D., 1994), p16 (Serrano, M. et al., 1993), p18 (Guan, K.L. et al., 1994; Hirai, H. et al., 1995) and p19 (Chan, F.K. et al., 1995; Hirai, H. et al., 1995), which specifically bind cdk4 or -6, preventing the formation of heterodimers with the D cyclins (Sherr, C.J., and Roberts, J.M.,
1999). The second one is composed by p21 or CDKN1A (Gu, Y. et al., 1993; Harper, J.W. et al., 1993; El-Deiry, W.S. et al., 1993; Xiong, Y. et al., 1993; Dulic, V. et al., 1994; Noda, A. et al., 1994), p27 or CDKN1B (Polyak, K. et al., 1994; Toyoshima, H., and Hunter, T., 1994) and p57 or CDKN1C (Lee, M.H. et al., 1995; Matsuoka, S. et al., 1995), which bind most cyclins and CDKs and their complexes (Harper, J.W., 1997).
The Cip/Kip inhibitors play an important role in the regulation of all the phases of the cell cycle because they have no particular specificity for one cyclin/kinase complex. These inhibitors are also able to facilitate the formation and the consequent activation of the same cyclin/CDK complex (Labear, J. et al., 1997). The balance between the activator and inhibitor activity is still a subject of debate. According to the most accredited theory, different roles can depend on the concentration of these molecules: low levels seem to stimulate, while high levels appear to inhibit the activity of cyclin-CDK complexes. It, also, seems that different sites of binding (on cyclin or on kinase) may be involved in different functions (Nabel, E.G., 2002). In any case, INK4 and Cip/Kip implement a negative control of cell proliferation and deregulative events against these molecules play an important role in carcinogenesis (Harper, J.W., and Elledge, S.J., 1996).
2.1.3. CHECKPOINTS
The aim of cell duplication is to generate two cells that possess the same genetic set. Unfortunately, the replicative machinery is prone to errors, which can alter genetic information and trigger a programmed cell death (apoptosis). The establishment of such alterations can lead to neoplastic transformation (Murray, 1992). To prevent these errors, cells have developed several “checkpoints”, which guarantee the successful completion of the current phase of the cell cycle before going through the next. Restriction Point (R-point)
is the first checkpoint which controls the passage through the G1
phase. It responds to growth factors and to the subsequent hyperphosphorylation of pRb by cyclin D/CDK4-6 complexes. R-
point is the crucial control point because determines whether a cell can enter the cell cycle (Foster, D.A. et al., 2010). The checkpoint
regulating the beginning of S phase (G1/S transition) is important for
the control of DNA integrity before its replication, while the
checkpoint that controls the entry into M phase (G2/M transition)
bypasses the start of mitosis when a damage is detected or when the DNA replication is incorrect (Agarwal, M.L., et al., 1995). Finally, the correct segregation of chromosomes is entrusted to the checkpoint of the M phase (metaphase/cytokinesis), which examines the correct interaction between the mitotic spindle and chromosomes and their proper alignment along the metaphase plate (Nigg, E.A., 2001). Any anomalies and aberrations must be promptly reported to allow the cell to repair DNA damages. This is possible through the interaction of three systems: the sensors, the transducers and the effectors (Zhou and Elledge, 2000). Briefly, the sensors perceive the damage and communicate it to the transducers, which amplify the signal and recruit effectors that block the cell cycle and, if it is possible, repair the damage. The immediate consequence of checkpoint activation is
therefore the cell cycle arrest in G1, S or G2/M, in relation to when
the damage occurred, allowing the cell to repair the error or activate apoptotic mechanisms, depending on the nature of the lesion and its extension (Lukas, J. et al., 2004).
2.2. NON-PROLIFERATIVE STATES
Most adult vertebrate cells spend most of their time in a non-
proliferative state, labeled as G0. Physiologically non-proliferating
cells can be found in three conditions: reversible quiescence, replicative senescence and postmitotic state; the latter characterizes and defines terminal differentiation.
2.2.1. QUIESCENCE
Quiescence is defined as a temporary, reversible absence of proliferation. This state can be induced by a variety of conditions,
including growth factor deprivation, contact inhibition, and loss of anchorage (Coller et al., 2006). Quiescence can be usually readily reverted by removing the conditions that determined it. The molecular mechanisms determining quiescence vary according to its causes. For example, lack of growth factors triggers a swift reduction in the expression of D-type cyclins. Such reduction is at least in part responsible for the cell cycle exit that follows growth factor deprivation (Ekholm and Reed, 2000). Lack of space for cell division, the in vitro phenomenon known as contact inhibition, determines growth arrest via an increase in p27 levels, with no reduction in cyclin D (Sherr and Roberts, 2004). Usually, removal of the condition that causes quiescence promptly abolishes its molecular underpinnings and reverts the cell to proliferation. As already described, the
functions of cyclins and CKIs in G1 phase modulate the proliferation-
restrictive activity of pocket proteins, more often that of pRb. Interestingly, pRb is not required for the induction of quiescence (Sage, J. et al., 2000), but its ablation reactivates quiescent and senescent cells (Sage, J. et al., 2003). Thus, the mechanisms establishing quiescence are partly distinct from those that maintain it.
2.2.2. REPLICATIVE SENESCENCE
A cell can divide a fixed limited number of times. This is the replicative senescence (Hayflick and Moorhead, 1961). Replicative senescence is, thus, a state in which cellular proliferation is permanently stopped. It appears to be a fundamental feature of somatic cells, with exception of most tumor cells and certain stem cells. More recent findings indicate that senescence (or cellular aging) is a response of the cell to different stress conditions. Among these: telomere attrition, DNA damage, and oncogene activation (Herbig and Sedivy, 2006). Senescent cells, unable to enter S phase, express constitutively high levels of cyclin D1 and cyclin E, independent of mitogenic stimulation (Dulic et al., 1993). These cyclins form complexes with CDKs, but such complexes are enzymatically inactive. Senescent cells express large amounts of p21 (Tahara et al., 1995) and p16 (Shenghui, 2017), and the absence
of either inhibitor before the beginning of senescence has been found to facilitate immortalization. Together, these findings, suggest that, in senescent cells, CDKs are held inactive by preponderant levels of CKIs, despite the presence of large amounts of cyclins. Indeed, suppression of either p21 or p16 has been shown to induce previously senescent cells to proliferate (Pajalunga et al., 2007).
2.2.3. TERMINAL DIFFERENTIATION
Terminal differentiation is empirically defined as a state characterized by specialized cell functions and permanent withdrawal from the cell cycle. In extreme cases, such as that of keratinocytes and erythrocytes, the terminally differentiated cells (TD) eliminate their nucleus, losing irreversibly the ability to divide. Other examples of TD cells are neurons, skeletal and heart muscle
cells, adipocytes. Expression of G1 phase cyclins is generally
downregulated in TD cells, while that of different CKIs, depending on cell type, is increased (Kranenburg et al., 1995). Thus, the balance of cell cycle regulation is markedly tilted toward growth arrest, making TD cells capable of resisting a variety of powerful mitogenic stimuli (Tiainen et al., 1996).
