Materiali
Ceppi batterici
DH5α: F-, φ80dlacZ∆M15 supE44, λ- ∆(lacZYA-argF) U169, deoR, endA1, gyrA96, hsdR17(rk-, mk+), phoA, thi-1, recA1, relA1.
Questo ceppo è stato utilizzato per amplificare i vettori plasmidici: è difettivo per la restrizione e porta le mutazioni recA1 e relA1 per migliorare la stabilità e la qualità dei plasmidi ricombinanti preparati dalle “mini-“ e “midi- preps”. Contiene inoltre il marcatore φ80lacZ∆M15 che permette la α-complementazione del gene della β-galattosidasi e la selezione dei ricombinanti in un test bianco-blu.
Vettori plasmidici
Vettori plasmidici sono stati utilizzati per trasportare ed amplificare sequenze geniche di interesse da usare per la trascrizione di DNA senso o antisenso.
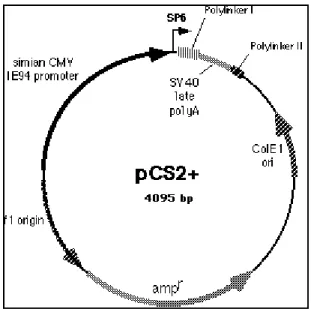
pCS2+ (Fig. 13) : è un vettore di espressione che consente vari usi. E’ progettato per l’espressione di proteine in embrioni di Xenopus, sia da RNA che da DNA microiniettati e si può usare in esperimenti di trascrizione/traduzione in vitro. Contiene un forte promotore virale (CMV IE94) seguito da un “polylinker” e dal sito di poliadenilazione del virus SV40. Un promotore SP6 consente la trascrizione di RNA da sequenze clonate nel “polylinker”; un promotore T7 inserito tra il “polylinker” e il sito polyA SV40 ad orientamento invertito consente la sintesi di sonde; un secondo polylinker successivo al sito polyA SV40 fornisce diversi siti di restrizione con cui linearizzare il vettore per la trascrizione SP6. La base del vettore è il plasmide pBluescript II KS+ e include il gene di resistenza all’ampicillina e un’origine f1 per produrre DNA a singolo filamento (Rupp et al., 1994; Turner e Weintraub, 1994).
Figura 13: rappresentazione schematica del plasmide pCS2+. Non sono visualizzati i siti di
restrizione.
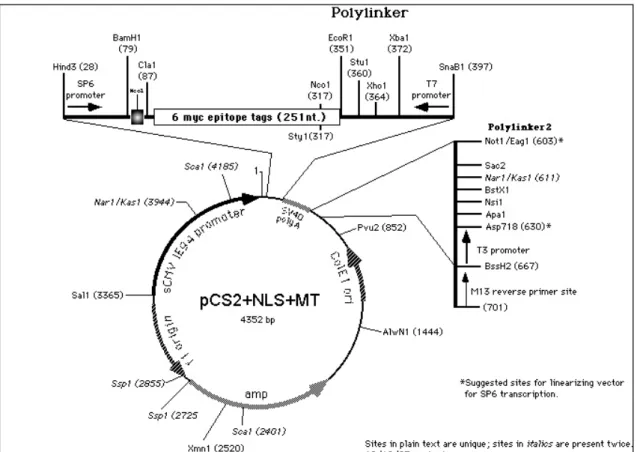
pCS2+MT (Fig. 14): studiato per produrre proteine di fusione, include 6 copie dell’epitopo myc riconosciuto dall’anticorpo monoclonale 9E10, clonate all’interno dei siti ClaI ed EcoRI del vettore pCS2+ (Rupp et al., 1994; Turner e Weintraub, 1994).
pCS2+NLS+MT (Fig. 15): possiede il segnale di localizzazione nucleare dell’antigene nucleare T grande del virus SV40, clonato all’interno dei siti BamHI e ClaI, a monte degli epitomi myc. Per il resto questo vettore è identico a pCS2+MT (Rupp et al., 1994; Turner e Weintraub, 1994).
Figura 15: rappresentazione schematica del plasmide pCS2+NLS+MT.
Cloni
Le sonde marcate con DIG-UTP sono state trascritte da vettori provenienti anche da altri laboratori, sotto forma di DNA adsorbito su carta da filtro, che sono stati amplificati, linearizzati e trascritti in base alle indicazioni disponibili.
Xbf1: linearizzato con XhoI; trascritto con SP6 RNA polimerasi.
Pax2: clone contenente un frammento di 850 pb; linearizzato con EcoRI; trascritto con SP6 RNA polimerasi.
Pax6: clone contenente un frammento di 450 pb; linearizzato con EcoRI; trascritto con SP6 RNA polimerasi.
Xotx5: frammento di 1.6 Kb clonato in pBluescript II SK- nei siti EcoRI e NotI; linearizzato con NotI; trascritto con T7 RNA polimerasi.
MC 19/11 (Xotx2): ; frammento di 860 pb clonato in pGem3 nei siti EcoRI e StuI; linearizzato con EcoRI; trascritto con SP6 RNA polimerasi (Pannese et al., 1995).
Xhairy2: clonato in pBluescript II SK+ nei siti BamHI (abolito) e XhoI; linearizzato con BamHI (taglia nel 5’ UTR); trascritto con T7 RNA polimerasi.
ElrC: clonato in pBluescript II SK; linearizzato con NotI; trascritto con T7 RNA polimerasi.
3.2/4 (Xrx1): frammento di 1485 pb clonato in pGem3 nel sito EcoRI; linearizzato con BamHI; trascritto con T7 RNA polimerasi.
Xanf-1: linearizzato con XbaI; trascritto con T7 RNA polimerasi.
pBS-KS-XNS7L (Xngnr-1): frammento di 1.4 Kb clonato in pBluescript II KS; linearizzato con BamHI; trascritto con T3 RNA polimerasi.
pBS-Cyclin D1: clonato in pBluescript II KS nei siti ClaI e BamHI; linearizzato con BamHI; trascritto con T3 RNA polimerasi.
Fez: clonato in pBluescript II SK nei siti XhoI e XbaI; linearizzato con XhoI; trascritto con T3 RNA polimerasi.
pCS2RtXic1 #5/#6 (p27Xic1): frammento di 4730 pb clonato in pCS2 nei siti EcoRI e BamHI; linearizzato con BamHI; trascritto con T7 RNA polimerasi.
XGadd45γ: clonato in pGemT; linearizzato con KspI; trascritto con Sp6 RNA polimerasi.
5.2XFGF-8bs (FGF-8): clonato in pBluescript II; linearizzato con XbaI; trascritto con T3 RNA polimerasi.
