MATERIALI E METODI
BIOINFORMATICA
Ricerca dei siti di Splicing
La sequenza nucleotidica del clone BC072971.1 è stata analizzata con SplicePort, un software bioinformatico che, basandosi sulle sequenze di splicing note per i geni umani immagazzinate in bancadati, riconosce eventuali siti di splicing in una qualsiasi sequenza immessa. Il software si può trovare al sito http://spliceport.cbcb.umd.edu/SplicingAnalyser.html.
Ricerca in bancadati NCBI
Al fine di cercare un cDNA della JHDM1D senza introne, è stato usato l'algoritmo BLASTn dell'NCBI (National Center for Biotechnology Information) accessibile all'indirizzo web http://blast.ncbi.nlm.nih.gov/Blast.cgi. Questo algoritmo ha permesso il confronto fra il clone IMAGE BC072971.1 (la JHDM1D full lenght con un introne) e tutte le sequenze contenute nel database dell'NCBI, disponibili in rete. In questo modo è stato trovato il clone XL043i05 (la JHDM1D senza introne) proveniente da una library di Xenopus laevis allo stadio di neurula (stadio 15), del consorzio giapponese NIBB, su cui si è incentrato il mio lavoro di tesi.
Disegno dei primer per il sequenziamento
Dal momento che in banca dati sono presenti solo le estremità 3' e 5' del cDNA XL043i05, è stato necessario sottoporre a sequenziamento tutta la sequenza. Inizialmente sono stati usati i primer universali T7 ed M13.
Una volta decodificato il tratto immediatamente successivo, ho disegnato nuovi primer con l'ausilio del software gratuito di analisi bioinformatica "NetPrimer",
sviluppato da Premier Biosoft, all'indirizzo http://www.premierbiosoft.com/netprimer/index.html. I primer sono stati disegnati rispettando dei criteri forniti dalla ditta milanese Primm, in modo da ottenere i migliori risultati col sequenziamento automatico. Gli oligo disegnati hanno tutti una temperatura di melting compresa fra i 50 e i 55 °C e hanno come ultima base al 3’ una guanina o una citosina (Tabella 1).
Primer Forward Primer Reverse
F2_JHDM CCGATGATTCGTTCTTCAC R2_JHDM GACAGCTTCTTGCTGATGTC F4_JHDM GTCGAGGAGGAAGGGAAC RJ3.5 CCTTTTTTCCATCTCGTAAC F4.5_JHDM CAGACTGTAAGACTACGGGGAG R4_JHDM TTCCCTTCCTCCTCGACC F5_JHDM GTGACCCTAATTTCCAGTGG R5_JHDM CTTTTATGGGGAGAATCAGG JHDM_F7 ACCCGCTTCTTTGTCTG JHDM_R6 GCGACTCCCAGTGCTC R7_JHDM AAATGATAAAAAAGGAACAGTG
Tabella 1. Primer usati per il sequenziamento automatico del clone XL043i05.
Assemblaggio della sequenza del gene JHDM1D
Dopo aver raccolto tutti i frammenti forward e reverse basandomi sulla bontà dell'elettroferogramma risultante dal sequenziamento automatico, ho assemblato separatamente il filamento forward e quello reverse e li ho confrontati in formato FASTA utilizzando il software Serial Cloner 1.2. Ho ottenuto così la sequenza codificante del clone NIBB XL043i05 pronta da immettere in bancadati.
TECNICHE DI BIOLOGIA MOLECOLARE
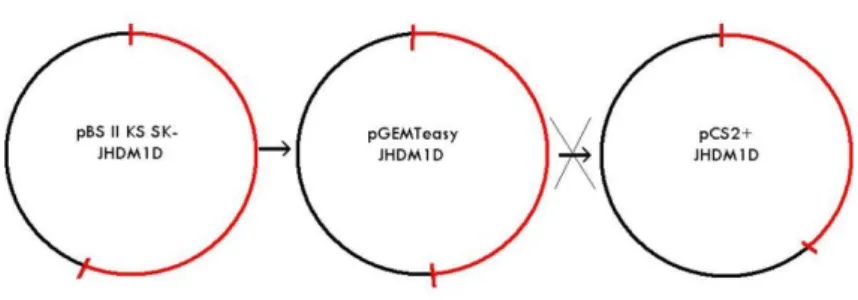
SUBCLONAGGIOAl fine di sovraesprimere la JHDM1D negli embrioni di Xenopus laevis, la demetilasi è stata subclonata a partire da un pBlueScript II SK-, nel vettore di
espressione pCS2+. Gli approcci utilizzati sono stati due, ma solo uno si è rivelato
vincente. Il primo ha previsto diversi tentativi di taglio e di ligazione a partire da un inserto dapprima trasferito nel vettore pGEMT-easy. Nello specifico, tale strategia comporta prima l’amplificazione dell’inserto per PCR con primer appositamente disegnati per contenere due diversi siti di restrizione al fine di ottenere un clonaggio direzionale. Poi, l'inserto così amplificato viene inserito in pGEMT easy grazie all'estremità sticky a singola base del vettore (una timidina) e dell'inserto (una adenina aggiunta tramite “tailing” al prodotto di PCR). La direzionalità dell'inserto in pGEMT easy viene saggiata tramite analisi di restrizione. Il clone così selezionato viene inviato a sequenziare, previo disegno di oligo forward e reverse specifici per la sequenza in esame, al fine di confermare il frame di lettura dopo il passaggio in PCR.