2.2.3.1 MUSCLE DIFFERENTIATION
Skeletal muscle constitutes a classic example of terminal differentiation and, for its ease of growth in vitro, it represents an excellent experimental model. Several myogenic cell lines (myoblast) are available and they can differentiate into myocytes and fuse into myotubes. Activation, proliferation and differentiation of muscle satellite cells (MSCs) are processes regulated by a family of transcription factors called myogenic regulation factors (MRFs), including MyoD, Myf5, Myogenin, Mrf4 (Charge, SBP, 2004) and numerous structural genes, such as the myosin heavy chain and muscle creatine kinase. MyoD is expressed in proliferating myoblasts at an early stage of muscle differentiation and induces the expression
of other genes involved in myogenesis, such as myogenin (Kim, J. et al., 2013). Following deprivation of mitogenic factors, MyoD exerts its transcriptional activity on specific muscle promoters, particularly on p21 and pRb. pRb, in skeletal muscle cells, plays a fundamental role in the induction of the postmitotic state, but not in its maintenance: the removal of the protein from myotubes does not suppress their postmitotic state (Camarda, G. et al., 2004). This observation allows to distinguish permanently TD cells from the quiescent and senescent ones, where the maintenance of the mitotic arrest depends on pRb. On the contrary, the presence of p21 is not indispensable for the induction of muscular differentiation (Figliola, R. and Maione, R., 2004), but it is fundamental for its myogenic maintenance (Pajalunga, D.et al., 2007). The action of MyoD mediates the exit of cells from the cell cycle. During the differentiation of skeletal muscle, the levels of some cyclins (D1, A, B) are lower (Ohkubo, Y. et al., 1994), while those of some CKI rise. The correlation is necessary for terminal differentiation of myotubes (Zabludoff, S. D. et al., 1998). This characteristic is present in the non-proliferative states (quiescence, replicative senescence and terminal differentiation): in all these three cases, in fact, it has been shown that by suppressing specific CKI, the cells re-enter into the cell cycle, even in the absence of factors of growth (Pajalunga, D.et al., 2007).
3.2.3.2. REENTRY INTO THE CELL
Unlike quiescent cells, TD cells are unable to proliferate, even in the presence of growth factors. In some TD cell types, such as myotubes, serum stimulation induces cell cycle entry, accompanied by the
expression of early G1 phase (c-myc, c-fos, c-jun, Id-1) and cyclin D.
The reactivated myotubes, however, are blocked in the middle of the
G1 phase, failing to progress in the cell cycle and to synthesize DNA
(Tiainen et al., 1999). TD cells are physiologically refractory to proliferation, but they can be forced to synthesize DNA if reactivated with DNA tumor virus oncogenes, such as the Large T of SV40 (Endo and Nadal-Ginard, 1998) or E1A of adenovirus (Crescenzi et
al., 1995). Some TD cell types, such as muscle cells, neurons and adipocytes, can start the S phase, if the exogenous overexpression of the cyclin D/CDK 4 and 6 complexes is induced (Latella et al., 2001). Another effective strategy to stimulate DNA synthesis in TD cells is the use of siRNA against CKIs. These inhibitors are expressed at high levels in many types of non-proliferative cells and contribute
BrdU Hoechst phase contrast
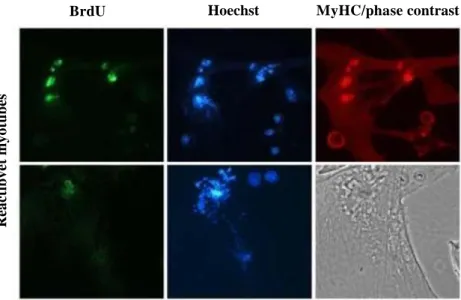
Figure 3.2 | Immunofluorescence for BrdU. Reactivated myotubes by siRNA against p21 and p27 proteins, show an altered pattern of BrdU incorporation (green) in comparison to the homogenous pattern of proliferating myoblast (Pajalunga, D. et al., 2010)
critically to arrest the cell cycle (Pajalunga et al., 2007). Cells in any non-proliferative state reenter in the cycle upon removal of appropriate CKIs, the most important of which is p21 (Pajalunga et al., 2007). In fact, it has been shown that RNA interference (RNAi) suppression of the appropriate CKIs triggers DNA synthesis and mitosis in myotubes, in quiescent human fibroblasts, and in senescent human embryonic renal cells (Pajalunga et al., 2007, Falcone et al., 2013). Hence, temporary or permanent arrest often requires the constant expression of CKIs. Reactivation of the cell cycle mediated by CKI suppression is due to the presence, even in permanently non- proliferating cells, of preassembled cyclin/cdk/CKI complexes, which spontaneously acquire activity after the removal of the inhibitory molecule (Pajalunga et al., 2007). TD cells, even if forced
my o b la st s R ea ct iv ed m y o tu b es
to reenter the cell cycle, generally cannot proliferate because they undergo incomplete DNA replication (Fig. 3.2), DNA damage and death by apoptosis (Fig. 3.3) (Pajalunga et al., 2010, Pajalunga et al., 2017). In contrast, quiescent and even senescent cells, after removal of the CKI inhibitor, proliferate despite the absence of growth factors (Pajalunga et al., 2007, Falcone et al., 2013). Recently, it was showed that the temporary suppression of p21 in vivo, through an adeno-associated virus (AVV) that expresses a shRNA directed against p21, triggers the proliferation of different cell types (Biferi et all., 2015).
