ContentslistsavailableatSciVerseScienceDirect
Journal
of
Molecular
Catalysis
A:
Chemical
j o ur na l ho me p ag e :w w w . e l s e v i e r . c o m / l o c a t e / m o l c a t a
Acid
catalyzed
alkylation
of
phenols
with
cyclohexene:
Comparison
between
homogeneous
and
heterogeneous
catalysis,
influence
of
cyclohexyl
phenyl
ether
equilibrium
and
of
the
substituent
on
reaction
rate
and
selectivity
L.
Ronchin
∗,
A.
Vavasori,
L.
Toniolo
DepartmentofMolecularScienceandNanosystems,UniversityCa’FoscariofVenice,Dorsoduro2137,30123Venice,Italy
a
r
t
i
c
l
e
i
n
f
o
Articlehistory: Received1August2011 Receivedinrevisedform 28November2011 Accepted7December2011 Available online 16 December 2011 Keywords: Acidcatalysis Sulfonatedresins Phenolsalkylation Alkylationselectivity
a
b
s
t
r
a
c
t
Thereactivityofseveralphenolstowardliquidphasealkylationwithcyclohexeneinthepresenceof heterogeneousandhomogeneousacidcatalystat358Kisstudied.ThecomparisonbetweenAmberlyst 15andCH3SO3H,asexamplesofheterogeneousandhomogeneoussystems,showsahigheractivityof
theformerwithdifferentbehaviorofselectivitybetweenthetwosystems,anyway,inbothsystems O-alkylationandringalkylationsoccur.Aremarkabledifferenceintheselectivityofthering alkyla-tionbetweenheterogeneousandhomogeneoussystemsisobserved:Amberlyst15showsaconstant ortho/pararatiocloseto2,whileinthepresenceofCH3SO3Hortho/paraisvariablefrom3to5,
sug-gestinganinvolvementofthecyclohexylphenyletherrearrangement.Thisisprovedalsobyadirect relationshipbetweentheortho/pararatioandtheconcentrationofthecyclohexylphenyletherwhen CH3SO3Hisusedasacatalyst.Theformationofcyclohexylarylethersisreversible;onthecontrary,ring
alkylationappearsirreversible.Thereactivityofthedimethylphenolsshowsastronginfluenceofthe sterichindranceofthesubstituentontheelectrophilicattackofthecyclohexylcation,whichispoorly influencedbytheinductiveeffectofthemethylgroup.
© 2011 Published by Elsevier B.V.
1. Introduction
Organicindustrial processesemploy acidcatalyzed reactions
such as alkylation, acylation, isomerization, cracking, nitration,
condensation,esterification,etc.Thegreentechnologiesinorder
to replacethe traditional polluting mineralacid catalysts with
solidonesarecontinuouslyimproved[1–6].Zeolites,acidtreated
clays,ionexchangeresinsandsupportedacidsareinvestigatedby
severalresearchersfortheirapplicationinpharmaceutical,
per-fumery, agro-chemicals, dye-stuffs, intermediates and specialty
chemicalindustries[5–12].Alkylationreactionsinparticularare
really importantin theindustrialsynthesis ofmany largescale
productioncompounds[11,12].
Alkylationof phenolwithcyclohexene hasattracted
consid-erableinterest becauseofitsindustrialand academicrelevance
[13–21]. This reaction leads to a variety of products such as
4-cyclohexylphenol, 2-cyclohexylphenol, and cyclohexyl phenyl
etherdependingonboththecatalystandthereactionconditions.
Theuseof solid acidcatalystsappearsa suitable alternativeto
theusualproceduresinhomogeneousphasewithcatalystssuch
asAlCl3,BF3,TiCl4,HF.Onconsideringthecurrentefforttoward
∗ Correspondingauthor.Fax:+390412348517. E-mailaddress:[email protected](L.Ronchin).
processinnovationaimedtoavoidenvironmentalconcerns,
ion-exchangeresinsappeartobeidealcatalyststoconvertpolluting
processesintogreenerones[2–6].
Inalargenumberofindustrialprocessesthecation-exchange
resinsareusedasacatalystsuchasinMTBEorTAMEsynthesis,
themanufactureofalkylphenolsandbisphenolA,the
esterifica-tionofavarietyofcarboxylicacids,thehydrationofalkenes,the
dimerizationofisobutene,etc.[4–6,19,22,23].
Themechanismofacidcatalyzedalkylationiswellknownfor
alongtimeanditiswidelyacceptedthecarboniumionattackto
theelectronicrichcenterasthekeystepofthereaction[24,25].
Cyclohexeneinthepresence ofacidgivesthecyclohexylcation
asatransientspeciesthatreadilyreactswithanucleophilegiving
thecorrespondingcyclohexylderivative.Therearrangementtothe
morestablemethylcyclopentylcationoccursonlyinanegligible
extentsincetheskeletonrearrangementisslowerthanthe
nucle-ophilicattack,whichoccurs,formanynucleophiles,atencounter
[26].