Successivamente l'inserto è stato escisso con enzimi di restrizione che lasciassero estremità sticky compatibili con le estremità del vettore di espressione finale, il pCS2+.
Quest’ultimo passaggio da pGEMT-easy a pCS2+, dopo svariati tentativi, non è
riuscito (Figura 17).
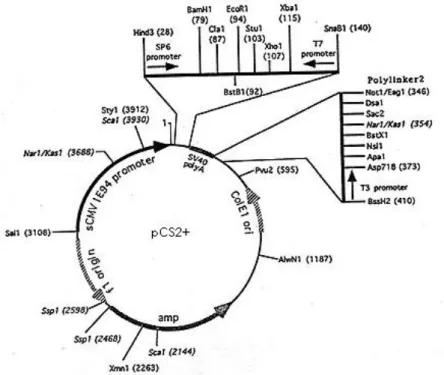
Il secondo approccio si è avvalso della tecnica di PCR per inserire il frammento neosintetizzato da una DNA polimerasi altamente fedele ed efficiente (la pfu DNA pol) su inneschi disegnati per un clonaggio direzionale, direttamente nel plasmide
pCS2+. Questo plasmide contiene un promotore forte eucariotico (CMV della scimmia
IE94), un gene di resistenza all'ampicillina, un polylinker fiancheggiato dai promotori
Figura 1. Rappresentazione schematica delle fasi successive di subclonaggio.
delle RNA polimerasi SP6 e T7, un sito di poliadenilazione SV40 e un secondo polylinker - a valle del sito SV40 - provvisto di siti di restrizione diversi e generalmente poco comuni nei geni eucariotici, per linearizzare il plasmide e trascrivere il messaggero con la RNA polimerasi SP6 (Figura 18). Una volta ottenuti inserto e vettore da ligare insieme, il clonaggio prevede due tappe diverse: la
ligation e la trasformazione.
Figura 2. Vettore pCS2, comunemente usato per
esperimenti funzionali nello Xenopus. Amp=ampicillina; ColE1 ori=origine di replicazione batterica.
Polymerase chain reaction (PCR)
Per subclonare la sequenza codificante della JHDM1D del clone NIBB XL043i05 dal plasmide pBluescript II SK- al vettore di espressione pCS2+, la JHDM1D è stata
amplificata per PCR (reazione a catena della Polimerasi). Tale tecnica amplifica selettivamente una regione di DNA compresa fra due oligonucleotidi detti primer, orientati in modo opposto e complementari a una determinata sequenza posta su ognuno dei due filamenti del DNA da amplificare. I primer Forward (JF1) e Reverse (JR1) sono stati disegnati inserendo il sito di restrizione ClaI per il primer Forward e il sito di restrizione XbaI per il primer Reverse (Tabella 2).
Primer per il clonaggio
GGATTATCGATATGGCCGGAG CTGTCTAGACAAAGAAGCG Tabella 2. Primer usati per il clonaggio. In celeste sono evidenziati i siti di restrizione
inseriti nel disegno dei primer: ClaI nel primer forward e XbaI nel primer reverse.
I primer per essere efficaci, sono stati disegnati seguendo i seguenti criteri: Lunghezza compresa fra 20-30 basi.
Massima specificità per la regione target e nessuna omologia con altre zone del clone.
Contenuto in GC compreso fra il 45-50%. Assenza di dATP al 3'.
Assenza di sequenze tra loro complementari oppure di sequenze ripetute invertite; si formerebbero infatti aggregati di primer (cosiddetti dimeri di
primer) o strutture a forcina.
