2. METODI E MATERIALI.
2.1. PLASMIDE.
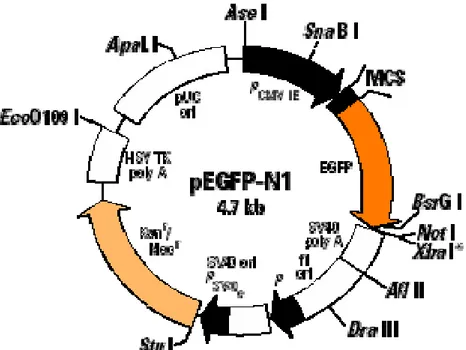
Il plamìsmide è un frammento di DNA presente nel citoplasma di numerosi batteri, capace di replicazione autonoma all’interno di una cellula (batterica o eucariotica). E’ di fatto un cromosoma indipendente che nella maggior parte dei casi è circolare. Possiedono delle caratteristiche peculiari come la resistenza agli antibiotici, resistenza a sostanze tossiche, capacità metaboliche aggiuntive che hanno a loro conferito un vantaggio evolutivo e ai ricercatori un ottimo strumento per la ricerca. Infatti i plasmodi sono strumenti fondamentali per il clonaggio genico e quindi nell’ingegneria genetica. Inoltre sono dotati di una porzione di DNA, contenente tutti i siti di taglio dei principali enzimi di restrizione, chiamata MCS (sito di multi clonaggio). Il plasmide che abbiamo utilizzato nel nostri esperimenti è il pEGFP-C2 ( PT3051, CLONTECH); la mappa è mostrata in fig 7
Le sequenze codificanti ERK1 ed ERK2 sono stato inserite in frame al 3’ della EGFP ( all’interno dell’MCS ) del plasmide.
2.2. AMPLIFICAZIONE DEL COSTRUTTO.
Cellule competenti di E.Coli sono state trasformate per shock termico con il plasmide codificante per ERK-GFP. La trasfezione è stata realizzata ponendo 50μl di cellule in ghiaccio per 30 minuti con 30ηg di plasmide. Questi poi vengono messi in stufa a 42 gradi per 30 secondi e 5 minuti in ghiaccio. Successivamente viene aggiunto 1ml di terreno SOC alle cellule competenti e messe su ruota per 1 ora a 37°centigradi.
Di tutto il preparato sono piastrati 100μl di batteri su Agar supplementato con Kanamicina.
2.3. COLTURE CELLULARI.
La linea cellulare di fibroblasti murini NHI 3T3, fatta crescere in DMEM/HIGH (Dulbecco’s Modified Eagle’s Medium HIGH glucose ECB7501L, Euroclone) con un supplemento del 10% di FBS (Fetal Bovin Serum CH4111L, HY CLONE)e di glutammina ed antibiotici (nelle concentrazioni standard), è stata utilizzata per i nostri esperimenti.
Queste sono state coltivate in piastre Petri con una confluenza dell’80%. Gli esperimenti sono stati condotti su cellule precedentemente private (circa 24 ore prima) di siero e mantenute in una soluzione fisiologica all’1% di siero e 0,5% di CO2.
2.4 TRASFEZIONE.
La trasfezione consiste nel trasferimento di molecole di DNA esogeno in cellule riceventi. Il DNA, una volta trasfettato può andare incontro a due destini alternativi: il costrutto viene mantenuto nel citoplasma per un determinato periodo di tempo, in genere due o tre giorni (trsfezione transiente) oppure viene integrato nel genoma (trasfezione stabile). L’isolamento delle cellule trasfettate richiede la selezione di un gene marcatore nel DNA esogeno che gli consenta di crescere in un terreno selettivo. Durante le 48 ore successive alla trasfezione, i geni esigeni sono soggetti a molte delle attività regolative che controllano l’espressione del materiale genetico della cellula. Successivamente a questo periodo, i plasmodi verranno persi per una combinazione di fattori quali diluizione e degradazione. Solo nel caso che questi vengano integrati stabimente nel genoma dell’ospite non vengono persi perché sono duplicati con il materiale genetico, quindi non possono essere diluiti, e non possono essere degradati. La trsfezione è un ottimo metodo di indagine utilizzato per la comprensione della strutture e della funzionalità di numerosi geni. Ci permette di identificare nuovi geni e di studiarne la loro regolazione.