ThestudyofRichardandcoworkersonthereactivityof
phe-nolasnucleophiletowardmethylphenylcarbocationshowedthat
therelativeratesforalkylationofphenolat OH,C-4andC-2are
230:20:1,respectively.Onthecontrary,thealkylationofthe
corre-spondingnucleophilicsitesofphenoxideion,whichisanencounter
reaction,showedtherelative rates of2:2:1 [27]. Otherauthors
pointedoutthattheselectivitytowardtheorthopositioninthe
1381-1169/$–seefrontmatter © 2011 Published by Elsevier B.V. doi:10.1016/j.molcata.2011.12.007
phenolalkylation is favored when theless hindered secondary
carboniumionsaretheelectrophiles,whiletertiarycarbocations
giveprevalentlyparaalkylation[28,29].ThestudiesofSharmaand
coworkers,carriedoutintheearlyninetyrelatingthereactivity
ofphenolinthepresenceofsulfonatedresins,pointed outthat
theortho–paraselectivityintheringalkylationofphenolisstrictly
relatedtothenatureoftheolefinemployed.Inparticular,propene
and1-butenegiveanortho–pararatiocloseto2,whileisobutene,
␣-methylstyreneand diisobutene give almost exclusivelypara
alkylation[29].Recently,BhattandPatelreportedthatsupported
12-tungstosilicicacidcatalyzesonlyringcyclohexylationofphenol
givinganortho–pararatiocloseto2[15].Morerecently,Hölderich
andcoworkersshowedthathighparaselectivityisobtainedinthe
alkylationofphenolwithisobuteneinthepresenceofcatalysts
withLewisorBrønstedacidsites,indifferently.Theselectivityisnot
sensibletothetypeofacidpresentinthecatalystbuttheactivity
isinfluencedbytheamountandthestrengthofthesites[30].
Thecomparisonofactivityandselectivitybetween
heteroge-neousandhomogeneousBF3/SiO2 and BF3·(H2O)2 catalystswas
studiedbyClarkandcoworkers[21].Theypointedoutthat
cyclo-hexylphenyl ether, and cyclohexyl phenols are formed in the
presenceofbothsystems,butonlybythehomogeneousBF3·(H2O)2
asacatalyst therearrangementoftheether toalkylphenols is
observed[21].YadavandKumarhaverecentlystudiedthekinetics
ofphenolcyclohexylationcatalyzedbydifferentsolidacids,which
catalyzetheformationofphenylcyclohexyletherandthe
prod-uctsof ringalkylationina ortho–pararatiocloseto2 [14].The
mechanisticaspectoftheelectrophilicattacktothephenolis
inves-tigatedfromatheoreticalpointofviewbyTangandcoworkers.
Theseauthorssuggestedthattheadditionofthesulfonicacidto
theolefinsoccursleadingtotheformationofasulfonicester
inter-mediate,which,inturns,reactswithphenoltoformtheproducts
ofalkylation[31].
Inthispaperwestudythecyclohexylationofsomephenolsand
thereactivityofcyclohexylphenyletherinthepresenceofboth
CH3SO3Handsulfonicresins.Inparticular,weinvestigatetherole
ofthecyclohexylphenyletherontheortho–paraselectivityandthe
reactivityofthedimethylphenolsinordertoaccountforthesteric
hindranceofthemethylgroupsontheelectrophilicattackofthe
cyclohexylcation.
2. Experimental
2.1. Materials
Reagentsandsolventswereusedafterpurificationofthe
com-merciallyavailablesamplesandtheirpuritywascheckedbythe
usualmethods(meltingpoint,TLC,HPLC,GCandGC–MS).The
sol-ventsweretreatedinadoublebedcolumn,filledwithH2SO4/SiO2
andSiO2toadsorbwaterandimpurities.Theresidualwatercontent
wascheckedbyHPLCanalysis[32].Commercialcatalysts:
macro-reticularsulfonatedstyrenedivinylbenzeneresinsAmberlyst15TM
andAmberlyst36TM(atrademarkofRohmandHaas)were
pur-chasedfromAldrich.
2.2. Reactions
The reactions and the kinetic runs were performed in a
stirred glass reactor thermostatted by a circulation bath at
358K,containingweighedsamplesofsolvent,reagentsand
cat-alyst at autogenous solvent pressure (122 and 158kPa for 1,2
dichloroethaneandbenzene,respectively).Inatypicalexperiment
10mLofsolutioncontaining10mmolofphenol,10mmolof
cyclo-hexeneplus5mmolofmethylcyclohexaneasinternalstandardand
thedesiredamountofcatalyst(100–500mg)wereplacedinthe
reactor.Alltheoperationswerecarriedoutintoagloveboxinorder
tominimizecatalystdeactivationbyairmoisture.Smallamounts
ofthesolutionweredrawnatdifferenttimesandthesampleswere
analyzedbyGC,andGC–MSusingaHP5capillarycolumn(300m
i.d.30mlong,95%methyl,5%phenylsiliconephase).Thesamples
werecheckedalsobyHPLCusingaPerkinElmerapparatusanda
Lichrosphere100(RP-18,5m)column.Thefirstderivativeattime
0ofathirdorderpolynomialfunction,obtainedbyfitting
cyclohex-eneconcentrationvs.timeat10%ofconversion,gavetheinitialrate
ofreaction.
Forareliablecomparisonoftheperformancesofdifferent
cat-alystsitisessentialtoknownifreactionratedataareaffectedby
diffusionphenomena.Thisisverifiedbystudyingtheinfluenceof
thegranulometryandofthecatalystamountonthereactionrate
catalyzedbythemostactivecatalyst(Amberlyst36)at373K.The
experimentalevidencessuggestthatthekineticsisnotinfluenced
bydiffusionphenomena,sincetherearenodifferencesinthe
ini-tialrateusingresinswithdifferentgranulometryandtheinitial
reactionratesarestrictlyproportionaltothecatalystamount.In
addition,theinspectionofCarberryandWheeler-Weisznumbers
showsvalueslowerthan0.1and0.4,respectively[33].