BrdU Hoechst MyHC/phase contrast
Figure 3.3 | Immunofluorescence. Mitotic catastrophes of reactivated myotubes by siRNA against p21 and p27 proteins. (Pajalunga, D. et al., 2010)
3.3 SATELLITE CELLS IN MUSCLE REGENERATION
In muscle repair, the most important role is played by satellite cells (SC), described for the first time by Mauro (Mauro, 1961). SCs comprise a small population (5% to 7% of all muscle nuclei) of undifferentiated and quiescent mononucleated cells that reside
Rea ct ibv et my o tub es
between the basal lamina and the outer membranes of skeletal muscle fibers, a microenvironment referred to as their niche (Mauro, 1961). These cells are quiescent and can be activated by appropriate stimuli following damage, resulting in regeneration of muscle tissue (Sutcu Haser Hasan and Ricchetti Miria, 2018). Other important morphological features of SC are high nuclear-to- cytoplasmic ratio, reduced organelle content, and smaller nuclei displaying increased amounts of heterochromatin, compared with fiber myonuclei (Mauro, 1961). SC express the transcription factor Pax7, a member of the paired-box gene family of transcription factors implicated in the development of the skeletal muscle of the trunk and limbs, as well as elements of the central nervous system. Pax7 activates the expression of myogenic factor 5 (Myf5) and myogenic differentiation (MyoD) genes. Myf5 and MyoD are required for myoblast specification (McKinnell et al., 2008), while differentiation into myotubes depends on the downstream transcription factor myogenin (Myog) (Knapp, J. R. et al., 2006). Mef2 is a family of myocyte enhancer factor 2 proteins that act in cooperation with Myog and MyoD to drive terminal muscle differentiation (Liu, N. et al.; 2014; Taylor MV and Hughes SM, 2017). During this process, mononuclear myocytes fuse to form mature multinucleated muscle fibers expressing hundreds of muscle- specific genes, prominent among which myosin heavy chain (Myh), myosin light chain (Myl) isoforms, and muscle creatine kinase (Ckm) (Abmayr et al., 2012). Muscle regeneration is characterized by two phases: a degenerative and a regenerative phase (Wosczyn and Rando, 2018). The initial event of muscle degeneration is necrosis of the muscle fibers. This phenomenon is generally triggered by disruption of the myofiber sarcolemma, resulting in increased myofiber permeability. The next event is the inflammatory process. In fact, the injured muscle starts to release factors that activate the inflammatory cells residing within the muscle itself. The most important event for muscle regeneration is cellular proliferation. SC, activated following damage, express both Pax7 and MyoD and assume a myoblast identity. After a few rounds of cell division, myoblasts undergo differentiation and fusion with the existing myofibers to recreate functional muscle tissue. SC also undergo asymmetric cell division to replenish the reservoir of
quiescent stem cells (Kuang S, et al., 2007). Histological characteristics are used to identify the mammalian skeletal muscle regeneration process. The new myofibers show a small caliber and centrally located myonuclei. Once fusion of myogenic cells is completed, newly formed myofibers increase in size, and the myonuclei move to the periphery of their muscle fiber. Under normal conditions, the regenerated muscle is morphologically and functionally indistinguishable from undamaged muscle.
3.4 Xenopus laevis
Xenopus is an aquatic anuran described by about 20 different species. The most known species is Xenopus laevis, commonly studied as a model organism in various disciplines such as genetics, biomedical research, developmental, cellular and molecular biology (Blow and Laskey, 2016; Lin et al., 2019). It is used in the laboratory as a research tool for in vivo studies, however it is mainly used to obtain acellular extracts of non-fertilized eggs (Xenopus Egg Extracts, XEE), which have a large spectrum of applications. XEE are used to reproduce in vitro the replication of genetic material at different levels of complexity. It contains all the factors necessary to synthesize the genome, such as: ATP, proteins of the pre-replicative complex, DNA polymerases etc. Xenopus unfertilized eggs arrested in metaphase of meiosis II are collected and crushed by sequential centrifugations. Low speed extracts (LSE) are prepared by a medium-speed centrifugation and contain the cytoplasm including light membranes, ribosomes and nuclear envelope, while high speed centrifugation producing high speed extracts (HSE) in which cytoplasm is separated from membranes and ribosomes. LSE extract can be interphasic, if unfertilized eggs, before crushing, are activated by calcium ionophore mimicking the fertilization, or mitotic, if the eggs remain inactive without calcium addition. In our laboratory LSE interphasic extract are used to study DNA and chromatin replication in terminally differentiated cells. In the absence of an intact cell, these extracts recapitulate the key nuclear transitions of the eukaryotic cell cycle in vitro under apparently the same controls that exist in vivo (Blow J.J.et al. 1988). They are able to duplicate
exogenous DNA molecules (Tutter, A., 2006) since they contain all the replication factors necessary to DNA synthesis. DNA added to the extracts is first assembled into chromatin and then into structures corresponding to interphase nuclei. Once nuclear assembly is complete, the DNA is efficiently replicated in a semi conservative way.
3. AIMS
Most cells of an adult vertebrate spend much of their time in a non-
proliferative state, termed G0. Physiologically, non-proliferating cells
can be found in three conditions: reversible quiescence, replicative senescence, and postmitotic state; the last one characterizes and defines terminal differentiation. Cells in any non-proliferative state can be induced to re-enter the cell cycle through different methods, one of these is the removal of appropriate CKIs. It was shown that the temporary removal of p21 in vivo triggers the proliferation of different cell types, leading to the increase in the number of muscles fibers, strength, and fatigue resistance (Biferi et all.; 2015). The first aim of this thesis is to study the deletion of p21 in quiescent muscle satellite cells (SC). To this purpose we have generated conditional p21 knockout mice, through a CRISPR/Cas9 strategy. To abrogate
p21 exclusively in Pax7+ SC, p21-LoxP mice were crossed with mice
expressing a tamoxifen-activatable Cre recombinase under the control of the Pax7 promoter. This system allows us to evaluate whether the fiber neogenesis and strength increase, showed by Biferi and colleagues, can be attributed solely to SC hyperproliferation. Besides, these mice enable us to verify if the absence of p21 in SC can improve muscle regeneration.
Skeletal muscle cells are a prototypic example of terminally differentiated cells (myotubes, Mt). Such cells can be forced to reentry the cell cycle, but cannot proliferate because they undergo incomplete DNA replication, DNA damage, and apoptosis (Pajalunga et al., 2010). Understanding the reasons behind the inability of Mt to fully replicate their genome is the objective of the second aim of this thesis. There are two classes of possible problems that can impair DNA replication: functional and structural obstacles. Defects in the replication machinery represent examples of functional problems, while any unusual conformation of the genetic material, that might physically prevent DNA replication, can be considered structural obstacles. We focus our attention on the latter. To
determine if a structural obstacle preventing DNA replication does exist in myotubes, we have used cell-free extracts from Xenopus laevis unfertilized eggs (XEE), that, autonomously, duplicate any type of genetic material. We will compare DNA replication in myotubes and myoblasts nuclei incubated in XEE. We reasoned that, since XEE provide the entire DNA replication machinery, incomplete DNA synthesis in Mt nuclei would imply the presence of a structural impediment. To dissect the nature of such hypothetical obstacle(s), we subdivided the nucleus into different levels of complexity: naked DNA, DNA arranged into nucleosomes and intact chromatin. Once purified, these distinct samples will be incubated in XEE to evaluate their replicability. In this way, it should be possible to identify the level at which the obstacle(s) lays.