In questo laboratorio viene eseguita la trasfezione transiente con lipofectamina. La lipofectamine sono una classe di molecole formate da una coda policationica a cui si lega il DNA e da una porzione lipidica che facilita il passaggio del complesso DNA-lipofectamina attraverso la membrana nucleare. Con questo metodo si ottimizza l’efficienza della trasfezione e si semplificano i passaggi sperimentali.
Il protocollo utilizzato è il seguente:
Il giorno precedente le cellule vengono divise e piastrate per la giornatasuccessiva. Una elevata efficienza di trasfezione richiede che le cellule raggiungono il 50-80% della confluenza.
Le cellule in piastrate in petri contenenti ciascuna 2 ml di DMEM, 10% FBS, glutammina e antibiotici.
Le cellule vengono mantenute in un incubatore termostatato a 37°C, umidità relativamente controllata e 5% CO2 per circa 24 ore
Il giorno della trasfezione il DNA viene diluito nel DMEM privo di antibiotici e siero ( 1 μg di DNA in 100 μg di terreno). A questa soluzione si aggiunge il reagente e si lascia in incubazione per 15 minuti a temperatura ambiente, in modo da favorire la formazione dei complessi reagente-DNA.
Nel frattempo si diluisce la lipofectamina, sempre in un terreno privo di siero e antibiotici ( 4 μl di lipofectamina in 100 μl di terreno).
Successivamente si unisce la lipofectamina ai complessi reagente-DNA, vortexando, e lasciando il tutto in incubazione per 30 minuti.
La soluzione viene poi portata ad un volume finale di 1 ml/petri.
A questo punto le cellule vengono private del terreno e lavate con 2 ml di terreno privo di siero e antibiotici per rimuovere degli eventuali residui di questi ultimi che comprometterebbero l’efficienza di trasfezione.
Il preparato è aggiunto alle cellule e posto in incubatore a 37°C per 5 minuti.
Dopo di che le cellule vengono private del mezzo, lavate con del terreno, è messo 1 ml di terreno completo per piastra e lasciate in incubatore 24 ore a 37°C.
2.5. Microscopia confocale.
Le cellule, precedentemente trasfettatte (con il plasmide codificante per la proteina EGFP-ERK) e successivamente private di siero, sono state osservate al microscopio confocale laser ( Olympus Fluoview BX50WI ). Le osservazioni sono state effettuate con un obbiettivo 60 x NA 0,9 ad acqua, immerso direttamente nel mezzo di coltura.
L’imaging è stato eseguito con una intensità laser pari al 1,5% di quella massima, con un valore di pinhole di 5 e un PMT di 675 mV.
Per la visualizzazione del processo di traslocazione è stata effettuata una scansione ( media di due immagini) ogni 20 secondi. L’attivazione della via degli ERK è stata ottenuta somministrando alle cellule due stimoli diversi: siero e FGF 4; il primo è stato disciolto in una soluzione extracellulare di registrazione ( con HEPES Bicarbonato) e somministrato tramite un sistema di per fusione continua con flusso di 1 ml/minuto. Quest’ultimo è stato generato per gravità ed il volume di soluzione all’interno della camera di registrazione è stato mantenuto costante da una pompa peristalsica. L’altro stimola, l’FGF, è stato aggiunto direttamente nella camera di registrazione ad una concentrazione di 80 ng/ml.
Le registrazioni sono state analizzate utilizzando il programma per l’analisi delle immagini Fluoview ( Olympus ). Per ogni cellula sono state disegnate delle aree relative al nucleo e citoplasma e ne è stato calcolato il valore medio di fluorescenza.
CYT TOT NUC
Figura 8
Rappresentazione del metodo di selezione della regione di interesse (ROI) entro la quale viene misurata l’intensità di fluorescienza
Durante la registrazione si possono verificare dei cambiamenti di messa a fuoco che contribuiscono alla rumorosità del segnale della fluorescenza riemessa.
La rumorosità del segnale detta rumore di fondo, può avere diverse origini tutte dovute alle difficoltà sperimentali, ad esempio:
La variazione della morfologia della cellula ( dovuta alla motilità di quest’ultima) durante la registrazione.
Il fenomeno di photobleaching provocato dalle ripetute esposizioni al laser.