3. Resultsanddiscussion
3.1. Influenceofsolventandcatalystonreactionrate,conversion
andselectivity
Table1reportstheactivityoftwosulfonatedresinsinthe
alkyla-tionofphenol.Maximumyield,asreportedinTable1,iscomprised
between20and26%at40–50%ofconversion.DespiteofAmberlyst
36promotesainitialrateofreactionhigherthanthatofAmberlyst
15,thelattergivesthehighestyieldintheether.Asamatteroffact,
Amberlyst36(5.5meq.H+g−1cat)showsahigheractivitythanthe
Amberlyst15(4.7meq.H+g−1cat),thisislikelyduetothehigher acidcontentoftheformer.Infact,theactivitiesofthetwocatalysts
arequitesimilarconsideringtheinitialturnoverfrequencyreferred
tothewholeH+sites(Table1).CH
3SO3H(inhomogeneousphase)is
theleastactivecatalystanditsTOFis20timeslowerthanthatofthe sulfonicresins,likelyduetothehigheracidityofthelatter[34].As
amatteroffact,byconsideringp-toluensulfonicacidassimplified
modelforthesulfonicresins,thepKaofthep-toluensulfonicacidsis
2.7pKaunitslowerthanthatofmethanesulfonicacid(−4.7and−2,
respectively)[35].InthepresenceofAlCl3thereactionisfaster,but
thecomparisonoftheactivitiesofsulfonatedresins,
methanesul-fonicacid(proticacids)andAlCl3(Lewisacid)iscumbersomedueto
thedifferentnatureoftheacidsite.Itisnoticeablethatthe
reactiv-ityoftheorthoandparapositionsofphenolisnotinfluencedbythe
typeofsulfonicresinsemployed.Ortho-andpara-positionsofthe
phenolshowsimilarrelativereactivitygivingortho–pararatio ∼= 2
ineitherbenzeneor1,2-dichloroethane.Incontrast,anot
negligi-blesolventeffectseemstobeplayedbynitromethane,sincethe
initialreactionratesarealmostoneorderofmagnitudelowerthan
thosemeasuredinbenzeneand1,2-dichloroethane.Inaddition,the
ortho–pararatioclearlydiminishes(o/p ∼= 1.5),thussuggestingan
influenceofthesolventontheelectrophilicattack[36].
DespiteofthelargedifferenceofactivitybetweenCH3SO3Hand
AlCl3inhomogeneousphase,thereactionsshowasimilaro/pratio
(4.2and4.5),thussuggestingsimilarrelativereactionrateforeach
stageinthishomogeneousreactions.
Theconcentration–timeprofilesreportedinFigs.1–3,relativeto
thereactionsinthepresenceofAmberlyst15,CH3SO3HandAlCl3,
respectively,showdifferenttrends.Fig.1reportsthereaction
cat-alyzedbyAmberlyst15.Itappearsthatcyclohexylphenyletheris
atransientspecies,whichisalmostcompletelyconsumedatthe
Table1
Alkylationofphenol:selectivityafter240minofreactionat358K.Runconditions:phenol1.1molL−1,cyclohexene1.1molL−1,catalyst400mg,reactionvolume10mL.
Catalyst Conv.(%) r0a TOFb Ethermaximum
yield(%)
Selectivityc(%) o/p
2-Cyclohexylphenol 4-Cyclohexylphenol Dicyclohexylphenols Cyclohexylcyclohexene Ratio Benzene Amb.15 42 15 5.3 25 29 16 13 15 1.9 Amb.36 48 18 5.8 23 38 21 14 18 1.9 1,2-Dichloroethane Amb.15 51 18 6.8 26 29 14 15 12 2.0 Amb.36 64 22 6.9 23 31 16 18 15 1.9 CH3SO3Hd 21 3.5 0.34 17 10 2.4 2.3 2 4.2 AlCl3d,e 44 58 7.7 15 5 1.1 4.6 1 4.5 Nitromethane
Amb.15 16 3.8 0.81 10 24 16 Traces Traces 1.5
Amb.36 19 6.0 1.1 10 24 16 Traces Traces 1.5
a(105molL−1s−1gcat−1).
bInitialturnoverfrequency(104s−1)
c Productsintraceamount,astheisomersofalkylatedthecyclohexylether,hasbeenneglected. d Homogeneousreactions. eT288K,AlCl 31mmol. 1200 1000 800 600 400 200 0 0.0 0.2 0.4 0.6 0.8 1.0 1.2 cyclohexene phenol
cyclohexyl phenyl ether
2-cyclohexylphenol 4-cyclohexylphenol co nc en tr a tio n (M ol L -1 )
time (miinutes)
1200 1000 800 600 400 200 0 0.00 0.02 0.04 0.06
cyclohexycyclohexene
di-cyclohexylphenol
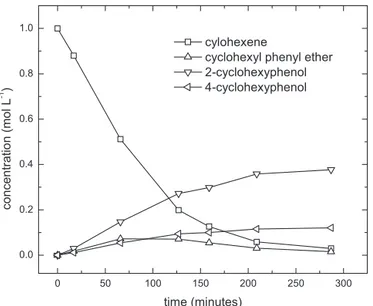
Fig.1.Reactionprofileofalkylationofphenolat358KcatalyzedbyAmberlyst 36.Runconditions:phenol1.1molL−1,cyclohexene1.2molL−1,catalyst400mg, solvent1,2-dichloroethane,reactionvolume10mL.