Oligonucleotidi antisenso
MoXrx1 :5’-TCAGGGAAGGGCTGTGCAGGTGCAT-3’ (Soluzione stock 1 mM)
MoC:
Terreni di coltura
Liquidi
Luria-Bertani Broth (LB):
NaCl 1%
bacto tryptone 1%
bacto yeast extract 0,5% (autoclavare)
Solidi
Bottom Agar:
agar sciolto in LB 1,5% (autoclavare)
Antibiotici usati per terreni selettivi:
Ampicillina 100 µg/µl
Soluzioni di uso comune
PBS NaCl 8 g KCl 0.2 g Na2PO4.16H2O 1.44 g KH2PO4 0.24 g pH 7.4 TBS NaCl 8 g KCl 0.2g Tris base 3 g pH 7.4 SSC 20x NaCl 3.0 M Na citrato 0.3 MSoluzioni per “MINI-prep”
Soluzione 1:EDTA 10 mM glucosio 50mM lisozima 10 mg/ml Soluzione 2: NaOH 0,2 M SDS 1% Soluzione 3 pH 4.8: CH3COOH 5 M KCI 3M TE : Tris (pH 8.0) 10 mM EDTA (pH 8.0) 1 mM
Soluzioni per la “MIDI-prep”
S1: RNAsi A 100 (g/ml Tris-HCI 50 mM EDTA (p H 8.0) 10 mM S2: NaOH 200 mM SDS 1 % S3 : KCH3COO (pH 5.1) 2.80 mM N2 : Tris 100 mM EtOH 15% KCl+H3PO4 (pH 6.3) 900 mM N3 : Tris 100 mM EtOH 15% KCl+H3PO4 (pH 6.3) 1150 mM N5 : Tris 100 mM EtOH 15%KCl+H3PO4 (pH 8.5) 1000 mM
Soluzioni per l’elettroforesi su gel di agarosio
TBE pH 8.0: Tris base 0.089 M Acido borico 0.089 M EDTA 0.002 M “Loading buffer” 6x : Glicerolo 5% Blu di bromofenolo 0.05% Xilene cianolo 0.05%“Loading buffer” 2x denaturante per RNA:
EDTA 18 mM Formammide 95 % SDS 0.025 % Blu di bromofenolo 0.025% Xilene cianolo 0.025% Gel di agarosio: Agarosio 1-2,5% (peso/vol) Et-Br c.f. 10 (g/ml) TBE 1X
“Marker” di lunghezza: Invitrogen 1 kb DNA ladder
Soluzioni per estrazione di proteine da embrioni di
Xenopus laevis
Tampone di estrazione: Tris HCl ph 7,5 50 mM NaCl 150 mM DTT 1mM Glicerolo 20% Mix inibitoridelle proteasi Sigma* 10x H20 mQ
DYE 3X : Tris-HCl pH 6.8 150mM SDS 6% β-mercaptoetanolo 10% Blu di bromofenolo 0,2% Glicerolo 20%
Reattivo di Bradford: Soluzione stock SIGMA B69-16
Soluzioni per elettroforesi su gel di acrilammide
(SDS-PAGE)
“Stacking gel” (per 4ml):
Mix acrilammide 30% 0,67 ml Tris pH 6.8 1.0 M 0,5 ml SDS 10% 0,04 ml APS 10% 0,04 ml TEMED 0,01 ml H2O 2,7 ml
“Resolving gel” 12% (per 5 ml): Mix acrilammide 30% 4 ml Tris pH 8.8 2,6 ml SDS 10% 0,10 ml APS 10% 0,10 ml TEMED 0,010 ml H2O 3.4 ml Tampone di corsa: Tris-base 25 mM Glicina 192 mM SDS 0,1% “Staining solution”:
Comassie brilliant blue 0,1% Et-OH assoluto 48%
Acido acetico 25%
“Destaining solution”:
Et-OH assoluto 10%
Ponceau S 10x
Ponceau S 2 g (Sigma P7767) Acido tricloroaceetico 30 g
Acido sulfosalicilico 30 g H2O fino a 100 ml
Soluzioni per “Western blotting”
Tampone di trasferimento Tris Base 24.8 mM Glicina 192 mM Metanolo 10% pH 8.3 “Blocking solution”: Tween 20 0.2% PBS 1xLatte scremato in polvere 5%
PBTw 0.05%
PBS 1x 500ml
Tween 20 250 µl
Soluzioni per embrioni di Xenopus laevis
MMR NaCl 0.1 M KCl 2 mM MgSO4 1 mM CaCl2 2 mM HEPES 5 mM pH 7.8 EDTA 0.1 mM Soluzione degelificante DTT 3.2 mM Tris-HCl 0.2 M pH 8,8Soluzione per il testicolo
MMR 1x
Heat inactivated lamb serum 10% Gentamicina 20 µg/ml
MEMFA
MOPS 0.1 M pH 7.4
EGTA 2 mM
MgSO4 1 mM
Formaldeide 3.7%
Generalmente si preparano “stocks” sterili di una soluzione 10x dei Sali (MEM) che viene diluita e addizionata di formaldeide al momento dell’uso.
Soluzioni per la reazione cromogenica della
β-galattosidasi
Soluzione di ferri K3Fe(CN)6 30 mM K4Fe(CN)6.3H2O 30 mM PBS 1x Stock Salmon-galSalmon-gal (Sigma) 5% in metanolo
Soluzioni per “whole mount in situ hybridization”
PBTw 0.1% PBS 1x Tween 20 0.1% TBSx TBS 1x Triton X-100 0.1% Paraformaldeide 20% • Sciogliere 5 g di paraformaldeide in 100 ml H2O RF a 60°C • Chiarificare con 10 µl NaOH 10 N, portare a volume e filtrare. • Conservare a -20°C. Una volta scongelata usare entro un mese.Tampone di ibridazione
Formammide 50%
SSC 5x
RNA di torula 1 mg/ml Eparina 100 µg/ml Boehringer blocking reagent 1 %
Tween 20 0.1 %
CHAPS 0.1 %
EDTA 10 mM
Soluzione stock di Proteinasi K Proteinasi K 20µg/µl
APB (“Alcaline phosphatase buffer”) Tris HCl pH 9.5 100 mM MgCl2 50 mM NaCl 100 mM Tween 20 0.1 % Tetramisole 2 mM “Egg extract”
• Omogeinizzare embrioni/uova conservati a -20°C in 1 volume di PBS 1x
• Centrifugare 10’ a 4°C a 12000 rpm, recuperare la fase acquosa. • Centrifugare 5x10’ a 4°C a 12000 rpm recuperando la fase
acquosa.
• Denaturare a 56°C per 15’, centrifugare per 10’ a 12000 rpm a 4°C. • Aliquotare e conservare a -20°C.
“Blocking buffer”
TBSx 1.6 ml
Heat inactivated lamb serum 300 µl
Egg extract 100 µl
Boehringer blocking reagent 400 µl
“Bleaching solution”
H2O2 1%-3%
SSC 0.5x
Metodi
I metodi usati sono stati ricavati sostanzialmente da Sambrook e Russell, 2001; di seguito vengono riportati i protocolli che è stato necessario adattare alle necessità sperimentali.
Preparazione di cellule competenti
Mediante il seguente protocollo le cellule di E. coli (ceppo DH5α) sono state rese competenti per la successiva trasformazione con vettori plasmidici. • Prelevare una colonia cresciuta ON su terreno solido e inocularla in 50 ml di
terreno liquido.
• Far procedere la crescita a 37°C in agitazione fino a che la densità ottica (OD) misurata a 600 nm raggiunge il valore di 0,2.
• Interrompere la crescita mantenendo 3’ in ghiaccio. • Centrifugare in tubo sterile per 10’ a 5000 rpm a 4°C.
• Eliminare il sopranatante e risospendere in ½ del volume iniziale (circa 25 ml) di RbCl 50 mM freddo.
• Mantenere 30’ in ghiaccio.