Un’alterazione consistente del segnale è dovuta all’introduzione del siero perché ha un certo grado di autofluorescenza e un indice di rifrazione molto diverso dal liquido di coltura e ciò causa un cambiamento del piano focale. Per questo motivo viene effettuata una correzione manuale del piano focale. Tutti questi inconvenienti possono essere ridimensionati trattando il segnale con di fluorescenza come rapporto N/C, questo parametro di riferimento descrive la distribuzione della fluorescenza tra i due compartimenti e rappresenta una stima del grado di traslocazione nucleare, in più offre il vantaggio di buona parte degli effetti dovuti al rumore sperimentale perché si ripercuotono, nelle condizioni di registrazione, nello stesso modosu tutte le tracce rilevate. In alcuni casi è stato sottratto anche il segnale di background.
2.5.1.Esperimenti di FRAP: trasporto nucleo-citoplasma
Ogni “photobleaching” è preceduto dall’acquisizione di un’immagine a basso rumore dell’intera cellua. Quest’ultima (pre-bleach) è usata per stimare la perdita di fluorescenza dovuta al processo di bleaching e per la normalizzazione dei dati. Il nucleo di una o due cellule per campo è stato fotoschiarito con scansioni ripetute lungo una linea centrata sul nucleo ad un’alta potenza di laser (0,7 mW di potenza totale). La scansione di schiarimento è stata applicata per circa 8 secondi ed è stato possibile notare come questo tempo sia sufficiente per schiarire la maggior parte della fluorescenza nucleare, indicando che il tempo caratteristico per la diffusione di ERK2-GFP nel compartimento nucleare è molto minore di 10 secondi. Il recupero è stato misurato facendo partire un’acquisizione di time-lapse entro 2 secondi dalla fine del photobleaching (60 immagini in un tempo di 5 secondi). Il recupero è descritto dalla funzione ) ( ) ( ) ( t t t F
F
F
F
F
Tot Nuc PB Nuc PB Tot ⋅ =ove Fpb indica la fluorescenza (corretta per il fondo) misurata per lo schiarimento nel nucleo (Nuc) o nell’intera cellula (Tot). Questo modo di esprimere il recupero corregge per qualsiasi schiarimento che potrebbe avvenire durante l’acquisizione in time-lapse (Phair, 2000). In assenza di frazioni immobili, la formula riporta un valore asintotico di 1. Il recupero di tutte le cellule acquisite è stato accuratamente adattato con un singolo esponenziale che riporta le costati di tempo del trasporto e dell’entità della frazione immobile (valore asintotico).
2.6. Saggio di poliadenilazione RACE-PAT.
Questo saggio consiste nell’amplificazione di cDNA, che nel mio caso è ottenuto dall’mRNA di ERK1 ed ERK2, tramite dei cicli di PCR utilizzando un primer foreward studiato sulla porzione terminale del 3’ UTR dei su detti RNA messaggeri; e come primer reverse lo stesso oligo dT utilizzato nella reazione di retrotrascrizione. Il prodotto di questo passaggio è costituito da un pool di cDNA di lunghezza variabile in funzione dell’ appaiamento casuale dell’oligo dT Anchor primer sulla coda di poliadenilazione del messaggero degli ERK. I possibili appaiamenti di questo ultimo primer sono casuali quindi il prodotto della PCR rappresente, complessivamente, la lunghezza della coda di poliadenilazione dell’RNA messaggero degli ERK.
Il prodotto finale della RACE-PAT viene corso su di un gel di agarosio al 2% e fotografato. Il profilo elettroforetico che osserviamo nella foto è costituito di uno smear che è tanto più esteso quanto più estesa è la coda di poliadenilazione del messaggero. La lunghezza della smear rappresenta lo stato di poliadenilazione dell’RNA degli ERK, quindi una differenza significativa tra gli smear dei due ERK e tra i campioni (di ciascun ERK) sottoposti a trattamento e quelli di controllo, indicano una differente regolazione tra ERK1 ed ERK2 e una relazione tra il trattamento e il grado di poliadenilazione del messaggero di ogni singolo ERK.