1200 1100 200 100 0 0.0 0.1 0.2 0.4 0.6 0.8 1.0 co n centr ation (mol L -1 )
time (minutes)
cyclohexene
cyclohexyl phenyl ether
2-cyclohexylphenol
4-cyclohexylphenol
cyclohexylcyclohexene
Fig.2.Reactionprofileofalkylationofphenolat358Kcatalyzedby methanesul-fonicacid.Runconditions:phenol1molL−1,cyclohexene1molL−1,catalyst400mg, solvent1,2-dichloroethane,reactionvolume10mL.
di-cyclohexyl phenols increasesmonotonically duringthe
reac-tioncourse.Formationofdi-cyclohexylphenolsisobservedalso
atverylowconversion,becauseofalkylphenolsarehighly
acti-vatedtowardtheelectrophilicattack[37].Under theconditions
usedthemainsidereactioniscyclohexenedimerization,whose
product(cyclohexylcyclohexene) showsanalmostlinear
mono-tonicincreaseduringreactioncourse.Itisnoteworthythatdimer
formationisstronglyinhibitedusingnitromethaneasthesolvent,
suggestinganinhibitingeffectofthesolventontheformationof
theelectrophile[36].
Fig.2showsthereactionprofileinthepresenceofCH3SO3H:
thereactionisalmosttentimeslowerthanthatinthepresence
ofAmberlyst15asacatalyst(Table1)andafter20hofreaction
all theproductsare still increasing, while theformation of
di-cylohexylphenolsisnegligible.Theconcentration–timeprofileof
phenolcyclohexylationcatalyzedbyAlCl3(Fig.3)evidencesafast
reactionalsoat288K(almosttwiceofthatmeasuredinthe
pres-enceofAmberlyst15at358K)butthereactioncompletelystops
after55min,withamodestconversionandwithanoticeableloss
ofthemassbalance.Suchabehaviorsuggestsafastcatalyst
deac-tivationduetotheformationofheavypitch,whichisconfirmedby
HPLCanalysis. 1200 1000 250 200 150 100 50 0 0.00 0.05 0.10 0.15 0.6 0.8 1.0 1200 1000 200 150 100 50 0 0.000 0.005 0.010 0.015 0.020 co nc e n tr a tio n ( m o l L -1) time (minutes) cyclohexene
cyclohexyl phenyl ether
2-cyclohexyphenol 4-cyclohexylphenol cyclohexylcyclohexene di-alkykphenol
Fig.3. ReactionprofileofalkylationofphenolcatalyzedbyAlCl3at288K.Run conditions:phenol1molL−1,cyclohexene1molL−1,catalyst140mg,solvent 1,2-dichloroethane,reactionvolume10mL.
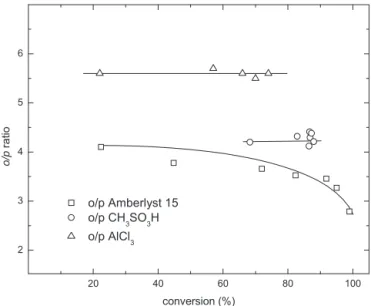
100 80 60 40 20 0 2 3 4 5 o/ p ra tio conversion (%) CH3SO3H Amberlyst 15 AlCl3
Fig.4.Comparisonofortho/pararatiovs.conversioninthecyclohexylationof phe-nolinthepresenceofliquidandsolidacidcatalysts.Runconditions:cyclohexene andphenol1molL−1,Amberlyst15andCH3SO3H1.8meqH+,AlCl310mmol,solvent 1,2-dichloroethane,reactionvolume10mLT358K.
InFig.4 thetrend oftheortho–pararatio vs. conversionfor
eachcatalyst isreported.EmployingAmberlyst15 asa catalyst
ortho–pararatioremainspracticallyconstant(1.9–2.1),whereas,
inthepresenceofCH3SO3H,monotonicallyincreasesfrom2.4to
4.2astheconversionincreases.AlsointhepresenceofAlCl3,the
ortho–pararatiovs.conversionincreasesfrom4.1to4.5.These
evi-dencessuggestadifferentnatureintheformationoftheorthoand
paraisomersasthecatalystnaturechange.Thisisconfirmedby
thetrendsoftheortho–pararatiovs.cyclohexylphenylether
con-centrationreportedinFig.5.Clearly,thereisalinearrelationship
betweentheetherconcentrationandtheformationoftheortho
iso-merinthepresenceofCH3SO3H,thussuggestingtheinvolvement
ofthecyclohexylphenyletherintheformationoftheorthoisomer,
likelyviarearrangement[37,38].Onthecontrary,inthepresenceof
Amberlyst15,thereisaverysmallinfluenceoftheconcentrationof
0.30 0.25 0.20 0.15 0.10 0.05 0.00 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 o/ p rat io
cylohexyl phenyl ether (mol L-1) o/p CH3SO3H
o/p Amberlyst 15 o/p AlCl3
Fig.5. Comparisonofortho/pararatiovs.cyclohexylphenyletherconcentration inthecyclohexylationofphenolinthepresenceofliquidandsolidacidcatalysts. Runconditions:cyclohexeneandphenol1molL−1,Amberlyst15,400mg,CH3SO3H 1.8meqH+andAlCl
31mmol,solvent1,2-dichloroethane,reactionvolume10mLT 358K. 1300 1200 1100 1000 200 100 0 0.00 0.05 0.10 0.15 0.20 1200 1000 200 0 0.00 0.02 0.04 0.06 0.08 0.10 co nc ent rat ion (mol L -1)
time (minutes)
cyclohexene
phenol
cyclohexyl phenyl ether
cyclohexylcyclohexene
2-cyclohexylphenol
4-cyclohexylphenol
di-alkylphenols
Fig.6.Reactivityofcyclohexylphenylether.Runconditions:T358KAmberlyst15, 400mg,solvent1,2-dichloroethane,reactionvolume10mL.