• Centrifugare in tubo sterile per 10’ a 5000 rpm a 4°C.
• Eliminare il sopranatante e risospendere in 1/50 del volume iniziale (circa 0,8 ml) di RbCl 50 mM freddo.
• Conservare in aliquote a –80°C.
Trasformazione batterica
Con questo procedimento è possibile trasformare cellule batteriche con DNA plasmidico.
• Aggiungere 5-20 ng di plasmide ad un’aliquota di 200 µl di cellule competenti.
• Incubare in ghiaccio per 30 min.
• Incubare in ghiaccio per 5 minuti.
• Aggiungere 800 µl di LB preriscaldato a 37°C e incubare in stufa per 60 minuti.
• Piastrare le cellule su terreno solido selettivo.
• Dopo incubazione a 37°C ON, sulla piastra Petri compaiono le colonie: l’antibiotico fa sì che crescano solo le cellule che hanno assunto il plasmide poiché esso contiene il gene che conferisce la resistenza.
• Ogni volta che è stata effettuata una trasformazione, bisogna è stata controllata l’efficacia dell’antibiotico piastrando, in parallelo, 100 µl di cellule batteriche sottoposte a uguale trattamento in assenza di DNA plasmidico.
Estrazione di DNA plasmidico su piccola scala
mediante lisi alcalina ("mini-prep”)
Tale metodo permette di estrarre ( 2-20 (g di DNA plasmidico da cellule trasformate per successivo sequenziamento o digestione diagnostica.
• Da una piastra di E. coli recanti il plasmide di interesse o da uno stock di batteri in glicerolo al 10%, viene effettuato in maniera sterile un inoculo di un clone che viene posto in un tubo batteriologico da 10-15 ml contenente 3 ml di brodo di coltura LB con ampicillina (100 (g/ml).
• Il tubo è incubato per 12-16 ore a 37°C in agitazione, affinché la coltura batterica raggiunga la fase di crescita stazionaria.
• La coltura viene quindi centrifugata a 12000 rpm per 1-3 min allo scopo di ottenere un “pellet” di cellule batteriche.
• Il pellet viene risospeso in 400 (g di “soluzione 1”.
• Alla suddetta sospensione si aggiungono 400 (g di “soluzione 2” e la miscela viene mescolata delicatamente invertendo il tubo.
• La reazione di lisi alcalina non deve procedere per una durata superiore a 5’, trascorsi i quali si aggiungono 400 (g di “soluzione 3”, necessaria a far precipitare le membrane e le pareti delle cellule lisate, insieme al DNA cromosomico e all’RNA ad alto peso molecolare che ad esse sono associati.
• Dopo una centrifugazione di 15’ a 12000 rpm viene recuperato il sopranatante, contenente il DNA plasmidico, l’RNA a basso peso molecolare, le proteine batteriche.
• Si aggiungono 0.7 volumi (V) di isopropanolo alla fase acquosa contenente il DNA plasmidico e si lascia 10’ a temperatura ambiente (RT).
• In questo modo il DNA plasmidico e l’RNA a basso peso molecolare precipitano e vengono recuperati mediante centrifugazione (10‘ a 12000 rpm).
• Seguono il lavaggio del “pellet”, ottenuto in etanolo (EtOH) al 70%, e la sua risospensione, in 20 (l di TE (o H2O mQ) contenente RNAsi A ad una concentrazione di 20 (l/ml, allo scopo di eliminare l’RNA a basso peso molecolare.
Estrazione di DNA plasmidico su media scala mediante
colonne NUCLEOBOND
©AX-100 (“midi-prep”)
Per poter disporre di quantità di plasmidi adeguate alle necessità sperimentali è stato necessario amplificarli: ciò è stato possibile tramite la trasformazione di cellule batteriche competenti all’acquisizione del plasmide e la successiva estrazione del DNA plasmidico per mezzo di colonnine cromatografiche “NUCLEOBOND©” a scambio anionico disponibili in commercio insieme alle soluzioni necessarie per il loro impiego. Con questa tecnica è possibile estrarre da 80 a 140 µg di DNA altamente purificato a partire da 100 ml di sospensione batterica satura.
• Da una piastra di E. coli recanti il plasmide di interesse o da uno stock di batteri in glicerolo al 10%, viene effettuato in maniera sterile un inoculo di un clone che viene posto in un tubo batteriologico da 10-15 ml contenente 3 ml di brodo di coltura LB con ampicillina (100 (g/ml).
• Il tubo è incubato per 12-16 ore a 37°C in agitazione, affinché la coltura batterica raggiunga la fase di crescita stazionaria.
• Si trasferisce 1ml di coltura in 100 ml brodo di coltura LB con ampicillina (100 (g/ml) e si incuba a 37°C in agitazione ON fino al raggiungimento della fase di crescita stazionaria..
• Si centrifuga la coltura a 3000 rpm per 20.
• Il pellet così ottenuto viene risospeso in 8 ml di soluzione S1; quindi si aggiungono 8 ml di S2, si capovolge delicatamente e si lascia a T ambiente per 5’.
• Dopo aver aggiunto 8 ml di soluzione S3 a 4°C ed aver capovolto delicatamente, si incuba in ghiaccio per 15’.
• Si filtra la sospensione e si recupera il sovranantante al fine di ottenere un lisato meno torbido possibile.
• Si equilibra due colonnine cromatografiche AX-100, caricandole con 3 ml di soluzione N2, permettendone lo svuotamento per gravità.
• Si caricano quindi la colonnine equilibrate con metà del lisato ciascuna; ciò risulta nell’immobilizzazione del DNA da parte della resina.
• Si libera il DNA dai sali e dall’RNA residuo mediante due lavaggi di 4 ml ciascuno con la soluzione N3.
• Si procede infine con l’eluizione del DNA dalla colonnina, con 5 ml di soluzione N5.
• L’eluato viene precipitato, dopo aggiunta di 0.7 V di isopropanolo, mediante centrifugazione a 4°C per 30’ a 12000 rpm.
• Il pellet viene lavato con EtOH al 70% preequilibrato a 4°C, centrifugato a 4°C per 10’ a 15000xg e risospeso in TE pH 8.0 (o in H2O mQ).
Elettroforesi su gel di agarosio
Si è fatto ricorso alla corsa elettroforetica su gel di agarosio all’1-2,5% (peso/volume) in tutti i casi in cui è stato necessario verificare la completezza di una digestione, valutare la purezza di un DNA estratto, di stimare la concentrazione o la lunghezza in paia di basi del DNA nelle varie preparazioni. • I gel sono preparati sciogliendo l’agarosio in TBE, portato alla temperatura di
• Prima che il gel polimerizzi si aggiunge EtBr (bromuro d’etidio) ad una concentrazione finale di 10 (g/ml.
• Il gel, lasciato brevemente a raffreddare viene colato in un lettino da elettroforesi di un apparato orizzontale.
• Una volta polimerizzato il gel è posto nell’apparato ed immerso in un tampone di corsa, TBE a pH 8.
• I campioni da analizzare vengono diluiti in acqua e “loading buffer”. Il “loading buffer” appesantisce il campione, facendolo andare sul fondo del pozzetto e consente al contempo di seguire la corsa elettroforetica.
• Si caricano i campioni su gel e si applica una differenza di potenziale di 50/120 V per un tempo variabile dai 5 ai 60 minuti.