4. RESULTS
I part
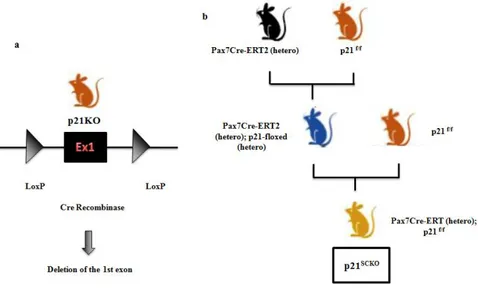
Generation of satellite-cell specific, conditional p21scko mice
Through a CRISPR/Cas9 strategy, we have generated conditional p21 KO mice, by encompassing exon 1 of the p21 gene within two
LoxP sites. These transgenic mice (referred to as p21f/f mice) were
bred with mice expressing a tamoxifen-activatable Cre recombinase
Figure 5.1 | Generation of satellite-cell specific, conditional p21scko mice (a) Through a
CRISPR/Cas9 strategy, we have generated conditional p21 KO mice, by encompassing exon 1
of the p21 gene within two LoxP sites. (b) These transgenic mice (referred to as p21f/f mice) were
crossed with mice expressing a tamoxifen-activable Cre recombinase under the control of the
Pax7 promoter, to knockout p21 exclusively in Pax7+ satellite cells (referred to as p21scko mice)
under the control of the Pax7 promoter, to knockout p21 exclusively
in Pax7+ satellite cells (referred to as p21scko mice) (Fig. 5.1). To
or untreated p21scko adult mice (n=2 for each group). After three
weeks in culture, DNA and RNA were extracted from the cells and analyzed by PCR and real-time PCR (qPCR) for p21. The results show that p21 was absent in genomic DNA and was not expressed at the mRNA level (Fig. 5.2 a, b). The expression of the p27 and p57 CKIs was also decreased. SC were shifted to differentiation medium for three days and RNA was analyzed through qPCR. Myotubes
derived from satellite cells of tamoxifen-treated p21scko mice showed
no detectable p21 expression and an increase in p57 mRNA levels (Fig. 5.2 c), compared to controls, probably due to a compensatory mechanism, similar to that described by Vaccarello (2006).
p21 ablation in SC does not induce cellular proliferation
We wished to determine whether the phenotype induced by muscle- wide p21 suppression can be mimicked by inducing proliferation of
a Tam No Tam 1663 bp b 1,2 1 0,8 400b p c 6 5 0,6 0,4 0,2 0 p21 p27 p57 msc+tam mscNotam 4 3 mt+tam mtNotam 2 1 0 p21 p27 p57
Figure 5.2 | Deletion of p21. (a)The presence of two lox-sites, flanked the exon 1 of the p21 gene, gives a synthetic allele amplicon of 1663 bp. When the Cre recombinase is activated by tamoxifen, its action causes the deletion of exon 1 of p21 and an amplicon of 400bp. (b) qPCR analysis for p21, p27, and p57 in SC. (c) mRNA levels in myotubes from SC isolated from tamoxifen-treated p21scko mice and control mice. SC isolated from mice were poolled and analysed (n=2 for each
m R N A l eve ls m R N A l eve ls
SC only. To address this point, we have used the p21scko mice. These
mice were injected intraperitoneally with 60 µg of tamoxifen per day per g of body weight for 8 days (days -8 to -1), to induce selective Cre-mediated deletion in SC cells. On days 5 and 10, we collected the tibialis anterior (TA) and soleus muscles from tamoxifen-treated and vehicle-injected, control mice. Hematoxylin and eosin (H&E)-stained muscle sections are similar (data not shown). There are no
evidencesshowing that the deletion of p21 in SC only is sufficient to
induce cellular proliferation in vivo.
b Tam
No tam
0 dpi 5 dpi 10 dpi 15 dpi
Figure 5.3 | Deletion of p21 in SC in muscle regeneration. (a) Following tamoxifen-mediated
Cre recombination, TA muscles of adult p21scko mice were injected with 35 µl of a 1.2% BaCl2
solution after which muscles were isolated and analyzed at 0, 5, 10 and 15 DPI. (b)
Representative photomicrographs of H&E-stained sections of injured TA muscle of p21scko mice
(Tam), compared with control p21scko mice (No Tam) after BaCl
2-mediated injury. N=3 in each
Deletion of p21 in SC in muscle regeneration
To assess if the lack of p21 in activated SC can have a role during
injury, we inflicted damage to TA muscles of p21scko mice. Barium
chloride (BaCl2) is a commonly used chemical agent that causes
muscle depolarization and myofiber death by stimulating exocytosis
while blocking the Ca2+
efflux (DalleDonne et al.; 1998). p21scko mice
were treated with daily tamoxifen injections (Tam) for eight days as
described above. On day 0, TA muscles of p21scko mice were injected
with 35 µl of a 1.2% BaCl2 solution; vehicle-injected p21scko mice
were used as controls (No Tam). Each group was composed by 3 mice. TA muscles were collected at 0, 5, 10, and 15 days from injury and analyzed by H&E staining (Fig 5.3). Preliminary data had shown an accelerated muscle regeneration in Tam compared to No Tam mice but the following experiments did not confirm this data. Together, these results suggest that, after injury, the absence of p21 in SC does not produce any effects on muscle regenerative capability.
II part
To investigate which impediment underlies myotube replicative inability, we used XEE to drive in vitro replication assays. These extracts contain all the factors necessary to replicate DNA in any form (Tutter AV, et al 2006). Accordingly, the self-sufficient XEE replication machinery should ignore biochemical defects underlying incomplete DNA replication in myotubes. Thus, if an incomplete DNA replication still exists in XEE condition it would represent a strong indication of the presence of structural obstacle(s).
Nuclei, isolated from proliferating (P) and quiescent (Q) myoblasts or myotubes, were incubated in XEE to quantify the amount of synthesized DNA during a period of 5 hours. Q nuclei represent the most appropriate control, since they come from a synchronous
population lying in G0 phase, like skeletal muscle TD cells. P nuclei
already gone through S phase in vivo and cannot re-replicate their DNA, thus P nuclei, on average, cannot attain 100% replication. Myotube nuclei started DNA replication more than 60 minutes later than Q ones. Furthermore, the myotube nuclei, after 5 hours of XEE incubation, replicate less than 50% of their genome, unlike the quiescent nuclei that reach almost complete replication already at 3 hours (Fig. 5.4).
Figure 5.4 | Myotube nuclei do not completely replicate their DNA in XEE. Quantification of replicated DNA (per nucleus) in nuclei isolated from myotube and quiescent or proliferating myoblast. Nuclei were incubated in XEE in presence of dTTP3H to follow replication kinetics. Aliquots were collected at indicated time points. The amount of radioactivity per sample is a measure of the replicated DNA; In parallel, the same analyses were performed with a fluorescent tracer (Cy3-dCTP), to visualize the percentage of nuclei replicating their DNA (typical range, 70- 90%). Radioactive measurements were normalized to the number of Cy3-dCTP+ nuclei and were plotted as percent of maximal DNA synthesis. Values are shown as means of three independent experiments with SE (error bars). At all time points, the differences between myotube and quiescent cell nuclei were statistically significant (p < 0.05 by Student’s test).