thecyclohexylphenylethertowardorthoandparaselectivity.Such
abehaviorsuggeststhat,inthepresenceofAmberlyst15,theortho
andparaselectivityismainlyinfluencedbythemesomericeffect
ofthehydroxylgroup[37–39].Besidesthereactioncatalyzedby
AlCl3showsasmallincreaseoftheortho/pararatiovs.cyclohexyl
phenyletherconcentration,butwithaneatprevalenceoftheortho
isomer.Thereasonsofsuchabehaviorarenotclearandatleast
twoeffectsmayconcurtogivethisresult:theetherrearrangement
andaspecificinteractionbetweenphenolandAlCl3,assuggested
bySartoriandcoworkers[40].
3.2. Reactivityofcyclohexylphenylether
Thereactivityofcyclohexylphenylether,inpresenceofacid
catalysts,isreportedinFigs.6–8andinTable2.Fig.6showsthe
concentration–timeprofileofthereactioncatalyzedbyAmberlyst
15:cyclohexenereachesamaximumafter2hofreaction,andthen
itdecreasestocompleteconsumption.Atthesametime,phenol
concentrationreaches toa maximum and subsequently
dimin-ishessmoothly(almosttoaplateau).Asamatteroffact,phenol
islessconvertedthancyclohexenebecauseallreactions,suchas
alkylation,dialkylationandcyclohexenedimerization,concurto
consumecyclohexene.Theinitialreactionrateof ether
decom-positioniscomparabletothatofthephenolcyclohexylationand
250 200 150 100 50 0 0.00 0.05 0.10 0.15 0.20 co nce n tr at io n ( m ol L -1 ) time (minutes)
cyclohexene
phenol
cyclohexyl phenyl ether
2-cyclohexyl phenol
4-cyclohexyl phenol
Fig.7.Reactivityofcyclohexylphenylether.Runconditions:cyclohexylphenyl ether0.21molL−1,T358K,CH3SO3H180mg,solvent1,2-dichloroethane,reaction volume10mL.
Table2
Reactivityofcyclohexylphenylether:selectivityafter240minofreactionat358K.Runconditions:cyclohexylphenylether0.2molL−1,solvent1,2-dichloroethane,reaction volume10mL.
Catalyst Conv.(%) r0a Selectivity(%) o/p
2-Cyclohexylphenol 4-CyclohexylPhenol Dicyclohexylphenols Ratio
Amberlyst15b 92 10 10 33 18 3.3 CH3SO3Hc 85 40 4 0.9 Traces 4.2 AlCl3d 74 75 19 3.4 22 5.6 a(105molL−1s−1g−1cat). bAmberlyst15,400mg. c CH 3SO3H180mg. d T288K,AlCl 30.2mmol.
consideringthewholereactionsteps,ringalkylationsarelikelythe
slowerstage,thusallowingaccumulationofphenoland
cyclohex-ene.InthepresenceofCH3SO3H(Fig.7)thereverseetherification
occurs with an initial reaction rate 4 times higher than when
Amberlyst15 isusedasa catalyst,and after50minofreaction
75%ofcyclohexylphenylether isconverted tocyclohexeneand
phenolin95%ofoverallselectivity.Infact,CH3SO3Hasacatalyst
doesnotgiveringalkylationproductsinhighyields,butallowsfast
decompositionoftheether.Fig.8showstheconcentration–time
profileofthereactivityofthecyclohexylphenyletherat288K,in
thepresenceofAlCl3,theinitialreactionrateismuchhigherthan
inthepresenceofAmberlyst15(7times)butitdoesnotreach
com-pletenessprobablybecauseofcatalystpoisoning,inagreementto
whatfoundinphenolalkylation.Inthiscase,theconcentrationof
cyclohexeneisnegligiblewithrespecttothatofphenol,duetoa
fastformationofcyclohexeneoligomerizationproducts,whichare
probablyoneofthereasonsofcatalystdeactivation.
Theortho/paraselectivity(seeTable2)withdifferentcatalysts
after4hofreactionisintherange3.3–5.6,and,asexpected,the
etherfavorstheortho-selectivity.Apparently,thetrendsoftheo/p
ratiovs.etherconversion,showedinFig.9,arenotinagreement
withthosefoundinthephenolcyclohexylation(Fig.5),because
bothAlCl3andCH3SO3Hshowaconstanto/pratio(5.3,4.3,
respec-tively),whileinthepresenceofAmberlyst15theo/pratiodecreases
from4.2to2.7.Despiteofthecomplexityofthereactionitisclear
thatcyclohexylphenyletherisinvolvedintheselectivityofthe
orthoandparaisomersofthealkylatedphenols,butfurther
inves-tigationsneedstohighlightsuchabehavior.