• Si visualizzano infine le bande degli acidi nucleici ponendo il gel sotto raggi UV: il bromuro di etidio che si è intercalato alle basi appare, in queste condizioni, fluorescente.
• Le dimensioni dei frammenti sono stimate in presenza di marcatori con lunghezza in paia di basi (pb) e peso molecolare noti (INVITROGEN 1kb DNA ladder).
Stima della concentrazione di DNA in soluzione
La stima della concentrazione dei campioni di DNA è stata effettuata mediante due metodi:
Elettroforesi su gel d’agarosio:
Si corre il campione in un gel di agarosio all’ 1% in presenza di bromuro di etidio e di adeguati marker di quantità a concentrazione nota: comparando la fluorescenza agli UV della banda di interesse con quella dei marker di quantità si può stimare approssimativamente la concentrazione del campione iniziale.
Spettrofotometria UV:
• Si tara lo spettrofotometro (Beckman DU-60) effettuando una misurazione con 600 µl di acqua mQ.
• Si diluiscono 2 µl di campione in 598 µl di acqua mQ (volume totale: 600 µl) in una cuvetta di quarzo e si misura l’assorbanza alla lunghezza d’onda di 260 nm: lo strumento restituisce direttamente il valore di concentrazione del campione e un’indicazione della purezza in base al rapporto OD260/OD280.(se maggiore o uguale a 1,8 il campione si ritiene privo di contaminanti quali Sali e proteine)
Estrazione di proteine da embrioni di Xenopus laevis
Il seguente protocollo è stato utilizzato per estrarre la frazione citoplasmatica di proteine da embrioni di Xenopus laevis a stadio 10, per la successiva analisi tramite SDS-PAGE.• Si prelevano 30 embrioni di Xenopus laevis a stadio 10 e si pongono in una provetta eppendorf insieme a 300 µl di soluzione di lisi (10 µl soluzione/embrione).
• Pestellare accuratamente e lasciare 15’ in agitazione orizzontale a 4°C. • Risospendere il contenuto della provetta e entrifugare 15’ a 13000 rpm a
4°C.
• Delle tre fasi visibili a questo punto prelevare quella acquosa evitando quella lipidica e il pellet.
• Centrifugare per 3 volte a 13000 rpm a 4°C, ogni volta prelevando la fase acquosa e ponendola in una nuova provetta eppendorf.
• Mantenere il lisato chiaro in ghiaccio e misurarne la concentrazione di un’aliquota allo spettrofotometro.
• Aliquotare il lisato, diluirlo in un buffer denaturante (Dye 3X) e denaturarlo per 5’ a 95°C.
• I campioni così preparati si possono caricare su gel di acrilammide o conservare a -20°C.
Quantificazione proteica: metodo di “Bradford”
Con questo metodo si può valutare la concentrazione proteica di un campione, sfruttando la relazione che la lega all’assorbanza (Legge di Lambert e Beer).
• Si diluisce il campione (in modo da rientrare nell’ordine di grandezza del campo dinamico dello strumento) e se ne aggiungono 20 µl a 1 ml di reattivo di Bradford.
• Si pone il preparato in una cuvetta e se ne misura l’assorbanza alla lunghezza d’onda di 595 nm allo spettrofotometro (Beckman DU-60), preventivamente tarato con uno standard.
• Lo strumento, in base ai valori di assorbanza misurati, fornisce direttamente i corrispondenti valori di concentrazione del campione.
Elettroforesi su gel di poliacrilammide (SDS-PAGE)
Questa tecnica permette di separare le proteine in base al loro peso molecolare.
L’SDS è un detergente carico negativamente che si lega alle regioni idrofobiche delle proteine, sopraffacendone la carica intrinseca e provocandone lo svolgimento in catene polipeptidiche estese. Inoltre, aggiungendo l’agente riducente β-mercaptoetanolo vengono ridotti i ponti disolfuro, contribuendo a separare le proteine da eventuali associazioni dovute a tale legame: in questo modo la proteina denaturata, se sottoposta ad un campo elettrico, può migrare al polo positivo in maniera dipendente esclusivamente dalle sue dimensioni. • Il gel di poliacrilammide viene colato tra due lastrine di vetro (12X12X0,3
cm) sovrapposti e separate da due spaziatori di teflon di spessore 0,8 mm in un apposito apparato bloccato in posizione verticale la cui estremità inferiore è sigillata.
• Viene colato per primo il “Resolving gel” fino a circa 2/3cm dalla superficie. • Per pareggiare il fronte del gel e per evitare che il contatto con l’ossigeno
atmosferico rallenti la polimerizzazione si versa sopra al gel uno strato di acqua che, conclusa la polimerizzazione, viene asciugata con carta 3MM.
• Si cola lo “Stacking gel” fino al bordo del vetro e si inserisce un pettine (spessore 0,8 mm) che modella la forma dei pozzetti dove sarà caricato il campione.
• Avvenuta la polimerizzazione (15-20’), si tolgono i pettini, si apre inferiormente l’apparato e si pone in una vasca da elettroforesi verticale (Amersham Hoefer MiniVE) in cui si versa il tampone di corsa: il gel deve essere in contatto con il tampone sui suoi lati superiore e inferiore.
• I campioni, preventivamente diluiti in un buffer denaturante (Dye 3X) e riscaldati a 95°C per 5’, si caricano nei pozzetti; in un pozzetto si carica un marcatore di peso molecolare pre-colorato (Invitrogen Benchmark© pre-stained protein ladder).
• La corsa elettroforetica inizia collegando l’apparato ad elettrodi e applicando una differenza di potenziale di 80 V, finchè il campione attraversa lo Stacking gel, e prosegue a 100-120 V lungo il “resolving gel”.
• Terminata la corsa si smonta l’apparato: il gel può essere sottoposto a colorazione con Coomassie o a “western blotting”.
Colorazione con Coomassie Brilliant Blue
• Si immerge il gel in una soluzione colorante (“Staining solution”) per circa 30’, dopodiché si decolora con la “Destainig solution” per un tempo variabile tra 2h a ON, avendo cura di sostituire la “Destaining solution” periodicamente.
• Il gel con le bande proteiche evidenziate in questo modo può essere essiccato in un “gel-dryer” sotto vuoto o analizzato per scansione al computer.
“Western blotting”
Questa tecnica consente di trasferire le proteine separate con la SDS-PAGE dal gel di acrilammide a una membrana di nitrocellulosa o PVDF
(polivinildifluoruro) e successivamente di evidenziare le bande proteiche di interesse tramite una reazione immunologica.
• Si ritaglia una membrana di dimensioni adeguate: se si usa una membrana in PVDF si preequilibra per 15’’ in metanolo e quindi per 2’ in H2O mQ.
• Si montano membrana e gel di acrilammide accollati e protetti da spugne spaziatrici in un apparato per Western (Gibco Mini V 8.10) riempito di tampone di trasferimento.
• Si impone una corrente costante di 150 mA per 2 ore: le proteine, cariche negativamente, si trasferiscono dal gel alla membrana con velocità inversamente proporzionale alle loro dimensioni.
• Si smonta l’apparato di trasferimento e si pone la membrana in “blocking solution” per 1h a RT oppure ON a 4°C. Il gel si colora con Blu di Coomassie per verificare l’avvenuto trasferimento delle bande.