Primer 3’UTR
specifico
Anchor dt primer
Profilo elettroforetico
caratterizzato da uno smear
sopra una banda minima
Lunghezza variabile della
coda di poli-adenosine
RT, PCR
2
AAAAAAAAAA
AAA
1
Primer 3’UTR specifico
Anchor dt primer
RT, PCR
Lunghezza costante della
coda di poli-adenosine
PAT
-RACE
Saggio dello stato di poliadenilazione dell’mRNA
Figura 9
2.7. Saggio di poliadenilazione LM-PAT.
Questa tecnica è una modificazione del metodo RACE-PAT, studiata per avere una sensibilità superiore rispetto alla lunghezza della coda di poliadenilazione. Il saggio consiste nell’incubare tutto l’RNA estratto con oligomeri di 12-18 nucleotidi di d-Timina fosforilati, che va ad ibridarsi con la coda di poli-A saturando la maggioranza dei siti disponibili dall’Anchor primer oligo dT. La ligasi T4 lega gli oligo dT alla temperatura di 42° C. Gli estremi 5’o 3’ dell’RNA restano liberi dall’appiaamento con il p(dT)12-18 nella maggior parte dei casi per via di condizioni sfavorevoli di ibridazione. A seguire, viene aggiunto il primer ancorato con un eccesso di 5X rispetto al p(dT)12-18 e pasta la miscela di reazione ad incubare per 12 ° centigradi, dove questo ultimo primer avrà una alta probabilità di appaiarsi in posizione terminale in 3’ della coda di poliadenilazione. Alla fine di questo periodo viene rialzata la temperatura per permettere alla ligasi di legare il 3’ del’oligo dT al 5’ dell’Anchor primer. Complessivamente il primer ancorato legato all’oligo dT serve da primer reverse nella successiva fase di retrotrascrizione. Il prodotto di questo processo è amplificato mediante PCR come nella RACE-PAT, corso su un gel al 2% di agarosio e fotografato. Il profilo elettroforetico presenta uno smear con un aspetto bandeggiato e meno omogeneo.
Saggio dello stato di poliadenilazione dell’mRNA
LM-PAT
Lunghezza costante della co Allungamento della coda di poli-adenosine
Figura 10
Rappresentazione schematica del saggio di LM-PAT.
AAAAA
AAAAAAAAAA
Oligo-dTTT
TT
TT
TT
TT
AAAAA
AAAAAAAAAAAAA
TT
TTTTTTTTT
RT, PCR
RT, PCR
Ligasi T4
1 2 Oligo-dT 1 2 Primer 3’UTRspecifico
Primer 3’UTRspecifico
Anchor dt primer
Anchor dtprimer
Profilo elettroforetico caratterizzato da una molteplicità di bande sopra una banda minima
2.8. Southern blot.
La tecnica del Southern blot è usata per rilevare la presenza di specifiche sequenze di DNA in una miscela complessa. Nel nostro caso ci siamo avvalsi di questa tecnica per trasferire il cDNA, prodotto dal saggio di RACE-PAT e LM-PAT, su di un foglio di nitrocellulosa, con lo scopo di sottoporlo ad ibridazione con una sonda specifica.
Nel nostro esperimento abbiamo caricato il prodotto della PCR su un gel al 2% di agarosio con un DNA ladder da 1 Kb. Durante l’elettroforesi i frammenti di DNA,che sono carichi negativamente a causa dei gruppi fosfato, vengono respinti dall’elettrodo negativo verso quello positivo e devono passare attraverso i pori del gel. I frammenti più piccoli si muovono più velocemente di quelli più grandi, separandoli in base alle dimensioni del frammento. Al termine della corsa viene fatta una foto al gel con una scala di riferimento; in questo modo abbiamo un metro di misura a cui paragonare il risultato dell’esperimento. Dopo di che il gel è: trattato con HCl (0,25 M) per 15 minuti su bascula, lavato con acqua distillata, immerso nel Denaturatine buffer per 30 minuti, nuovamente lavato con acqua distillata e sottoposto a due lavaggi (ciascuno di 15 minuti) con Neutralization buffer su bascula. Il passaggio successivo è il trasferimento del DNA dal gel ad un foglio di nitrocellulosa, dove saranno immobilizzati sulla membrana in modo da avere una copia fedele della separazione per dimensione attenuta con l’elettroforesi. Per far ciò è allestito un apparato per il Southern blot utilizzando un foglio di nitrocellulosa e SSC 20X come transfer buffer. Il filtro è rimosso, dopo blotting overnight, sciacquato con SSC 20X, asciugato e sottoposto a cross-linking in stufa a 80°C per 2 ore, avvolto in carta 3MM e in stagnola.