250 200 150 100 50 0 0.00 0.05 0.10 0.15 0.20 concent rat ion (mol L -1 )
time (minutes)
cyclohexyl phenyl ether
cyclohexene
phenol
2-cyclohexyl phenol
4-cyclohexyl phenol
Fig.8.Reactivityofcyclohexylphenylether.Runconditions:cyclohexylphenyl ether0.21molL−1,T358K,AlCl31mmol,solvent1,2-dichloroethane,reaction vol-ume10mL.
3.3. Reactivityofdimethylphenolsand2,4,6-trimethylphenol
Furtherinsightonthereactivityofthecyclohexylcationas
elec-trophiletowardphenolsmaybegainedbystudyingthereactivity
ofdimethylphenolsandofthe2,4,6-trimethylphenol,inorderto
test theinfluence ofthemethyl substituentboth for its
induc-tiveeffectaswellasforthesterichindrance.InFigs.10and11,
theconcentration–timeprofileofthecyclohexylationofthe
2,3-dimethylphenolcatalyzedbybothAmberlyst15andCH3SO3Hare
shown.Thegeneraltrendobservedinthis caseissimilarfor all
phenols,thussuggestingtheinvolvementofasamereactionpath.
Inagreementwiththatobservedforthephenoltheactivityofthe
Amberlyst15is,inanycase,higherthanthatofCH3SO3H(Table3),
whichislikelyduetothesuperiorprotonationabilityofthesolid
acidwithrespecttotheliquid one [34,35]. Theactivityof
3,5-dimethylphenoland2,6-dimethylphenolarelowerthanthatof
neatphenol(Table1),thephenomenonmaybeascribedtothe
sterichindranceofthesubstituents, whichslowdownthe
elec-trophilicattack[37,38].Suchaneffectismorepronouncedonthe
initialreactionrateofthe2,6-isomerthanthe3,5-one.Thesmall
increase of the reaction rateof the 2,3-dimethyl-phenol
cyclo-hexylationwithrespecttothatofneatphenolmaybeduetothe
inductiveeffectofmethylgroups,eventhoughthesterichindrance
mayplaya non-negligibleeffect.Infact,theinitialreactionrate
of2,4-dimethyl-phenol isequivalenttothatof phenol,the
rea-sonofthisbehaviorisnotclear,butitmightbeascribedtothe
100 80 60 40 20 2 3 4 5 6 o/p r a tio conversion (%) o/p Amberlyst 15 o/p CH3SO3H o/p AlCl3
Fig.9.Comparisonofortho/pararatiovs.conversioninthecyclohexylationof phenolandintherearrangementofcyclohexylphenylether.Runconditions: cyclo-hexeneandphenol1molL−1,Amberlyst15,CH3SO3H1.8meqH+andAlCl31mmol ascatalysts,solvent1,2-dichloroethane,reactionvolume10mLT358K.Cyclohexyl phenyletherrearrangementarecarriedoutwiththesamerunconditionsexcept theinitialconcentrationof0.2molL−1.
Table3
Alkylationofdimethylphenolsand2,4,6-trimethylphenol:selectivityafter240minofreactionat358K.Runconditions:dimethylphenols1.2molL−1,cyclohexene1.1molL−1, Amberlyst15,400mgorCH3SO3H180mg,solvent1,2-dichloroethane,reactionvolume10mL.
Catalyst Conv.(%) r0a Selectivity(%)
Ether 2-CyclohexylDMPb 3-CyclohexylDMPb 4-CyclohexylDMPb Cyclohexyl cyclohexene 2,6-Dimethylphenol Amberlyst15 15 2.6 26 – 5 15 13 CH3SO3H 7.2 0.9 18 – 11 16 55 3,5-Dimethylphenol Amberlyst15 35 16 24 27 – 5 10 CH3SO3H 14 4.1 29 14 – 0.1 13 2,3-Dimethylphenol Amberlyst15 95 44 4 38 5 12 4 CH3SO3H 13 3.1 18 19 – 5 55 2,4-Dimethylphenol Amberlyst15 80 30 10 42 10c – 5 CH3SO3H 12 2.1 12 10 2c – 62 2,5-Dimethylphenol Amberlyst15 57 14 16 16 – 16 2 CH3SO3H 16 1.5 16 9 – 4 15 2,4,6-Trimethylphenol Amberlyst15c 1 0.2 – – – – 50 CH3SO3Hc 5 0.6 – – – – 50 a(105molL−1s−1gcat−1). b DMP=dimethylphenol.
c Betweenthetwopossibleisomer(in3or5position)ithasnotverifiedwhatisformed,butthe5isomerismoreplausible,duetothelowersterichindranceofthe5 position.
dLargepartoftheproductsarepeach,formationoftracesoftheetherandthem-isomerhasbeenobservedafter20hofreaction.
simultaneouspresenceoftwocontraryeffects:thesterichindrance
andtheinductiveeffectofthemethylgroups.Thereactivityof
2,5-dimethyl-phenolis clearlyinfluencedbythesterichindranceof
themethylgroups,inparticulartheonein2-positionsslowsdown
theattackof thecyclohexyl cationtoward thehydroxyl group,
whilethatin5-positionsimultaneouslyhamperstheattacktoboth
orthoandparapositions.Asamatteroffact,2,4,6-trimethyl-phenol
showsanegligiblereactivitytowardelectrophilicattack,thus
sug-gestingthecyclohexylcation isgreatly influencedby thesteric
hindrancebothontheattacktothehydroxyl groupandtothe
phenylring.