• Si effettuano 2 lavaggi di 5’con PBTw 0.05%.
• Si pone la membrana in “blocking solution” in cui è stato diluito l’anticorpo primario in modo che sia ricoperta da un film di soluzione.
• Si effettuano 5 lavaggi di 5’ con PBTw 0.05%
• Si pone la membrana in “blocking solution” in cui è stato diluito l’anticorpo secondario in modo che sia ricoperta da un film di soluzione.
• Si effettuano 5 lavaggi di 5’ con PBTw 0.05% • Si effettuano due lavaggi di 5’ in PBS.
Rivelazione di bande proteiche
Rivelazione tramite Ponceau S
• Dopo lo “western blot” si incuba il filtro in una soluzione 1x di Ponceau S finchè le bande non siano visibili.
Rivelazione tramite diaminobenzidina (DAB)
Questo protocollo fa uso di un kit commerciale (Sigma Fast DAB tablets): • Si sciolgono le pasticche del kit in 5 mlH2O mQ.
• Si pone la membrana nella soluzione: la perossidasi coniugata all’anticorpo secondario reagisce con l’ H2O2 presente in soluzione, libera O2 che provoca l’ossidazione della diaminobenzidina che produce un precipitato rosso scuro in corrispondenza delle bande di interesse.
Rivelazione per chemoluminescenza
Questo protocollo fa uso di un kit commerciale (Pierce SuperSignal® West Pico Chemiluminescent Substrate):
• Si miscelano i due substrati (SuperSignal® West Pico Luminol/Enhancer Solution e SuperSignal® West Pico Stable Peroxide Solution) con un rapporto 1:1 per un volume finale di 5-10 ml.
• Si pone la membrana nella soluzione: la perossidasi coniugata all’anticorpo secondario reagisce con l’ H2O2 presente in soluzione, libera O2 che
provoca la reazione luminosa del luminolo in corrispondenza delle bande di interesse.
• Si protegge la membrana con un foglio trasparente e la si usa per esporre una lastra fotografica
Reazione di PCR
La tecnica della reazione a catena della DNA polimerasi (PCR) permette di amplificare un frammento di DNA utilizzando specifici “primers”, sfruttando le proprietà di sintesi della Taq-polimerasi.
I “primers” sono oligonucleotidi sintetici lunghi alcune decine di deossinucleotidi trifosfato (dNTP) complementari alle estremità del frammento di DNA da amplificare, legandosi al quale permettono alla Taq polimerasi di sintetizzare stampi complementari.
Questa tecnica prevede un numero variabile di cicli di amplificazione (25-35), ciascuno dei quali composto da più fasi a temperature diverse, dipendenti
dalla composizione nucleotidica dei primers e dal tipo di Taq polimerasi utilizzata.
Il passaggio da una temperatura all’altra è permesso dall’uso di un “cycler” termico Bio-Rad iCycler.
Durante ogni ciclo i due filamenti di DNA vengono denaturati per permettere il successivo appaiamento dei primers (“annealing”), necessario per la fase di allungamento catalizzata dalla Taq polimerasi (“elongation”). Terminati i cicli di amplificazione si ha un ultimo “step” di circa 7-10’ per consentire alla Taq polimerasi di allungare i filamenti di DNA rimasti incompleti.
I “primers” utilizzati sono stati progettati per amplificare selettivamente la sequenza codificante del gene Xrx1 dal clone 3.2/4 in modo da dotarla alle estremita’ di sequenze di riconoscimento per enzimi di restrizione (evidenziate nel testo) che non tagliano al suo interno: ciò ha consentito di digerirne le estremità in modo da poterla clonare all’interno di un vettore linearizzato con gli stessi enzimi di restrizione.
In particolare i “primers” FORRXBAM e REVRXBAM sono progettati in modo tale da rimuovere il codone di stop della sequenza e da aggiungere anche una sequenza consensus ‘Kozak’ (CCGCCACC) alla sua estremità 5’ (Kozak, 1987). • FORRXBAM: 5’-gaGGATCCccgccaccatgcacctgcacagccc-3’ Tm≈61.8°C BamHI • REVRXBAM: 5’-gaGGATCCccaaggcttgccaataaactg-3’ Tm≈61.7°C BamHI • RXRV: 5’-cagcaGATATCatgcacctgcacagccc-3’ Tm≈64.3°C EcoRV • XRX1STOPXHO : 5’-gtcCTCGAGttaccaaggcttgccaataaactg-3’ Tm≈63.4°C XhoI
Le reazioni di PCR sono state allestite come segue: • AccuTaq© Taq LA polimerase 0.05 U/µl
• 10x AccuTaq©buffer 1x • dNTP mix (10 mM ciascuno) 500 µM
• DNA stampo 10 ng
• “Primer forward” 400 nM • “Primer reverse” 400 nM • H2O fino a un volume totale di 50 µl.
Per ogni reazione sono stati assemblati corrispondenti controlli negativi (eliminando il DNA stampo) e positivi (usando un DNA stampo già usato in precedenza).
Aliquote dei prodotti di reazione sono statte sottoposte a elettroforesi su gel d’agarosio 1% con EtBr insieme a un marcatore di lunghezza per stimare la dimensione del frammento amplificato e valutarne la purezza, oltre che per valutare la specificità delle condizioni di reazione o eventuali contaminazioni dei reagenti.
Digestione di DNA con enzimi di restrizione
L’uso di enzimi di restrizione è stato necessario per effettuare digestioni diagnostiche, rimuovere sequenze non desiderate, verificare la presenza e l’orientamento di un inserto all’interno di un vettore, linearizzare plasmidi per un clonaggio o per usarli come DNA stampo lineare in esperimenti di trascrizione in vitro. Per quest’ultima applicazione è stata fatta particolare attenzione ad usare enzimi che lasciassero un’estremita 5’ protrudente o ‘blunt’, in modo che l’RNA non venisse trascritto dal filamento complementare a quello usato.
Le reazioni di digestione sono state assemblate di norma con 3 unità/µg di DNA da digerire di enzima di restrizione e con il suo tampone specifico, facendo attenzione che il volume di enzima non eccedesse il 10% del volume finale di reazione: in questo modo il glicerolo presente negli stock di enzimi non interferisce con l’attività enzimatica. L’incubazione è stata protratta da 2h a ON a seconda dell’enzima e della quantità di DNA da digerire, a una temperatura scelta in base alle specifiche del singolo enzima.
La completezza della reazione è stata valutata sottoponendo un’aliquota della miscela di reazione a elettroforesi su gel d’agarosio 1% colorato con bromuro d’etidio e confrontando la banda risultante con quella generata da un’aliquota di corrispondente DNA non digerito: un plasmide linearizzato ad esempio, corre più lentamente del suo corrispondente non digerito, circolare, superavvolto e, quindi, più compatto.
Quando è stato necessario effettuare doppie digestioni, si è preferito assemblare due reazioni in sequenza, purificando il DNA dopo la prima digestione e procedendo con la seconda.
“Ligation”
La reazione di “ligation” consente di saldare le estremità di un plasmide linearizzato a quelle di un frammento lineare di DNA grazie all’attività dell’enzima DNA ligasi, rendendo così’ possibile il clonaggio di frammenti di DNA all’interno di vettori plasmidici.