These data reveal that the inability of myotubes to complete DNA replication is due, at least in part, to structural obstacles.
embedded in the nuclear structure, myotubes and myoblasts nuclei were purified and treated with 0,5 M NaCl. High salt concentration is used to preferentially detach proteins not tightly bound to DNA, keeping histones in their place. To prove the results of such selective detachment, we performed an immunofluorescence to histone H1, the histone most weakly bound to DNA, and to pRB, one of the non- histone proteins most strongly attached to DNA (Fig. 5.5). The presence of histone H1 and the absence of pRB confirmed a successful preparation.
The following results were obtained by studying the replication kinetics in XEE of salt-treated nuclei, using microfluorimetry to quantitate DNA.
Figure 5.5 | High salt treatment detaches pRB protein from DNA maintaining H1 bound to chromatin. Immunofluorescent for pRB (green) and H1 (red) proteins. DAPI in blue.
NaCl-treated nuclei from proliferating myoblasts displayed similar percentages of DNA replication in comparison with nuclei from proliferating myoblast without salt, as well as nuclei from myotubes with salt have similar DNA replication of nuclei from myotubes without salt (Fig. 5.6). The data obtained suggests that the treatment with salt does not eliminate the differences between the nuclei of proliferating and terminally differentiated cells.
To determine the molecular impediment that hinders replication, we conceptually subdivide the nucleus into different levels of complexity: naked DNA, DNA arranged into nucleosomes and intact chromatin. To identify at which of these levels this obstacle lies, we thought to study replication kinetics in XEE of the different preparation of genetic material from myotubes and myoblasts (Fig. 5.7).
Figure 5.6 | Salt treated nuclei from myotubes and proliferating cells show different replication kinetic in XEE. Nuclei were extracted from the above mentioned cells. They were treated with 0,5M NaCl and then incubated in XEE. Aliquots were collected at indicated time points. DAPI fluorescence intensity proportional to DNA content was misurated. Untreated nuclei from myoblasts and myotubes were used as controls. Values are shown as means of two independent experiments with SD (error bars). Proliferating msc: myoblast nuclei; Myotubes: myotubes nuclei; Proliferating msc+0,5M NaCl: myoblast nuclei treated with 0,5M NaCl; Myotubes+0,5 M NaCl: myotube nuclei treated with 0,5M NaCl.
The incubation of naked DNAs from myoblasts and myotubes in XEE produced a similar amount of DNA synthesis (Fig. 5.8). So, at the level of naked DNA, there is no barrier prohibiting full DNA replication.
Figure 5.7 | Schematic illustration of different levels of complexity of nucleus
Figure 5.8 | Myotubes necked DNA replicate as much as proliferating samples in XEE. Densitometry of replicated DNA from myotubes and proliferating myoblast. Necked DNA was incubated in XEE in the presence of biotinylated-dUTP to follow replication kinetics. Aliquots were collected at the indicated time points. Dot blot, incubation with streptavidin-peroxidase and chemiluminescent analyses were performed. Mytubes: Myotubes DNA; proliferating myoblast: myoblast DNA. Values are shown as means of three independent experiments with ESs (error bars). At all time points the differences between myotubes and proliferating myoblasts were not statistically significant (p > 0.05 by Student’s test).
The next level of complexity was represented by DNA + histones. To obtain this genetic material, during the preparation, we have used high concentration of NaCl, like that used for nuclei (0,5 M). Incubating DNA + histones from myoblasts or myotubes in XEE, the percentage of DNA replication is similar between the two samples, to any points considered. This means that myoblasts and myotubes do not have differences neither at the level of DNA sequence, nor of histone proteins.
Figure 5.9 | DNA+histones from myotubes replicates as much as proliferating samples in XEE. Densitometry of replicated DNA+histones from myotubes and proliferating myoblast. DNA+histones was incubated in XEE in the presence of biotinylated-dUTP to follow replication kinetics. Aliquots were collected at the indicated time points. Dot blot, incubation with streptavidin-peroxidase and chemiluminescent analyses were performed. Values are shown as means of six independent experiments with ESs (error bars). At all time points the differences between myotube and proliferating DNA+histones were not statistically significant (p > 0.05 by Student’s test)
The last level that we have analyzed is the intact chromatin. Using the same protocol for the extraction of DNA + histones but reducing the concentration of NaCl from 0,5 M to 150 mM, we obtained an intact chromatin. In this case the XEE assay revealed a similar DNA replication after 1,5 h of incubation, but the newly replicated DNA was increased after 3 h of incubation in myoblasts versus myotubes.
This suggests that a possible structure obstacle exists, and it seems to be associated to non-histone proteins.
Figure 5.10 | Intact chromatin from myotubes do not completely replicate DNA in XEE. Densitometry of replicated intact chromatin from myotubes and proliferating myoblast. Intact chromatin was incubated in XEE in the presence of biotinylated-dUTP to follow replication kinetics. Aliquots were collected at the indicated time points. Dot blot, incubation with streptavidin-peroxidase and chemiluminescent analyses were performed. Values are shown as means of three independent experiments with ESs (error bars). (p =0.06 by Student’s test).
5. DISCUSSION
Our laboratory has shown that acute p21 ablation in the context of intact skeletal muscle tissue leads to intense proliferation of multiple cell types (Biferi et al.; 2015). However, it remains unclear if this process is driven by SC alone or other cell types are necessarily involved. In the first part of my Ph.D. work, I have shown that the deletion of p21 exclusively in SC is not sufficient to trigger cellular proliferation. Hence, I suppose that other interstitial cells concur with SC in determining the previously described phenotype.
SC are primarily a quiescent cell population in adult skeletal muscle. To support repair and regeneration, these cells, after traumatic muscle injury, undergo a few rounds of cell division. The results of a preliminary experiment showed a faster regeneration in mice deleted of p21 but the following experiments have shown very similar, complete muscle regeneration 15 DPI in both tamoxifen-treated and control mice (Fig. 5.3). Thus, the absence of p21 in satellite cells have no effect during the regeneration process.
I wish to underline that our experimental model has a high degree of flexibility and allows to induce p21 KO in various tissues. In future developments, by crossing the p21 KO mice with animals expressing Cre under the control of appropriate, tissue-specific promoters, it will be possible to delete p21 in specific organs and tissues. This should allow to probe the applicability of cell cycle stimulation to heal, regenerate, and improve organs other than muscles.
The second purpose of my thesis was to understand if does exist a structural impediment to full DNA replication in myotubes. In the presence of all components necessary for DNA synthesis, provided by XEE, myotube nuclei replicate only half of their genetic material (Fig 5.4). The treatment with salt does not eliminate the differences between the nuclei of proliferating and terminally differentiated cells (Fig. 5.6). To determine the molecular impediment that hinders replication, we conceptually subdivide the nucleus into different
levels of complexity: naked DNA, DNA arranged into nucleosomes and intact chromatin.