Il kit usato è Pfu DNA Polymerase-mediated PCR amplification della Promega. La Pfu DNA Polimerasi è un enzima termostabile di circa 92kDa con attività polimerasica 5' -3'. Durante la polimerizzazione, errori di sintesi sono corretti efficientemente dalla Pfu DNA polimerasi, dotata di attività esonucleasica 3'-5' (polimerasi proofreading) indispensabile per reazioni che richiedano alta fedeltà di scrittura, come nel mio caso specifico. La miscela di reazione di PCR è composta da:
Buffer 10X
dNTPs mix 10mM Primer Fw (JF1) Primer Rv (JR1)
pBS II KS SK-/JHDM1D
Pfu DNA Polimerasi (Promega) 2,5u/µl
La miscela viene inizialmente portata a 95°C per 2 minuti. Seguono poi 25 cicli di amplificazione del DNA, composto ciascuno di tre fasi. La prima è la denaturazione, a 95°C per 30 secondi, che separa i due filamenti di DNA. La seconda è l'annealing, in cui la temperatura raggiunge il valore di melting dei primer (50°C per 30 secondi) per permettere a questi di appaiarsi alle sequenze complementari sul DNA. A questo punto lo stampo, con l'innesco dei primer è pronto per la Pfu DNA polimerasi che, nella terza fase detta di "prolungamento" (a 72°C per 8 minuti), procederà con la sintesi di un nuovo filamento. Il ciclo ricomincerà dalla fase di denaturazione (30 secondi a 95°C) per separare il filamento originario da quello di nuova sintesi. Entrambi fungeranno da stampo per il prossimo ciclo. Alla fine dei 25 cicli di sintesi la temperatura viene mantenuta per ulteriori 5 minuti a 72°C per permettere alla Polimerasi di completare la sintesi; la reazione viene fermata a 18°C fino alla successiva purificazione.
Purificazione del prodotto di PCR
L'intera miscela di PCR viene caricata su gel di agarosio all'1% e fatta correre a 90 Volt per circa mezzora. Con la lampada agli UV accesa e con tutti i dispositivi di protezione appropriati, si ritaglia la banda del prodotto di PCR dal gel di agarosio usando una lama da bisturi sterile.
Il DNA verrà liberato dalle maglie del gel di agarosio e purificato dai reagenti della
reazione di PCR usando il kit Gen EluteTMPCR Clean-Up (SIGMA), seguendo le
indicazioni fornite dal produttore, ad eccezione del volume di risospensione finale di 40µl invece che 50µl di acqua mQ fresca.
Ligation
Il frammento ottenuto per PCR e il vettore pCS2+ vuoto vengono sottoposti a
doppia digestione enzimatica da parte delle endonucleasi ClaI e XbaI e poi ligati insieme, in vitro, dalla ligasi T4. Come controllo si sottopone anche il solo vettore vuoto a una reazione di ligation "self". Se tutto ha funzionato bene, infatti, le estremità
rilasciate dai due diversi enzimi di restrizione selezionati per il sub clonaggio direzionale, non sono reciprocamente compatibili. La reazione di ligation permette l'inserimento della JHDM1D nel plasmide tramite formazione di ponti fosfodiesterici promossi dalla T4 DNA ligasi. Le miscele, tenute over night (ON) a 22°C sono:
Ligation pCS2+/JHDM1D Ligation self
2 µl pCS2+ 2 µl pCS2+
6 µl JHDM1D 2 µl Buffer 10X
2 µl Buffer 10X 2 µl T4 DNA ligasi
2 µl T4 DNA ligasi 14 µl acqua mQ
8 µl acqua mQ
Segue poi uno shock a 68°C per 10 minuti per inattivare l'enzima.
Preparazione di cellule competenti
Il ceppo di E. Coli usato nel nostro laboratorio è il DH5α. Le cellule sono rese competenti, cioè in grado di accettare DNA plasmidico esogeno, chimicamente tramite il metodo del cloruro di rubidio. Il protocollo prevede di:
1. Prelevare una colonia cresciuta ON su terreno solido e inocularla in 50ml di terreno liquido.
2. Far crescere i batteri a 37°C in agitazione (150rpm) finché la densità ottica (OD) misurata a 600 nm segna il valore 0,2.
3. Interrompere la crescita mantenendo 3 minuti in ghiaccio. 4. Centrifugare in tubo sterile per 10 minuti a 5000 rpm a 4°C.
5. Eliminare il sopranatante e risospendere in ½ del volume iniziale (circa 25ml) di RbCl 50 mM freddo.
6. Mantenere 30 minuti in ghiaccio.
7. Centrifugare in tubo da batteri sterile per 10 minuti a 5000 rpm a 4°C. 8. Eliminare il sopranatante e risospendere in 1/50 del volume iniziale (circa
0,8ml) di RbCl 50 mM freddo.
Trasformazione di cellule E. Coli DH5α
La trasformazione è avvenuta usando quantità diverse di plasmide derivante dalla
ligation pCS2+/JHDM1D e dalla ligation self aggiunte, separatamente, a 100µl di
cellule E. Coli DH5α competenti. La trasformazione batterica consta di diverse fasi: 1. Lasciare la miscela di ligation più le cellule competenti in ghiaccio per mezzora. 2. Sottoporre le cellule a uno shock termico a 42°C per 45 secondi e subito di nuovo in
ghiaccio per 2 minuti circa.
3. Aggiungere 900µl di LB e incubare le cellule in stufa, a 37°C per un'ora, in agitazione.
4. Centrifugare a 12000 rpm per 2 minuti.
5. Eliminare il sopranatante e risospendere le cellule in 20µl circa di LB.
6. Piastrare le cellule su terreno solido selettivo di LB con agar e ampicillina 50µg/ml. Per completare l'esperimento, vengono piastrati anche due controlli negativi, ossia cellule che sono state trattate solo con acqua mQ e cellule trasformate con ligation self.