Ogni reazione di “ligation” è stata allestita come segue: • T4 DNA ligasi (Invitrogen) 1 unità
• T4 DNA ligasi buffer 5x (Invitrogen) 2 µl
• Plasmide linearizzato 40-80 ng • quantità variabili di inserto
• H2O mQ fino a un volume di 10 µl • Incubazione ON a 18°C
Per ciascuna clonazione è stato valutato di volta in volta il rapporto vettore/inserto in grado di spostare efficacemente la reazione verso la saldatura tra DNA lineare e DNA plasmidico; una miscela di reazione priva dell’inserto è stata inoltre utilizzata come controllo per valutare la capacità del vettore di richiudersi su se’ stesso. Le miscele di reazione sono state utilizzate interamente per trasformare cellule E.coli DH5α; dal confronto delle piastre si è ricavata una valutazione della efficienza di “ligation”: quando le piastre di controllo contenevano una quantità inferiore di colonie rispetto alle altre si è assunto che il vettore fosse scarsamente capace di richiudersi su se’ stesso e che di conseguenza la maggior parte delle colonie sviluppatesi contenesse il vettore con l’inserto.
Da ogni piastra sono state prelevate 30-50 colonie: il plasmide estratto da ciascuna tramite “mini-prep” è stato analizzato per restrizione, per verificare la presenza dell’inserto e il suo verso.
Plasmidi recanti l’inserto con il verso corretto sono stati successivamente sequenziati per controllare la correttezza della sua sequenza.
Defosforilazione con SAP (“shrimp alcaline
phosphatase”)
Quando non è stato possibile effettuare clonaggi direzionali (vettore digerito con due differenti enzimi di restrizione per forzare l’inserto a saldarsi in un verso noto), per impedire la risaldatura spontanea del vettore si è reso necessario defosforilarne le estremità mediante SAP (“shrimp alkaline phosphatase”, Roche) con il seguente protocollo:
• Assemblare la reazione con: o DNA linearizzato
o SAP 10 unità
o SAP buffer 10x 10 µl
o H2O mQ fino a un volume di 100 µl o Incubare 1h a 37°C
Il DNA è stato successivamente purificato e usato nella reazione di “ligation” come descritto precedentemente.
Trascrizione in vitro di mRNA per microiniezioni
Per trascrivere mRNA senso da iniettare in embrioni di Xenopus laevis a partire da plasmidi linearizzati contenenti la sequenza di interesse clonata a valle del promotore SP6 è stato utilizzato il kit commerciale Roche SP6 Cap-Scribe:
• Assemblare in ghiaccio la reazione con: o 0.5-1 µg di DNA linearizzato o 4µl di Buffer Cap-Scribe 5x o 2µl SP6 RNA polimerasi
o portare a un volume finale di 20 µl con H2O mQ • Incubare 1h a 37°C.
• Prelevare 1µl del volume di reazione e conservare in ghiaccio per la successiva corsa di controllo.
• Aggiungere 2µ di DNase/RNase free (Invitrogen) e incubare per 15’ a 37°C. • Aggiungere 2µl EDTA 0.2M pH 8 in ghiaccio per fermare la reazione.
• Portare a un volume di 160 µl con H2O mQ, aggiungere 160 µl di fenolo-cloroformio-isoamil alcool 25:24:1 pH 7.5, e agitare con vortex.
• Centrifugare per 5’ a 14000 rpm a RT e recuperare la fase acquosa.
• Precipitare il trascritto con 0.7 V (volumi) di NH4Ac e 4.5 V di EtOH 100% ghiacciato. Rimescolare e lasciare 30’ a RT.
• Centrifugare per 20’ a 13000 rpm a 4°C, eliminare il sovranantante e lavare il pellet con EtOH 70% ghiacciato (non meno di 100 µl).
• Centrifugare 5’ a 13000 rpm a 4°C, eliminare il sovranatante, asciugare il pellet e risospenderlo in 25 µl H2O mQ.
• Prelevare 1 µl del campione e caricarlo in un gel di agarosio 1% con “loading buffer” tipo II insieme al campione prelevato in precedenza e a soluzioni di tRNA a concentrazione nota usate come marcatori di quantità. • L’assenza della banda del DNA stampo successiva al trattamento con
DNasi indica il buon fine della reazione; il confronto con i marcatori da’ una stima della concentrazione dell’mRNA prodotto.
• L’mRNA va suddiviso in aliquote di 1-2 µl e conservato a -80°C.
Trascrizione in vitro di RNA marcato con DIG-UTP.
Questo protocollo è stato usato nella trascrizione di RNA antisenso marcato con DIG-UTP (digossigenina-UTP) da usare come sonda negli esperimenti di “whole mount in situ hybridization”: l’ RNA polimerasi necessaria (SP6, T3 o T7) è stata scelta in base al verso di clonaggio della sequenza di interesse nel vettore, che è stato linearizzato di conseguenza.
• Assemblare la reazione con 1-1.5 µg di DNA linearizzato, 2 µl DTT 100 mM, 4 µl transcription buffer 5x, 2 µl DIG-labeling mix (Roche), 1 µl RNase OUT (Invitrogen), 2 µl RNA polimerasi. Portare a un volume finale di 20 µl con H2O mQ.
• Incubare 2h a 37°C.
• Prelevare 1µl del volume di reazione e conservare in ghiaccio per la successiva corsa di controllo.
• Aggiungere 2µ di DNase/RNase free (Invitrogen) e incubare per 15’ a 37°C. • Aggiungere 1 µl EDTA 0.5 M, 1/10 Vol di NH4Ac, 1 Vol isopropanolo.
• Centrifugare per 15’ a 13000 rpm a 4°C e rimuovere il sovranatante.
• Lavare con EtOH 75% (non meno di 100 µl) e centrifugare 3’ a 13000 rpm RT.
• Eliminare il sovranantante e risospendere in 22 µl H2O mQ.
• Prelevare 1 µl del campione e caricarlo in un gel di agarosio 1% con “loading buffer” tipo II insieme al campione prelevato in precedenza e a soluzioni di tRNA a concentrazione nota usate come marcatori di quantità. • L’assenza della banda del DNA stampo successiva al trattamento con
DNasi indica il buon fine della reazione; il confronto con i marcatori da’ una stima della concentrazione dell’mRNA prodotto.
Purificazione del DNA
I seguenti protocolli sono stati utilizzati per purificare campioni di DNA in seguito a reazioni quali digestioni con enzimi di restrizione, “ligation”, reazioni di PCR.
Eluizione da gel di agarosio con kit Nucleospin 2in1 Extract
©(Macherey-Nagel
)
Questo protocollo prevede l’uso di colonnine cromatografiche, commercializzate insieme alle soluzioni per il loro uso sotto il nome Nucleospin Extract© (Macherey-Nagel), ed è stato applicato tutte le volte che si è reso necessario estrarre una singola banda di DNA da un gel di agarosio successivamente a una corsa elettroforetica.
• Ritagliare dal gel un tassello contenente la banda di interesse, trasferirla in un tubo Eppendorf e stimarne il peso.
• Per ogni 100 mg di gel di agarosio aggiungere 300 µl di soluzione NT1 e incubare a 50°C per circa 10’, agitando su vortex ogni 2-3’ per facilitare lo scioglimento del gel.
• Porre il campione nella colonnina Nucleospin e centrifugare per 1’ a 8000 x g. Il DNA è trattenuto dalla colonnina.