Per controllare l’avvenuto trasferimento, dopo gli opportuni lavaggi, viene preso il gel e osservato al transilluminatore; se tutto è avvenuto correttamente non si osserva niente.
2.9. Ibridazione su filtro.
Trasferito il profilo elettroforetico sul filtro, questo viene inserito in un tubo da 40 o 80 ml(a seconda delle dimensioni del filtro) dove viene aggiunto rispettivamente 40 o 80 ml di Rapid Hyb Buffer e successivamente messo in agitazione su di un dispositivo ruotante in stufa a 65°C per una ora. Al termine di questo periodo di tempo viene aggiunto al buffer nel tubo un volume di sonda che è pari ad una attività di 1x106 CPM per ogni millilitro di soluzione di ibridazione e rimesso sulla ruota per tutta la notte. Successivamente a questo passaggio si effettuano due lavaggi da 30 minuti ciascuno in stufa a 65°C su un supporto rotante, dopo di che i filtri vengono sigillati in canta 3MM per non farli asciugare.
2.10. Autoradiografia.
L’autoradiografia è un metodo per visualizzare bande di DNA separate sul gel e trasferite su un filtro di nitrocellulosa. Questa tecnica è utilizzata per evidenziare strutture intra- o extracellulari marcate con isotopi redioattivi. In questa metodica sperimentale una lastra fotografica è messa ad impressionare, per un periodo di tempo variabile, all’interno di un apposito contenitore nella camera oscura. Dopo di che questa viene sviluppata in una apposita macchina, sempre in camera oscura, ed analizzata. Quello che si ottiene è un tracciato di bande che indicano la presenza di DNA, le dimensioni dei frammenti di DNA che costituiscono la banda e come nel nostro caso l’attivazione di meccanismi molecolari come la poliadenilazione citoplasmatica dell’mRNA. Questa tecnica possiede una vasta gamma di applicazioni perché può essere utilizzata per evidenziare il legame di farmaci con recettori. Oppure in biologia molecolare è comunemente utilizzata per evidenziare l’ibridazione di una sonda radioattiva con RNA o DNA denaturati, in
vari esperimenti quali il Southern blot, Northern blot, ibridazione di colonie batteriche o di placche fagiche.
2.11. APPENDICE.
TERRENI PER LA CRESCITA BATTERICA.
SOC medium Bacto-triptone 20g Estratto di bacto-lievito 5g NaCl 0,5g Glucosio 20g H2O deionizzata 950ml LB medium Bacto-triptone 12g Estratto di bacto-lievito 24g Glicerolo 4ml H2O deionizzata 950ml
SOLUZIONE DI REGISTRAZIONE Soluzione extracellulare Sulfimilpirazone 0.0162g/21 Acido ascorbico 0.3522g/21 Mio inositolo 0.1802g/21 Piruvato 0.4400g/21 NaCl 132.80ml/21 KCl 3.10ml/21 CaCl 8.00ml/21 MgCl2 2.00ml/21 K2HPO4 2.00ml/21 HEPES/NaOH 40.00ml/21 NaHCO3 16.00ml/21 Glucosio 1.8020ml/21
CONDIZIONI DI REAZIONE DELLA 3’ RACE
La reazione di PCR viene condotta nelle seguenti condizioni
93°C/ 5 ‘ 93°C / 30’’ 58°C / 30’’ 72°C / 1’ 72°C / 7’ 4°C ad libitum
CONDIZIONI DI REAZIONE LM-PAT
La reazione viene condotta nelle seguenti condizioni:
93°C/ 5 ‘ 93°C / 30’’ 58°C / 30’’ 72°C / 1’ 72°C / 7’ 4°C ad libitum
35 cicli
GENE IMAGES 3’-OLIGOLABELING KIT
Oliginucleotidi 10μM 1μl Fluorescein-11-dUTP 0.5μl Cacodylate buffer 0.8μl Acqua 5.3μl Terminal trasferase 0.4μl Soluzioni. 5x SSC 0,1% (w/v) SDS 0,5% destran solfato (mw 500000)
GENE IMAGES CDP-START DETECTION KIT Baffer A
100mM Tris-HCl 12,119 gr 300mM NaCl pH 9,5 17,539 gr
Si aggiunge acqua fino al volume di 1 litro e autoclavata. Bovine serum albumin (BSA) fraction V