7. Dopo incubazione delle piastre a 37°C per tutta la notte, sulla piastra Petri
compaiono colonie su tutte le piastre, tranne che nei controlli negativi. L'antibiotico, infatti, seleziona solo quelle colonie che hanno incorporato il plasmide pCS2+
richiuso, contenente il gene che conferisce resistenza all'ampicillina.
Terreni di coltura
Luria-Bertani Broth (LB): NaCl 1%
bacto tryptone 1% bacto yeast extract 1% Bottom agar:
agar sciolto in LB 1,5%
Estrazione di DNA plasmidico su piccola scala mediante lisi alcalina (miniprep)
Ottenuti i cloni sul terreno di coltura selettivo, alcuni di essi vengono “screenati” e selezionati come positivi. Viene effettuato in condizioni sterili, un inoculo di una singola colonia in 4ml di brodo LB con ampicillina (100µg/ml) in un tubo da batteri da 15ml. La colonia viene fatta crescere per circa 12-16 ore a 37°C in agitazione, condizioni necessarie a raggiungere la fase di crescita stazionaria. Dopodiché si procede con il protocollo dell’estrazione di DNA plasmidico su piccola scala:
1. La coltura viene centrifugata a 12000rpm per 2 minuti, ottenendo così un pellet di cellule batteriche che verrà risospeso, vortexando, in 400µl di soluzione S1. 2. Segue il passaggio della lisi alcalina, aggiungendo 400µl di soluzione di lisi S2
e mescolando il tubo 3-4 volte delicatamente, per non più di 5 minuti.
3. Trascorsi i 5 minuti, si aggiungono 400µl di soluzione neutralizzante S3, che permette la precipitazione delle membrane e delle pareti cellulari lisate, insieme al DNA genomico e all'RNA ad alto peso molecolare che restano a esse agganciati.
4. Dopo una centrifuga di 15 minuti a 12000rpm si recupera il sovranatante contenente il DNA plasmidico, l'RNA a basso peso molecolare e le proteine batteriche.
5. Per eliminare la contaminazione proteica si procede con la precipitazione a temperatura ambiente per 5-10 minuti con isopropanolo (0,7 volumi), cui segue una centrifuga di 15 minuti a 14000rpm.
6. Per eliminare l'RNA a basso peso molecolare precipitato insieme al DNA, il pellet ottenuto viene lavato in etanolo (EtOH) al 70% e risospeso in 20-25µl di acqua ultrapura, contenente RnasiA (100µg/ml).
Questa procedura di estrazione, consente di ottenere circa 2µg di DNA plasmidico. La concentrazione viene stimata per corsa elettroforetica, confrontando un'aliquota con altre a concentrazioni note che fungono da standard.
Soluzioni Soluzione 1 Tris-HCl 25mM pH8 EDTA 10Mm Glucosio 50mM Lisozima 10mg/ml Soluzione 2 NaOH 0,2M SDS 1% Soluzione 3 CH3COOH 5M KCOOH 3M
Stima della concentrazione di DNA.
Elettroforesi su gel di agarosio. Allo scopo di valutare l'efficienza del processo di
estrazione del DNA plasmidico in termini di purezza e di quantità, ho utilizzato dei gel di agarosio allo 0,8%, preparati facendo sciogliere l'agarosio in TBE 1X tramite ebollizione. Prima che il gel polimerizzi, viene aggiunto bromuro di etidio (1:20000) che, intercalandosi tra le basi, permette di evidenziare il DNA agli UV. Si cola il gel in un lettino da elettroforesi orizzontale e si aspetta che polimerizzi. Una volta pronto, il gel viene adagiato in un apparato da elettroforesi pieno di tampone di corsa TBE 1X. La migrazione varia dagli 80 ai 120 Volt per un tempo massimo di un'ora. I campioni vengono caricati aggiungendo 1/6 del volume finale di loading buffer di tipo III, un colorante che appesantisce il DNA, permettendo che questo si depositi sul fondo del pozzetto e che permette la visualizzazione a occhio nudo della corsa.