• Scartare l’eluato, aggiungere 500 µl di soluzione NT2 che rimuove eventuali contaminazioni e centrifugare per 1’ a 11000 x g.
• Scartare l’eluato, aggiungere 600 µl di soluzione NT3 e centrifugare per 1’ a 11000 x g.
• Scartare l’eluato, aggiungere 200 µl di soluzione NT3 e centrifugare per 2’ a 11000 x g.
• Eluire con 30 µl di soluzione NE o con H2O mQ.
Precipitazione in Fenolo-Cloroformio isoamilico
• Portare il campione a un volume totale di 300 µl con H2O mQ.
• Aggiungere 300 µl (ugual volume del campione) di fenolo-cloroformio-isoamil alcool 25:24:1 pH 8 e agitare con vortex accuratamente fino a ottenere un’emulsione.
• Centrifugare 15’ a 11000 rpm RT.
• Recuperare il sovranatante avendo cura di evitare l’interfaccia e misurare il volume recuperato.
• Aggiungere 0.1 volumi di NaOAc pH 5,2 3M.
• Aggiungere 3 volumi di EtOH assoluto e precipitare ON a -20°C oppure 1 h a -80°C.
• Centrifugare 1’ a 12000 rpm a RT oppure 20’ a 15000 x g a -4°C e scartare il sovranatante.
• Lavare il pellet con EtOH ghiacciato e centrifugare 5’ a 11000 rpm a -4°C. • Eliminare l’EtOH e risospendere il pellet in H2O mQ o TE pH 8.0.
Purificazione con colonnine Nucleospin 2in1 Extract
©(Macherey-Nagel)
Questo protocollo prevede l’uso di colonnine cromatografiche, commercializzate insieme alle soluzioni per il loro uso sotto il nome Nucleospin 2in1© (Macherey-Nagel):
• Portare se necessario il volume del campione a 50 µl con TE pH 7.5
• Aggiungere 4 volumi di soluzione NT2 e caricare il campione sulla colonnina.
• Centrifugare 1’ a 12000 rpm e scartare l’eluato.
• Aggiungere 600 µl di soluzione NT3, centrifugare 1’ a 12000 rpm e scartare l’eluato.
• Aggiungere 200 µl di soluzione NT3, centrifugare 2’ a 12000 rpm per rimuovere NT3 quantitativamente e scartare l’eluato.
• Aggiungere 25-50 µl di soluzione NE, lasciare per 1’ a RT per aumentare la resa di DNA e centrifugare 1’ a 12000 rpm.
Sequenziamento
Il sequenziamento automatico impiega nucleotidi marcati con quattro fluorofori differenti che eccitati da un raggio laser emettono luce ad una lunghezza d’onda nota. Inoltre, tali nucleotidi sono modificati in ddNDT (2’,3’ dideossinucleosidi 5’ fosfato) e questo fa sì che là dove si inseriscono si interrompa la polimerizzazione del filamento nascente.
• Effettuando una PCR con una miscela di dNTP e di ddNTP marcati (oltre ad un appropriato primer per l’innesco della sintesi) si ottiene una serie di frammenti di amplificazione più o meno lunghi, a seconda del momento di incorporazione del nucleotide modificato.
• I frammenti marcati vengono separati automaticamente all’interno di un polimero e ciascuna banda viene analizzata per l’emissione di fluorescenza della lunghezza d’onda specifica per ogni tracciante.
• I picchi di emissione risultanti (rappresentanti ciascuno una singola base) sono poi elaborati da un software che fornisce l’elettroferogramma da cui si ricava la sequenza nucleotidica del frammento.
I risultati del sequenziamento presenti in questo lavoro di tesi sono stati forniti dal Servizio Sequenziamento della M-Medical GENENCO-Laboratori di Roma.
Metodi bioinformatici
Per l’analisi delle sequenze è stato usato il software DNAStrider Ver 1.1 . Per il confronto delle sequenze è stato usato il “software” per allineamenti multipli CLUSTAL W, disponibile “ondine” presso il seguente URL:
http://www.ebi.ac.uk/clustalw/index.html
Le sequenze sono state scaricate dal “database online Genbank”, gestito dai National Institutes of Health:
http://www.ncbi.nlm.nih.gov/
Embrioni di Xenopus laevis
Gli embrioni di Xenopus laevis per gli esperimenti sono stati ottenuti mediante fecondazione in vitro.
Prima di essere operato per la rimozione dei testicoli, il maschio è stato anestetizzato immergendolo in una soluzione 0.1% di MSS (metansulfonato dell’estere etilico dell’acido 3-aminobenzoico) disposta in ghiaccio per abbassare rapidamente il metabolismo dell’animale. I testicoli si possono conservare per 3-5 giorni a 4°C immersi nella soluzione per testicoli. Dopo l’operazione il maschio viene sacrificato. Lo stoccaggio avviene secondo la vigente normativa veterinaria.
Le femmine di Xenopus laevis sono state pre-stimolate con 100 UI di Folligon Intervet per uso veterinario da 4 a 11 giorni prima della deposizione e con 1000 UI di Profasi HP 5000 Serono (gonadotropina corionica) 10-12 h prima della deposizione: entrambi gli ormoni sono stati somministrati mediante iniezione nel sacco perilinfatico.
Le uova da fecondare sono state ottenute esercitando una leggera pressione sull’addome degli animali e raccolte in piastre Petri: questa operazione può essere ripetuta a intervalli di 1 ora. La fecondazione è stata effettuata bagnando le uova con una sospensione ottenuta sminuzzando un frammento di testicolo in MMR 1x e lasciandole in poco MMR 0.1x +gentamicina.
Dopo almeno 30 minuti o quando comunque fosse ben rilevabile la rotazione corticale degli embrioni, effetto dell’avvenuta fecondazione, gli embrioni sono stati privati del loro rivestimento gelatinoso ricoprendoli di soluzione degelificante e lasciandoveli 5-10 minuti e comunque fino a che non fosse evidente la perdita dl suddetto rivestimento. La soluzione degelificante è stata eliminata sciacquando gli embrioni in MMR 0,1%.
Gli embrioni vengono fatti sviluppare in MMR 0.1x fino allo stadio desiderato, valutato secondo i criteri di Nieuwkoop e Faber (Hubrecht-Laboratorium (Embryologisch Instituut) et al., 1967).
Gli embrioni sono stati di norma fissati in MEMFA 1h a RT o ON a 4°C e conservati in metanolo 100% a -20°C.
Microiniezione di embrioni di Xenopus laevis
Per gli esperimenti di microiniezione sono stati usati embrioni pigmentati in cui è possibile distinguere il polo animale da quello vegetativo e i blastomeri dorsali da quelli ventrali.
Al momento della microiniezione gli embrioni degellificati vengono trasferiti in una piastra Petri del diametro di 5 cm sul fondo della quale è stata fissata una reticella di plastica, con maglie di 1 mm, che ne limita gli spostamenti. Inoltre gli embrioni sono immersi in una soluzione di Ficoll al 4% (peso/volume) sciolto in MMR 0.1x: il Ficoll è uno zucchero viscoso, permette agli embrioni di mantenere la forma sferica durante la microiniezione e ne limita la perdita di citoplasma successivamente.