The analyses on naked DNA showed that there are no differences at this level between myoblasts and myotubes. This result reveals that terminal differentiation process does not introduce any chemical modifications in the DNA molecules able to impair DNA synthesis (Fig. 5.8). Rising the level of complexity to DNA wrapped with histones helped us to verify if DNA replication defects lay in the nucleosomes. The obtained results demonstrated that also at this level no differences were measured (Fig. 5.9). Finally, studying intact chromatin, potential alterations at the level of accessory proteins between myotubes and myoblasts are emerged (Fig. 5.10). Future studies of proteomic analysis will be performed by mass spectrometry. The goal is to identify the factors that will enable terminally differentiated cells to replicate and then to proliferate.
6. MATERIALS AND METHODS
Generation of satellite cell-specific p21 knockout mice. Through a
CRISPR/Cas9 strategy, we have generated conditional p21 KO mice, by encompassing exon 1 of the p21 gene within two LoxP sites
(referred as to p21f/f mice). Satellite cell-specific p21 knockout mice
(referred as to p21scko) were generated by crossing Pax7CreER mice
(obtained from Dr. Ombretta Guardiola CNR, Naples) with p21 floxed mice (p21f/f mice). All mice were in the C57BL6 background and their genotype was determined by PCR from tail DNA. To detect the presence of two lox-sites, flanked the exon 1 of the p21 gene, the primers used were:
fwd AACTCAGAAATCCGCCTGCCTCTG rev GTGTGAACATCCCAAATA CGT GTG.
To genotype Pax7CreER mice we used these primers: fwd CCACACCTCCCCCTGAACCTGAAACATAAA rev GAATTCCCCGGGGAGTCGCATCCTGCGG.
For conditional p21 knockout in satellite cells, p21scko mice were
injected intraperitoneally with 60 µg of tamoxifen (TMX) (Calbiochem) per day per g of body weight for 8 days to induce Cre-
mediated deletion. p21scko mice not subjected to injection of
tamoxifen were used as control.
SC isolation. SC were isolated from tamoxifen-treated or not-treated
p21scko adult mice. Fore-limb and hindlimb muscles were dissected, mechanically cut, and enzymatically digested at 37°C under constant shaking with 10 ml/limb of 0,1 mg/ml Collagenase I (Sigma-Aldrich) for 30 min. After a centrifuge at 50 x g, the supernatant was removed and the pellet resuspended in 10 ml of 1 mg/ml Collagenase/Dispase (ROCHE) and incubated in water bath at 37 °C for 30 min on rocker.
An equal volume of plating media (Ham plus 20% FBS, 3% chick embryo extract, bFGF 2,5ng/ml, 5% Penicillin/Streptomycin) was added, and all the solution passed through a 40 µm filter. After a centrifuge at 470 x g for 10 min, the supernatant was aspirate and the pellet was resuspended in plating media and preplated in 100 mm petri dishes for 1 hr. After pre-plating, the non-adherent, myoblast enriched population was collected and plated in dishes collagen coated.
Animals and skeletal muscle injury. Ministerial decree n° 12/2005
of the 4th of February 2005 authorized the central animal breeding
department of “Istituto Superiore di Sanità” to the GMO facility. Mice were housed in individual cages in an environmentally controlled room (23 °C, 12-h light–dark cycle) with ad libitum
access to food and water (authorization n° 92/2016-PR, of 29th of
January 2016). Animal care followed the directives of the EU (86/609/EEC). The experimental protocol was approved by the National Committee of the Italian Ministry of Health. Replacement, Reduction, Refinement were followed. All experiments were performed on 8– 10-week-old male (24–28 g) and female mice (21– 25 g). Mice of different sex were randomly assigned to experimental groups. For muscle injury experiments, 35ul of 1.2% BaCl2 (Sigma Chemical Co.) dissolved in PBS was injected into the TA muscle of mice, before anesthetized with Avertin. At various time points, TA muscle was collected from mice for biochemical and histology studies. For this study were used 80 mice.
Histology. Tissues from mice were dissected and frozen immediately
in an O.C.T. (optimum cutting temperature) compound. Frozen muscles were cross-sectioned (9 um) using a Leica CMV 1860 UV cryostat. The slides were subjected to histological H&E staining.
DNA extraction. SC isolated from tamoxifen-treated or not-treated
p21scko adult mice, after three weeks in colture, were centrifuge at 300 x g for 5 min. After the centrifuge the supernatant was aspirate and the pellet was resuspended in PBS with 0.2 mg/ml RNAase and 0,2%
Tween 20 for 1 hour at RT. After a centrifuge at 300 x g for 5 min, the pellet was resuspended in a lysis buffer (1M NaCl, 10mM EDTA pH=8, 50mM Tris-HCl pH=8, 0,5% SDS, and 0,4 mg/ml proteinase K) and incubated ON at 37 °C. Sample was subjected to the classical phenol/chloroform extraction, followed by precipitation in ethanol for 1h at -80°C. Precipitates were washed with 75% of ethanol, dried and resuspended in distillated water.
RNA isolation and quantitative qPCR. qPCR was performed
starting from RNA extracts obtained from muscle tissues homogenized and extracted through Direct-zol RNA Mini Prep-Plus (Zymo, R2070) following manufacturer’s instructions. After treatment with DNase I (New England BioLabs), RNA quality was verified by gel electrophoresis. RNA (1 μg) was retro-transcribed to complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies) and 10ng of cDNA was used for each sample. Gene expression was quantified by comparative CT method, using GAPDH as a reference. Primers used are:
p21 fwd TGAGCGGCCTGAAGATTCCC
p21 rev GATAGAAATCTGTCAGGCTGGTCTGC
GAPDH fwd CCTCGTCCCGTAGACAAAATG GAPDH rev TCTCCACTTTGCCACTGCAA
p27 fwd CTCGTCAGACAATCCGGCTG
p27 rev GAAGAATCTTCTGCAGCAGGTCG
p57 fwd GGGTGTCCCTCTCCAAACGT
Cell culture. Muscle satellite cells (MSC) were isolated as described
above. The MSC were grown on gelatin-coated dishes in growth medium (GM): Ham’s F-10 medium (Life Technologies) supplemented with 20% fetal bovine serum (FBS), 3% chicken embryo extract, and 2.5 ng/ml basic-FGF, antibiotics (10,000 units/ml of Penicillin and 10,000 g/ml of streptomycin, GIBCO). Differentiation was induced by plating cells on gelatin-coated dishes in differentiation medium (DM): Dulbecco’s Modified Eagle Medium (Life Technologies) supplemented with 10% FBS and antibiotics. After 48 hours, most of the cells was differentiated (differentiation index higher than 95%).
MSC were made quiescent by culturing them in suspension on semisolid medium (GM with 0.7% Noble Agar (Life Technologies) for 22 hrs. Cell cycle reentry was triggered by replating them on gelatin-coated dishes in liquid GM.