Theinfluenceofthemethylgroupsontheringalkylation
selec-tivityisstrictlyrelatedtothepositionofthesubstituentratherthan
theirinductiveeffect.Forinstance,3,5-and2,3-dimethylphenol
show,inthepresenceofAmberlyst15asacatalyst,anortho–para
ratioof5.4and3.2,respectively.Asamatteroffact,suchabehavior
suggeststhattherelativereactivityoftheparapositionsofthese
compoundsisabout3timeslowerthantheorthoones.The
compar-isonoftheseresultswiththealmostequalrelativereactivityofthe
orthoandparapositions,observedinthecyclohexylationof
phe-nolinthepresenceofAmberlyst15asacatalyst,suggestsastrong
effectofthesterichindranceofthemethylgroupsonthe
selectiv-OH + O OH OH + OH Cy Cyisomers
acid catalyst
acid cata lyst acidca talyst + OCy Cy isomers + acid catalyst
n
acid catalyst300 250 200 150 100 50 0 0.0 0.2 0.4 0.6 0.8 1.0 concent rat io n (mol L -1 )
time (minutes)
cylohexene
cyclohexyl phenyl ether
2-cyclohexyphenol
4-cyclohexyphenol
Fig.10.Reactivityof2,3-dimethylphenol.Runconditions:T358K,solvent 1,2-dichloroethane,Amberlyst15,400mg,reactionvolume10mL.
ity.Furthersupporttothisistheequalreactivityoftheorthoand
parapositionsofthe2,5-dimethylphenol.Inthiscase,itislikely
thatthemethylin5-positionhasthesameeffectonthereactivity
oftheorthoandparapositionwiththeconsequentequalrelative
reactivity.
When CH3SO3H is used the ortho selectivity increases as
already observed for phenol. For instance, cyclohexylation of
3,5-dimethylphenol catalyzed byCH3SO3H shows an very high
ortho–pararatio(o/p=140)andinanycase,foreachphenolwith
unsubstituted orthoand para position, there is a neat increase
oftheorthoselectivityinthepresenceofCH3SO3Hcomparedto
Amberlyst15.Sucha behaviorisnotstraightforward,sinceitis
not clear what are the reasonsof such a specific ortho
direct-ingaction of theCH3SO3H, however,either themechanism via
cyclohexyl phenyl ether rearrangement [37–39], or that via a
methanesulfonic–phenolcomplex[40]canberesponsibleforthis
behavior. 250 200 150 100 50 0 0.00 0.05 0.8 0.9 1.0 cyclohexene (mol/ L) time (minutes)
cyclohexene
cyclohexylphenyl ether
2 cyclohexyl phenol
4 cyclohexyl phenol
Fig.11.Reactivityof2,3-dimethylphenol.Runconditions:T358K,solvent 1,2-dichloroethane,CH3SO3H180mg,reactionvolume10mL.
3.4. Reactionpathproposedforthereactions
Alkylation, etherification and olefin oligomerization are the
reactionsbetweenphenolsandcyclohexeneobservedinthe
pres-enceofacidcatalysts.Alltheexperimentalevidencessuggestthe
reactionpathdepictedinScheme1.Therearethreeparallelandtwo
consecutiveacidcatalyzedreactions.Inparticular,theformation
ofcyclohexylphenyletherisreversible(alsotheformationofthe
aliphaticetherisreversible[41]),whileringalkylationofphenol,
cyclohexeneoligomerizationcyclohexylphenylether
rearrange-mentand alkylphenolsisomerizationarepracticallyirreversible
[39].
4. Conclusions
Thereactionbetweenphenolandcyclohexeneoccursviaa
com-plexpath,whichischaracterizedbytheformationofthecyclohexyl
phenyletherasreversibleintermediateanditscomplete
conver-siontotheproductsofringalkylationattheendofthereaction,
whenevercatalystdeactivationdoesnotoccur.Thereactionsof
O-alkylation,ringalkylation,etherrearrangementandcyclohexene
oligomerizationoccursimultaneously,butthelatterispractically
negligiblebyselectingthepropersolventorcarryingoutthe
reac-tionin excessof phenols. Inthepresence of Amberlyst15 and
36resinstheselectivityofringalkylationofphenolseemstobe
drivenbythetypicalortho/paraorientingeffectofthehydroxyl
group.Onthecontrary,aspecificactionofhomogeneoussystems
(CH3SO3HandAlCl3)towardformationoftheorthoisomershas
beenobserved,butitisnotclearwhatisthereasonofsucha
behav-ior.The electrophilicattackof thecyclohexylcation isstrongly
influencedbythesterichindranceofthemethylgroupasamatter
offact,thealkylationof2,4,6-trimethylphenolpracticallydoesnot
occur,andtheactivation,duetotheinductiveeffecttothe
con-tiguouspositionsofthemethylgroup,isnegligiblecomparedto
thedeactivationinducedbythesterichindrance.
Acknowledgments
FinancialsupportbyCa’FoscariUniversityofVeniceisgratefully
acknowledged(Ateneofund2009).AthanktoDr.DavideMontin
forsomepreliminaryexperimentscarriedoutduringhisdegreein
IndustrialChemistry.Finally,aspecialthanktoMr.ClaudioTortato
forthehelpfuldiscussions.