Spettrofotometria UV. Per la quantificazione precisa di DNA plasmidico estratto, si
misura l'assorbanza alla lunghezza d'onda di 260nm allo spettrofotometro, assicurandosi che il raggio di misurazione non sia fuori dal range di sensibilità dello strumento. Sono infatti considerate attendibili le misurazioni di assorbanza comprese fra 0,08 e 2 per lo spettrofotometro utilizzato nel nostro laboratorio, il GeneQuant
Soluzioni
TBE 10X pH 8
Tris base 0,89M
Acido borico 0,89M
EDTA 0,02M
Loading buffer tipo III
Glicerolo 30%
blu di bromo fenolo 0,25%
xilene cianolo 0,25%
Gel di agarosio
Agarosio 0,8%(perso/volume)
EtBr 1:20000
TBE 1X
Marker di lunghezza: Invitrogen 1 kb DNA ladder
Digestione del DNA plasmidico con enzimi di restrizione
Dopo aver stimato la concentrazione del DNA plasmidico estratto, si procede con lo screening del plasmide mediante analisi di restrizione. Sono state fatte 4 diverse digestioni diagnostiche, ognuna in un volume finale di 20µl contenente l'enzima, il buffer specifico e il DNA plasmidico candidato. La quantità di enzima impiegato è pari a 4 unità enzimatiche (UE) per µg di DNA e non supera il 10% del volume totale, poiché il glicerolo in cui gli enzimi sono conservati ha il potere di disturbare l'attività endonucleasica dell'enzima di restrizione stesso. La reazione così preparata viene incubata a 37°C per 2 ore, poi caricata su gel insieme a marker di lunghezza nota e al DNA plasmidico non digerito.
Estrazione di DNA plasmidico su media scala (midiprep)
La tecnica si basa sull'uso di colonne cromatografiche NUCLEOBOND a scambio ionico, disponibili in commercio insieme alle soluzioni ad hoc per questo tipo di colonna. E' un metodo altamente efficiente che dà prodotti ben purificati e concentrati.
1. Dopo aver lasciato crescere per 48 ore in 100ml di LB con ampicillina diluita 1:2000 (50µg/ml come concentrazione finale) l'inoculo con il plasmide di interesse, da glicerolato, si centrifuga il brodo di crescita per 10 minuti a 3000rpm.
2. Il pellet ottenuto viene risospeso in 4ml di soluzione S1, a cui si aggiungono 4ml di soluzione S2. Si mescola delicatamente e si lascia agire la soluzione per non oltre 5 minuti.
3. Si aggiungono 4ml di soluzione S3, si mescola piano e si incuba in ghiaccio per 15 minuti; segue una centrifugazione a 4°C per 30 minuti a 12000rpm.
4. Intanto la colonna cromatografica AX-100 viene equilibrata bagnandola con 4ml di soluzione N2. Dopo che la colonna si è svuotata per gravità, si carica sulla colonna, poco alla volta, il sopranatante contenente il lisato batterico. In questo passaggio il DNA, l’RNA e i sali si legano alla resina.
5. Per purificare il DNA dai sali e dall'RNA, si lava la colonna per due volte con 4ml di soluzione N3.
6. L'eluizione del DNA viene effettuata con 2 ml di soluzione N5.
7. L'eluato viene precipitato con aggiunta di 0,7 volumi di isopropanolo, mediante centrifugazione a 4°C per 30 minuti a 12000rpm.
8. Il pellet così ottenuto viene lavato con EtOH al 70% e risospeso in acqua mQ. Con questa procedura si ricavano 80-150µg di DNA plasmidico con purezza 1,8 (valore ricavato dal rapporto OD260/OD280). La concentrazione è stata stimata sia
per corsa elettroforetica, sia per analisi spettrofotometrica (valore di assorbanza alla lunghezza d'onda di 260nm).
Soluzioni
Rnasi A 100µg/ml Tris-Hcl50mM EDTA10mM pH 8.0 S2 NaOH200mM SDS1% S3 KCH3COOH2,8 mM pH 5,1 N2 Tris100mM EtOH 15% KCl + H3PO4 900mM pH 6,3 N3 Tris 100mM EtOH15% KCl + H3PO4 1150mM pH 6,3 N5 Tris 100mM EtOH15% KCl + H3PO4 1000mM pH 8,5
COSTRUZIONE DI MESSAGGERI E SONDE ANTISENSO
Trascrizione dell'mRNA cappato
Al fine di valutare l'effetto della sovraespressione della JHDM1D sullo sviluppo embrionale dello Xenopus laevis e di monitorarne la localizzazione, ho eseguito esperimenti funzionali co-iniettando l'RNA messaggero del mio gene di studio e il messaggero del gene LacZ come reporter, negli embrioni a stadi precoci di sviluppo. Per valutare, invece, l'effetto diretto della sovraespressione della JHDM1D in modo specifico sui progenitori retinici, ho co-microiniettato il messaggero della JHDM1D e quello della GFP in un blastomero retinogenico allo stadio di 16 cellule.
La trascrizione dei messaggeri avviene in vitro ed è ottimizzata per rendere l'mRNA stabile e non aggredibile dalle esonucleasi delle cellule dell'embrione.
I trascritti da microiniettare devono contenere quindi, strutture alle estremità 5' e 3' che ne aumentino la stabilità e l'efficienza di traduzione nella cellula eucariotica. A questo scopo la JHDM1D è stata subclonata nel vettore di espressione pCS2+,
plasmide che contiene elementi stabilizzanti l'RNA. Inoltre per aumentare l'efficienza di traduzione, si aggiunge alla miscela di trascrizione, una "terminal cap structure" (cap) al 5'. Da qui il nome di mRNA cappato.