The plates were kept at room temperature (38°C) and at 6.7% of CO2. The differentiated cells were detached from the culture plate by treatment with 1X trypsin (Gibco) for 2 minutes at 37°C.
Immunofluorescence. Immunofluorescence was performed on cells
fixed with 4% formaldehyde and permeabilized with 0.25% Triton X-100 (Sigma Aldrich). Primary mouse monoclonal antibodies were used to detect: pRB (1:100 Neo Marker) and H1 (1:100 Upstate); AlexaFluor 488- or 594-conjugated secondary antisera to mouse IgG (Life Technologies) were used. Nuclei were counterstained with 0.2
g/ml DAPI for 20 min, after immunofluorescence.
Preparation of Xenopus laevis interphase egg extracts (XEE).
XEE were provided in the context of a collaboration with Dr. Vincenzo Costanzo. Female frogs were injected with 300U and 600U of Human Chorionic Gonadotropin respectively 24h and 16h before eggs collection and let in a 0.1M NaCl bath. After collection eggs were dejellied by several washes with the Dejellying Buffer (20mM Tris-HCl pH 8.5, 110 mM NaCl, 5 mM DTT) and then rinsed 3 times with MMR buffer (5 mM Hepes-NaOH pH7.5, 100 mM NaCl, 0.5 mM KCl, 0.25 mM MgSO4, 0.5 mM CaCl2,
25 μM EDTA). Dejellied eggs were then incubated with 2 µl of
10 mM Calcium ionophore A23187 (Sigma). After 5-
10 minutes eggs were washed three times with MMR buffer and then rinsed twice with ice-cold S buffer (50 mM Hepes-NaOH pH 7.5, 50 mM KCl, 2.5 mM MgCl2, 250 mM sucrose) freshly
supplemented with 2 mM β-mercaptoethanol and
15 μg/ml Leupeptin. Activated eggs were packed by centrifugation (few seconds at 3300 x g in a microcentrifuge) to get rid of excess buffer and then crushed by centrifugation for 10 minutes at 16100. Crude extract was separated from yolk and insoluble material, supplemented with 40µg/ml of Cytochalasin B and then ultracentrifuged for 18 min at 70000 rpm at 4°C with a TLA100 rotor (Beckman). The final extract, obtained by mixing the clarified protein extract and the membranes fraction, was supplemented with 200 μg/ml Cycloheximide (Calbiochem).
Nuclei preparation. Cells were collected and centrifugated to obtain
a pellet. The pellet was resuspended with IPO Buffer (10 mM K- Hepes (pH 7.5), 2 mM KCl, 2mM MgCl2,1 mM DDT, 1 mM PMSF, 1 mM protease inhibitors)). Samples were incubated for 60 min on ice and then centrifuged at 250 g for 4 min at + 4°C. To eliminate cellular debris, cells were incubated at room temperature (2 min for myoblasts and 4 min for myotubes) with trypsin (Gibco, 0.0025% and 0.00375% respectively for the myoblasts and myotubes). At the end of the process, nuclei were permeabilized with 0.2% Triton X- 100 for 5 min. After centrifugation at 250 g for 3 min at + 4°C, nuclei were washed with ISO Buffer (10 mM K-Hepes (pH 7,5), 25 mM KCl, 2 mM MgCl2, 75 mM sucrose, 0,01% of NP-40 (nonyl phenoxypolyethoxylethanol)) and centrifuged at 900g at + 4°C for 3 min. Nuclei were counted (throught Burker chambers) and supplemented with 10% DMSO (dimethylsulfoxide). They were, then, stored at -80°C.
Cell fractionation. Msc and Mt cells were fractionated by
differential centrifugation as described previously with some minor modifications (Dimauro et all., 2012; Fittipaldi at all., 2015). Briefly, cells were homogenized in the presence of STM buffer (250 mM sucrose, 50 mM Tris–HCl, pH 7.4, 5 mM MgCl2) and protease and
phosphatase inhibitor cocktails. The homogenate was maintained on ice for 30 min, passed 10–20 times through a 27-gauge needle, vortexed at maximum speed for 15 s, and then centrifuged at 800 g for 15 min. To obtain the nuclear fraction, the pellet was rehomogenized in STM buffer and centrifuged (500 g at 4 °C for 5 min). To increase nuclear fraction purity, after supernatant removal, STM was further added to the nuclear pellet, passed 10–20 times through a 27-gauge needle vortexed at maximum speed for 15 s, and then centrifuged at 1000 g for 15 min. The obtained pellet was resuspended in NET buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 150mM or 0,5M NaCl, 20% glycerol, 1% Triton-X-100, and protease and phosphatase inhibitors), vortexed at maximum speed for 15 s, and incubated on ice for 30 min. To increase the purity of lysed nuclei, the homogenate was passed 10–20 times through a 27-gauge needle and centrifuged at 9000 g for 30 min to remove the buffer.
Pellet was resuspended in PBS with 0,9mM CaCl2 and 0,5mM MgCl2
and sonicated at 10% for 50 sec, 25% for 70 sec (x2), 35% per 20%, 50% for 30sec with 30 s pauses while being kept on ice throughout. The lysate was centrifuged at 9000 g for 30 min (at 4 °C) and quantified with Picogreen.
Replication assay in XEE. The genomic material (nuclei, DNA or
chromatin) was added to a mix (replication mix) consisting of:
30l XEE
2l Energy Mix (20x; 0,2 mg/ml Creatine
phosphokinase (Sigma), 2 mg/ml Phosphocreatine (Sigma), 20 mM MgCl2, 2 mM EGTA)
2l dTTP-3H (Perkinelmer, 1mM)
The Mix was incubated at 23°C for the time of interest.
DNA replication quantification in XEE. 16 l of Stop Buffer (80
mM of Tris-HCl (pH 8), 8 mM di EDTA (pH 8), 0,1% H3PO4, 5% SDS, 50% Glycerol, 0.025% Blue-Bromofenolo, 1 mg/ml Proteinasi
K, 200 μg/ml Rnase) were added to 4l from the replication mix (see
‘replication assay in XEE’). The samples were incubated overnight at 37°C.
For radioactive material: the samples were transferred on glass microfilters (Whataman GF/C 2.5). The filters were incubated with 5% cold trichloroacetic acid (TCA) and 2% cold inorganic pyrophosphate (PPi) for 20 min at +4°C. After 4 washings in 5% cold TCA, the samples were treated with 100% ethanol and transferred to vials with liquid scintillation (Optifluor, Perkinelmer). Radioactivity (index of the quantity of DNA replicated) was evaluated with Betacounter (Beckman). The protocol of Menut et al., 1999, was used to quantity the DNA replicated.