Gli embrioni microiniettati sono stati lasciati sviluppare ON in MMR 0.1%-ficoll 4%, trasferiti in MMR 0.1x finchè gli embrioni di controllo non inietttati non avessero raggiunto lo stadio desiderato e quindi fissati.
Le microiniezioni sono state eseguite con un microiniettore Drummond Nanoject, che consente l’iniezione di volumi compresi tra 4.6 nl e 73.6 nl ad incrementi discreti. L’iniettore è dotato di un micromanipolatore che ne permette lo spostamento macrometrico nelle tre dimensioni e di un movimento micrometrico controllato idraulicamente lungo una direzione predefinita.
Gli aghi sono stati preparati per tiratura a caldo a partire da capillari forniti da Drummond: la loro bontà è stata controllata allo stereoscopio. Prima di essere montati sul microiniettore gli aghi sono stati riempiti di olio minerale con una siringa. Il caricamento degli aghi con la soluzione da iniettare è eseguito dal microiniettore stesso: alla soluzione da iniettare è stato aggiunto Fast Green, un colorante vitale, utile sia seguire sia il caricamento dell’ago che la microiniezione.
Reazione cromogenica della β-galattosidasi
Quando è stato necessario, negli esperimenti di microiniezione è stato co-iniettato mRNA per β-galattosidasi citoplasmatica: ciò ha permesso di valutare la bontà della microiniezione attraverso la reazione catalizzata dall’enzima, che in presenza di un substrato cromogeno produce un precipitato colorato. Come substrato cromogeno è stato utilizzato il Salmon-gal, che produce un precipitato di colore rosso, seguendo il seguente protocollo:
• Si fissano gli embrioni microiniettati in MEMFA per 15’-20’. • 2 x 5’ lavaggi in PBS 1x (si può lasciare a 4°C fino a 48 h)
• Rivelare in soluzione di ferri: 5 µl di stock Salmon-gal in 500 µl di soluzione di ferri.
• Incubare a 37°C e controllare periodicamente la rivelazione. • 2 x 5’ lavaggi in PBS 1x.
• Fissare in MEMFA per 30’-45’ e trasferire in metanolo. Conservare a -20°C.
“Whole mount in situ hybridization”
L’ibridazione in situ su embrioni interi permette di studiare il “pattern” di espressione di uno o più geni nei diversi stadi embrionali di Xenopus laevis mediante l’ibridazione di sonde marcate con DIG-UTP ai trascritti presenti nei tessuti; il protocollo seguito è sostanzialmente quello descritto da Harland (Harland, 1991) :
• Gli embrioni vengono posti in vials da 4 ml RF (RNase free).
• Reidratare gli embrioni conservati in metanolo, con passaggi di 5’ ciascuno in:
o Metanolo 75 % PBTw 25 % o Metanolo 50 % PBTw 50 o Metanolo 25 % PBTw 75 o PBTw 100%
• Lavare 2x5’ in PBTw con agitazione orizzontale.
• Aggiungere 1 ml/vial di soluzione di proteinasi K in PBTw 1:2000; incubare 5’ senza agitazione.
• Effettuare due brevi lavaggi con PBTw con agitazione manuale.
• Sostituire il PBTw con paraformaldeide al 4% in PBS, 2 ml per vial; incubare 20’ a RT con agitazione manuale occasionale.
• Effettuare due brevi lavaggi con PBTw con agitazione manuale, quindi 4x5’ lavaggi in PBTw con agitazione orizzontale.
• Sostituire il PBTw con tampone di ibridazione al 50% in PBS, incubare 3’ quindi sostituire con tampone di ibridazione 100%.
• Incubare 3’, sostituire con tampone di ibridazione 100% e pre-ibridare 2-3 h a 60°C.
• Denaturare 50 ng di sonda di RNA marcato in 10 µl di H2O distillata per 2’ a 95°C, passare rapidamente in ghiaccio e aggiungere 600 µl di tampone di ibridazione (concentrazione finale della sonda: 83.3 ng/µl). Aggiungere la miscela alle vials e ibridare ON a 60°C.
• Preparare una soluzione SSC 2x/0.1% CHAPS e lasciarla ON a 37°C. • Preparare una soluzione SSC 0.2x/0.1% CHAPS e lasciarla ON a 60°C. • Recuperare la miscela con la sonda e sostituirla con 50% tampone di
ibridazione/50% (SSC 2x/0.1% CHAPS); incubare per 10’. • Incubare 2x30’ con SSC 2x/0.1% CHAPS preequilibrato a 37°C. • Incubare 2x30’ con SSC 0.2x/0.1% CHAPS preequilibrato a 60°C.
• Sostituire con 50% (SSC 0.2x/0.1% CHAPS)/50% TBS 1x a RT per 5’, quindi TBS 1x per 5’.
• Sostituire con 1ml/vial di blocking buffer, incubare 2h a 4°C con agitazione orizzontale.
• Diluire l’anticorpo AP Fab Anti-DiG (Roche) 1:3000 in blocking buffer e incubare 2h a 4°C con agitazione orizzontale.
• Sostituire il “blocking buffer” con 500 µl/vial di soluzione di anticorpo diluito e incubare 4 h a RT con agitazione orizzontale.
• Eliminare la soluzione di anticorpo e effettuare 5x1h lavaggi in TBSx a RT (uno può essere ON a 4°C) con agitazione orizzontale.
• Effettuare 2x5’ lavaggi con APB.
• Sostituire l’APB con BM Purple e incubare a RT al buio (incubando a 14°C il segnale risulta più pulito, ma è necessario un tempo più lungo; a 4°C la reazione si blocca): seguire la rivelazione, sostituire il substrato quando necessario e quando la marcatura è adeguata sostituire il BM Purple con TBSx per 10’ per bloccare la reazione.
La fosfatasi alcalina coniugata all’anticorpo scinde il substrato cromogenico (BM Purple) generando un precipitato colorato che evidenzia le zone in cui la sonda si è ibridata, dove il gene in esame è stato trascritto.
• Fissare gli embrioni in MEMFA 1 h a RT oppure ON a 4°C e conservarli in metanolo a -20°C.
Depigmentazione degli embrioni (“bleaching”).
Per evidenziare meglio il segnale dell’ibridazione gli embrioni sono stati sottoposti a depigmentazione:
• Sostituire il metanolo in cui sono conservati gli embrioni con metanolo 70% in H2O per 5’ a RT in agitazione orizzontale.
• Sostituire con 50% metanolo/ 50% SSC 1x per 5’ a RT in agitazione orizzontale.
• Sostituire con “bleaching solution” e porre le vials in agitazione orizzontale sotto una lampada fluorescente, controllando costantemente la depigmentazione.
• Fermare la depigmentazione passando in metanolo 70% per 5’ e successivamente in metanolo 100%. Conservare gli embrioni a -20°C.
Fotografie
Le fotografie degli embrioni sono state realizzate mediante una fotocamera digitale Roper Coolsnap CF montata in asse focale ad uno stereoscopio Nikon SMZ1500 con l’ausilio di una coppia di fibre ottiche Gli/136P.
Il trattamento delle immagini è stato realizzato con l’ausilio del software Adobe Photoshop CS.
Nota: gli animali utilizzati durante gli esperimenti sono stati trattati nel rispetto della vigente normativa d’Ateneo stabilita dal C.A.S.A. (Comitato di Ateneo per la Sperimentazione Animale).