Il cap al 5', tipico degli mRNA cellulari destinati alla traduzione e trascritti in vivo, consiste in una 7-metil-guanosina trifosfato, che si lega mediante un ponte fosfodiesterico 5'-5' all'RNA trascritto in vitro (Figura 19). Per ottenere trascritti forniti di cap con alta resa, si fa avvenire la reazione di trascrizione in presenza di una concentrazione di GTP pari a 1/4 di quella della 7-metil-guanosina trifosfato.
Il template è il plasmide linearizzato in corrispondenza di NotI, un sito di restrizione a valle del sito di poliadenilazione virale portato dal plasmide stesso
Figura 3. Un trascritto con elementi stabilizzanti alle estremità. Al 5’ è presente
(Figura 18) e purificato in modo che non ci siano tracce di plasmide circolare. Un plasmide chiuso, infatti, usato come stampo, genera trascritti eterogenei e molto lunghi a causa della processività della RNA polimerasi. Gli enzimi di restrizione usati, inoltre, non devono lasciare estremità 3' protruding poiché la resa del trascritto desiderato risulterebbe molto più bassa.
A partire dal pCS2+JHDM1D o dal pCS2+GFP o dal pCS2+LacZ linearizzati, si
assembla la reazione di trascrizione in vitro con il Kit mMESSAGE mMACHINE® SP6 dell'Ambion®. Si prepara la reazione di trascrizione in 20μl totali:
H2O nuclease-free
2X NTP/CAP
10X Reaction Buffer
1 μg di DNA stampo linearizzato
Enzyme Mix
I reagenti forniti dal kit sono mantenuti in ghiaccio, ma la reazione viene assemblata a temperatura ambiente (TA) per la presenza della spermidina nel Reaction Buffer che coprecipita col DNA a basse temperature. Una volta assemblata la reazione di trascrizione, si procede col protocollo:
1. Picchiettare sulla eppendorf per mescolare i reagenti e microcentrifugare. 2. Incubare il mix di reazione a 37°C per 2 ore.
3. Aggiungere 1μl di TURBO DNase RNasi-free, mescolare bene, microcentrifugare e incubare per 15 minuti a 37°C. La DNasi, fornita dal kit, degraderà il DNA stampo senza bersagliare l'mRNAcap appena trascritto. 4. Seguono l'estrazione con fenolo:cloroformio e la precipitazione con
isopropanolo. Questo è il metodo più rigoroso per purificare trascritti e rimuove dal mix di reazione tutti gli enzimi e i nucleotidi non incorporati nel trascritto. 5. Dopo aver lasciato l'mRNAcap in isopropanolo per 15-40 minuti a -20°C (non
oltre per evitare il rischio che precipitino anche i sali), si centrifuga alla massima velocità per 15 minuti a 4°C, si elimina il sopranatante e si risospende l'RNA in acqua mQ RNase-free.
6. Si valutano la qualità e la quantità del trascritto per corsa elettroforetica comparandolo a tRNA di peso molecolare noto e si preparano aliquote da 100ng/μl da congelare rapidamente in un bagnetto di alcool denaturato a -80°C.
Trascrizione in vitro di sonde antisenso
Al fine di visualizzare l'espressione spaziale dei geni, si costruiscono sonde a RNA antisenso che vadano a ibridizzare con l'mRNA del gene corrispondente. Le basi Uracili delle sonde sono marcate con digossigenina, uno steroide di origine vegetale.
Per prima cosa si linearizza il plasmide tagliandolo con enzimi di restrizione 5' protruding che riconoscano sequenze al di fuori dell'inserto. E' importante non lasciare l'estremità 3' sporgente per evitare che l'RNA venga trascritto a partire dal filamento complementare a quello desiderato.
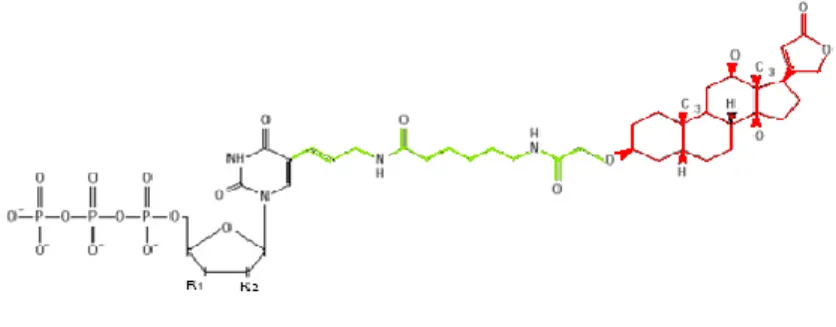
Alla digestione segue la purificazione del plasmide linearizzato che fungerà da stampo per la reazione di trascrizione in vitro. La RNA polimerasi usata varia a seconda del promotore al 3' del frammento e procederà sul filmaneto senso in modo da ottenere una sonda antisenso. La marcatura con digossigenina (DIG) deriva dal fatto che nella miscela di nucleotidi c'è un dUTP-DIG (Figura 20), un ribonucleotide a cui è legata la digossigenina.