For biotinylated material: the samples were transferred on a membrane (Optitrain BA-S 85”, 0,45 μm) through a dot blot microfiltration apparatus (BIO-RAD). The membrane was washed with 0,16M HCl for 3 times. The membrane was crosslinked (+80°C for 2 hours) and incubated with 1% Bovine Serum Albumin (BSA, Sigma) for 1hour. Streptavidin-Peroxidase from Stepromyces avicinii (Sigma, 1:10.000) coupled to horseradish peroxidase (HRP) was used to detect biotinylated material. The membrane was later treated with WesternBright ECL and visualized through the ImageQuant Las 4000 (EG Healthcare). The densitometric analyses were made with ImageJ (NIH software).
Chromatin replication quantification in XEE. 40 l of Stop
Buffer (0,1M of Tris-HCl (pH 8), 0,5 % IGEPAL, Tween 20 (0,5%), 8 mM
added to 8l from the replication mix (see ‘replication assay in XEE’). The samples were incubated overnight at 37°C.
For biotinylated material: the samples were transferred on a membrane (Hybond-N+, 0,45 μm) through a dot blot microfiltration apparatus (BIO-RAD). The membrane was dried for 30 min and incubated with 1% Bovine Serum Albumin (BSA, Sigma) for 30min.
Streptavidin-Peroxidase from Stepromyces avicinii (Sigma,
1:10.000) coupled to horseradish peroxidase (HRP) was used to detect biotinylated material. The membrane was later treated with WesternBright ECL and visualized through the ImageQuant Las 4000 (EG Healthcare). The densitometric analyses were made with ImageJ (NIH software).
Visualization of replicating nuclei in fluorescence. 1l of Cy3-
dCTP (GE Healthcare, 25nmol) (mixed to the replication mix) was
used to follow the replication kinetics. At different time points 5l
from the samples were taken and mixed with 5l of XEE fixative (3x
Buffer A (45 mM Pipes pH 7,2 + 45 mM NaCl + 240 mM KCl), 4% formaldehyde, 0,02 mg/ml DAPI, 50% glycerol). Samples were transferred on cover slips. Nuclei were visualized using an Axioskop 2 Plus fluorescence microscope. The percentage of nuclei incorporating Cy3-dCTP was counted.
DNA quantitation. Microfluorimetry was used to quantitate DNA
in myotube and myoblast nuclei stained with DAPI. Random nuclei images were captured at room temperature, using an Axioskop 2 Plus fluorescence microscope (Carl Zeiss) equipped with a Neofluar 40X objective (NA 0.75), and an AxioCam camera (Carl Zeiss). Images were capured with the ZEN 2012 software (Carl Zeiss). The images were analyzed with the public domain software ImageJ (v. 1.43u). DAPI fluorescence intensity (proportional to DNA content) was measured in all well-defined individual nuclei in the captured images (about 100 nuclei for each point), with local background subtraction. The average of fluorescence intensity for each sample was calculated. In each experiment, the fluorescence intensity of control Msc nuclei was
made equal to 100% and the percent in DNA content in the other samples was calculated.
Salting out with NaCl. Nuclei were treated with 0,5 M NaCl for 10
min. To remove NaCl, samples were washed with ISO Buffer (see ‘Nuclei Preparation’) and centrifugated at 250 g for 3 min.
Data Analysis. Analyses were conducted with Student’s t test, with
a one-tail distribution. Experimental data are represented as mean±ES. Comparisons with p values < 0.05 were considered significant.
REFERENCES
Abmayr, S. M. & Pavlath, G. K. (2012). Myoblast fusion: lessons from flies and mice. Development 139: 641–656
Alcorta, D.A., Xiong, Y., Phelps, D., Hannon, G., Beach, D., and Barrett, J.C. (1996). Involvement of the cyclin- dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A 93, 13742-13747.
Agarwal, M.L. , Agarwal, A. , Taylor, W. R. and Stark, G.R. (1995). p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc. Natl. Acad. Sci. USA. 92, 8493-7
Biferi M.G., Carmine Nicoletti, Germana Falcone, Eleonora MR Puggioni, Nunzia Passaro, Alessia Mazzola, Deborah Pajalunga, Germana Zaccagnini, Emanuele Rizzuto, Alberto Auricchio, Lorena Zentilin, Gabriele De Luca, Mauro Giacca, Fabio Martelli, Antonio Musio, Antonio Musarò and Marco Crescenzi (2015). Proliferation of Multiple Cell Types in the Skeletal Muscle Tissue Elicited by Acute p21 Suppression. The American Society of Gene & Cell Therapy 23: 885–895.
Blow JJ, Laskey RA. (1988). A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature 7;332(6164):546-8
Blow J. and Ronald A. Laskey. (2016). Xenopus cell-free extracts and their contribution to the study of DNA replication and other complex biological processes. Int. J. Dev. Biol. 60: 201-207
Camarda, G., Siepi, F., Pajalunga, D., Bernardini, C., Rossi, R., Montecucco, A., Meccia, E., and Crescenzi, M. (2004). A pRb- independent mechanism preserves the postmitotic state in terminally differentiated skeletal muscle cells. J. Cell. Biol. 167, 417-423.
Chan, F. K., Zhang, J., Cheng, L., Shapiro, D. N., and Winoto, A. (1995). Identification of human and mouse p19, a novel CDK4 and CDK6 inhibitor with homology to p16ink4. Mol Cell Biol 15, 2682- 2688.
Charge, S.B.P. (2004). Cellular and Molecular Regulation of Muscle Regeneration. Physiological Reviews 84, 209–238.
Coller, H.A., Sang, L., and Roberts, J.M. (2006). A new description of cellular quiescence. PLoS Biol 4, e83.
Crescenzi, M., Soddu, S., and Tato, F. (1995). Mitotic cycle reactivation in terminally differentiated cells by adenovirus infection. J. Cell. Physiol. 162, 26-35.
Deng, C., Zhang, P., Harper, J.W., Elledge, S.J., and Leder, P. (1995). Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82, 675-684.
Dimauro, T. Pearson, D. Caporossi, M.J. Jackson, A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue, BMC Res Notes 5 (2012) 513.
Dulic, V., Drullinger, L.F., Lees, E., Reed, S.I., and Stein, G.H. (1993). Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E- Cdk2 and cyclin D1- Cdk2 complexes. Proc Natl Acad Sci U S A 90, 11034-11038
Dulic, V., Kaufmann, W. K., Wilson, S. J., Tlsty, T. D., Lees, E., Harper, J. W., Elledge, S. J., and Reed, S. I. (1994). p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 76, 1013-1023.
Ekholm, S. V., and Reed, S. I. (2000). Regulation of G(1) cyclin- dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 12, 676-684.
El-Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B., Parsons, R., Trent, J. M., Lin, D., Mercer, W. E., Kinzler, K. W., and Vogelstein, B. (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817-825.
Endo, T., and Nadal-Ginard, B. (1998). Reversal of myogenic terminal differentiation by SV40 large T antigen results in mitosis and apoptosis. J Cell Sci 111 (Pt 8), 1081-1093.