References
[1]K.Komiya,S.Fukuoka,M.Aminaka,K.Hasegawa,H.Hachiya,H.Okamoto,T. Watanabe,H.Yoneda,I.Fukawa,T.Dozono,in:P.T.Anastas,T.C.Williamson (Eds.),GreenChemistry:DesigningChemistryfortheEnvironment,American ChemicalSociety,Washington,DC,1996,p.20.
[2]V.C.Malshe,E.S.Sujatha,React.Funct.Polym.43(2000)183–194. [3]A.Sato,I.Shimizu,E.Matsuzaka,US4144279.
[4]M.A.Harmer,Q.Sun,Appl.Catal.A:Gen.221(2001)45–62. [5]W.F.Hölderich,G.Heitmann,Catal.Today38(1997)227–233. [6]A.Mitsutani,Catal.Today73(2002)57–63.
[7]K.G.Chandra,M.M.Sharma,Catal.Lett.19(1993)309–317.
[8]H.Zhang,S.M.Mahajani,M.M.Sharma,T.Sridhar,Chem.Eng.Sci.57(2002) 315–322.
[9]A.deAngelis,C.Flego,P.Ingallina,L.Montanari,M.G.Clerici,C.Carati,C.Perego, Catal.Today65(2001)363–371.
[10]R.A.Rajadhyaksha,D.D.Chaudhari,Ind.Eng.Chem.Res.26(1987)1276–1280. [11]R.H.Rosenwald,AlkylationinKirkOhtmerEncyclopediaofChemical
Technol-ogy,vol.2,Wiley,1978,p.50.
[12]H.W.B.Bird,AlkylphenolsinKirkOhtmerEncyclopediaofChemical Technol-ogy,vol.2,Wiley,1978,p.73.
[13]G.D.Yadav,S.Ganesh,Pathre,Ind.Eng.Chem.Res.46(2007)3119–3127. [14]G.D.Yadav,P.Kumar,Appl.Catal.A:Gen.286(2005)61–70.
[15]N.Bhatt,A.Patel,J.Mol.Catal.A:Chem.264(2007)214–219. [16]R.Anand,K.U.Gore,B.S.Rao,Catal.Lett.81(2002)33–41.
[17] R.Amandi,K.Scovell,P.Licence,T.J.Lotz,M.Poliakoff,GreenChem.9(2007) 797–801.
[19]M.M.Sharma,React.Funct.Polym.26(1995)3–23. [20] A.J.Hoefnagel,H.vanBekkum,Catal.Lett.85(2003)7–11.
[21]K.Wilson,D.J.Adams,G.Rothenberg,J.H.Clark,J.Mol.Catal.A:Chem.159 (2000)309–314.
[22]P.F.Siril,H.E.Cross,D.R.Brown,J.Mol.Catal.A:Chem.279(2008)63–68. [23]A.Akelah,A.Moet,FunctionalizedPolymersandTheirApplications,Chapman
andHall,1990.
[24] G.A.Olah,A.M.White,D.H.O’Brien,Chem.Rev.70(1970)561–591. [25]G.A.Olah,G.K.SuryaPrakash,J.Sommer,Superacids”,J.Wiley,1985,p.90. [26] N.S.Isaacs,PhysicalOrganicChemistry,Longman,1987,p.395.
[27]Y.Tsuji,M.M.Toteva,H.A.Garth,J.P.Richard,J.Am.Chem.Soc.125(2003) 15455–15466.
[28]R.Klimkiewicz,H.Grabowska,H.Teterycz,Appl.Catal.A:Gen.246(2003) 125–136.
[29]B.Chaudhuri,M.M.Sharma,Ind.Eng.Chem.Res.30(1991)227–231. [30] E.Modrogan,M.H.Valkenberg,W.F.Hoelderich,J.Catal.261(2009)177–187. [31]Q.Ma,D.Chakraborty,F.Faglioni,R.P.Muller,W.A.Goddard,T.Harris,C.
Camp-bell,Y.Tang,J.Phys.Chem.A110(2006)2246–2252.
[32]B.Bjoerkqvist,H.Toivonen,J.Chromatogr.178(1979)271–276.
[33]G.W.Roberts,in:P.N.Rylander,H.Greenfield(Eds.),CatalysisinOrganic Syn-thesis,AcademicPress,1976,p.1.
[34]S.Kajount,B.M.Kierman,D.R.Brown,H.G.M.Edwards,J.A.Dale,S.Plant,Catal. Lett.85(2003)33–40.
[35]N.C.Marziano,A.tommasin,C.Tortato,J.Chem.Soc.Trans.2(1991)1575–1580. [36]C.Reichardt,SolventsandSolventEffectsinOrganicChemistry,2nded.,VCH,
Weinheim,1988,p.22.
[37]K.Schofield,AromaticNitration,CambridgeUniversityPress,Cambridge,1980. [38] P.B.D.DeLaMare,J.H.Ridd,AromaticSubstitution,ButtherworthsScientific
Pubblications,London,1959.
[39] C.B.Campbell,A.Onopchenko,D.C.Young,Ind.Eng.Chem.Res.29(1990) 642–647.
[40] G.Sartori,F.Bigi,R.Maggi,A.Arienti,J.Chem.Soc.PerkinTrans.1(1997) 257–260.
[41] J.F.Izquierdo,F.Cunill,M.Vila,M.Jhorra,J.Tejero,Ind.Eng.Chem.Res.33(1994) 2830–2835.