Figura 4. Struttura generale di un uracile marcato con digossigenina. La
digossigenina (in rosso) è legata a un nucleotide uridinico (in nero) in posizione 5 dell’anello pirimidinico mediante una catena spaziatrice di 11 atomi di carbonio (in verde). R1=OH; R2=H.
DNA linearizzato (CycD1, JHDM1D, N-tubulina, Otx2, Pax6, Rx1, Sox2, Zic-2) 1,2 µg
Tampone di reazione 10X2 µl DTT 100 mM2 µl
Rnasi Out 20UE/µl1 µl RNA pol2 µl
Nucleotidi-DIG 2,5 mM2 µl H2O RNasi free (RF)a volume
1. La miscela viene incubata per 2 ore a 37°C.
2. Per eliminare il DNA stampo si aggiungono 2µl di Dnasi I (1mg/ml) e la si lascia agire per 15 minuti a 37°C.
3. La reazione viene bloccata aggiungendo EDTA 0,5M pH 8.0 RNasi Free (RF). Si effettua una precipitazione alcoolica aggiungendo 1/10 del volume di ammonio acetato 5M sterile e 1 volume di Isopropanolo sterile. Si mescola il tutto bene e si pone il tubo a -20°C per 40 minuti circa.
4. Si centrifuga per 15 minuti a 12000 rpm a 4°C.
5. Si lava con etanolo (EtOH) RF al 70% ghiacciato e si centrifuga nuovamente a 12000 rpm a 4°C.
6. Quando l'EtOH è evaporato, il pellet viene risospeso in 22 µl di H2O RF e
conservato a -20°C. La stima della quantità dell'RNA ottenuto viene fatta su gel di agarosio – in condizioni RF – confrontando 1µl di sonda con tRNA di peso molecolare noto.
ESPERIMENTI FUNZIONALI – esperimenti in
vivo su Xenopus laevis
TECNICHE DI MANIPOLAZIONE
Raccolta di embrioni di Xenopus laevis
Gli embrioni si sviluppano a partire dall'incontro fra cellule uovo e spermatozoi, usando la tecnica della fecondazione in vitro. Gli spermatozoi si ottengono dalle gonadi del maschio, che viene anestetizzato e operato per estrarre le due gonadi interne in due momenti diversi. L'anestetico usato è una soluzione di metan-sulfonato dell'estere etilico dell'acido aminobenzoico (MS222) 0,1%. Il testicolo estratto viene conservato per qualche giorno a 4°C in una soluzione contenente MMR 1X, siero di pecora e l'antibiotico gentamicina.
La femmina di Xenopus deve essere stimolata 6 giorni prima della deposizione con 100UI di Folligon Intervet per uso veterinario e, 10-12 ore prima della deposizione, con 800/1000UI di Profasi HP 2000 Serono (gonadotropina corionica). Entrambi gli ormoni sono iniettati nel sacco perilinfatico della femmina. Questo serve a sincronizzare la maturazione di tutte le uova, in modo da averne il maggior numero possibile al momento della deposizione. Questa avviene applicando una leggera pressione con le dita sull'addome della femmina.
Le uova sono raccolte su una piastra Petri e vengono subito fecondate passando sopra di esse un frammento di testicolo che può essere ulteriormente lasciato in mezzo alle uova per 5minuti; si versa poi nella Petri MMR 0,1X. La raccolta delle uova può essere fatta ogni 2 ore.
Dopo un minimo di 30 minuti, gli embrioni sono trattati con una soluzione degellificante di DTT che rimuove il loro rivestimento gelatinoso e poi risciacquate più volte con MMR 0,1%. Gli embrioni vengono lasciati in MMR 0,1% fino allo stadio desiderato: 4 o 16 cellule per le microiniezioni, stadio 16 per le lipofezioni e stadi più avanzati per gli esperimenti di ibridazione in situ. Gli stadi sono classificati secondo i criteri di Nieuwkoop e Faber (Nieuwkoop et al., 1967).
Soluzioni
Siero di agnello inattivato al calore 10% Gentamicina 50 µg/ml 10 µl MMR 1X a volume MMR NaCl 0,1 M KCl2 mM MgSO41 mM CaCl22 mM HEPES 5 mM pH 7,8 EDTA 0,1 M Soluzione degellificante DDT 3,2 M Tris-HCl 0,2 M pH 8,8 Microiniezione
Per gli esperimenti di microiniezioni sono stati usati embrioni pigmentati che permettessero di distinguere i due emisferi animale e vegetativo e i blastomeri dorsali e ventrali, in modo da indirizzare la sovraespressione della JHDM1D nei territori dorsali (i blastomeri animali con pigmentazione più chiara). Al momento della microiniezione, gli embrioni degellificati, vengono trasferiti in una piccola piastra Petri sul fondo della quale è fissata una reticella nera di plastica con maglie di circa 1mm, per facilitare l'immobilizzazione e la conta degli embrioni. Gli embrioni da microieniettare sono immersi in Ficoll al 3,5% (peso/volume) sciolto in MMR 0,1X, uno zucchero viscoso che aiuta la cauterizzazione della puntura evitando la perdita di citoplasma da parte dell'embrione nel punto di entrata dell’ago. Il Ficoll inoltre, mantiene la forma sferica degli embrioni aumentando le probabilità che avvengano segmentazioni simmetriche che aiutino l’individuazione del blastomero di interesse.
Gli embrioni microiniettati sono lasciati sviluppare nella soluzione di Ficoll al 3,5% per 24 ore e poi trasferiti in MMR 0,1X. Raggiunto lo stadio desiderato, si
fissano controlli e iniettati per un'ora in MEMFA e si conservano in EtOH a -20°C fino ai successivi esperimenti di ibridazione.
Ho eseguito le microiniezioni con un microiniettore "Drummond Nanoject", che consente iniezioni di volumi compresi tra 4,6 nl e 73,6 nl a incrementi discreti. L'iniettore è dotato di un macromanipolatore che ne permette lo spostamento macrometrico nelle tre dimensioni e di un micromanipolatore per lo spostamento più fine lungo una direzione predefinita.
Gli aghi per l'iniezione sono preparati per tiratura a caldo da capillari forniti da "Drummond". Prima di essere montato sul microiniettore, l'ago deve essere riempito con olio minerale per poi essere svuotato quasi del tutto dal pistone del microieniettore stesso. Un po’ di olio infatti deve rimanere nell’ago per permettere il corretto funzionamento del pistone. L'RNA da microiniettare viene caricato dal microiniettore stesso, tramite la funzione "fill".
Per il mio progetto di tesi sperimentale ho iniettato dosi diverse di RNA cappato della JHDM1D nel blastomero animale dorsale (1AD) allo stadio di 4 cellule (4C), insieme alla dose fissa di 300 pg di β-galattosidasi e 300 pg di JHDM1D nel blastomero animale dorsale 1.2 (D1.2) allo stadio di 16 cellule (16C).
Dosi iniettate: 5 pg di JHDM1D + 300 pg di β-galattosidasi (1AD/4C) 12,5 pg di JHDM1D + 300 pg di β-galattosidasi (1AD/4C) 25 pg di JHDM1D + 300 pg di β-galattosidasi (1AD/4C) 50 pg di JHDM1D + 300 pg di β-galattosidasi (1AD/4C) 100 pg di JHDM1D + 300 pg di β-galattosidasi (1AD/4C) 300 pg di JHDM1D + 300 pg di β-galattosidasi (1AD/4C) 500 pg di JHDM1D + 300 pg di β-galattosidasi (1AD/4C) 700 pg di JHDM1D + 300 pg di β-galattosidasi (1AD/4C) 300 pg di β-galattosidasi (1AD/4C)
300 pg di JHDM1D + 300 pg di GFP (D1.2/16C) 300 pg di GFP (D1.2/16C)
Lipofezione
La lipofezione è una tecnica che permette di introdurre all’interno di cellule embrinali cDNA opportunamente clonati in vettori di espressione. Le cellule che assumono il costrutto hanno un destino differenziativo più limitato rispetto ai blastomeri bersaglio della microiniezione. La tecnica prevede una internalizzazione del cDNA in una micella creata da un materiale lipofilico, il DOTAP, seguito da una fusione della particella stessa con la membrana della cellula bersaglio.
La miscela di cDNA lipofettata è costituita dal costrutto del gene di interesse e da un marcatore fluorescente di cellule effettivamente lipofettate, la GFP. Le lipofezioni sono state effettuate a stadio embrionale 18, a livello delle regioni di localizzazione delle vescicole ottiche. Alla miscela di cDNA da lipofettare viene aggiunto un tracciante di colore verde (Fast Green).
Le lipofezioni sono state eseguite con un microiniettore Drummond Nanoject che consente l’iniezione di volumi compresi tra 4,6 nl e 73,6 nl ad incrementi discreti. Gli aghi sono stati preparati per tiratura a caldo a partire da capillari forniti da Drummond: la loro bontà è stata controllata allo stereomicroscopio. Prima di essere montati sul microiniettore gli aghi sono stati riempiti di olio minerale con una siringa. Il caricamento degli aghi con la soluzione da iniettare è eseguito dal microiniettore stesso.
Lo scopo della lipofezione è andare a visualizzare l’effetto della espressione ectopica di un cDNA sulla proliferazione e sulla morfologia delle cellule retiniche.
Mix di reazione
pCS2+-JHDM1D 2µg
pCS2+-GFP 0,5 µg
DOTAP 7,5 µl (3µl ogni µg di DNA) Fast Green 1X
pCS2+-GFP 0,5 µg
pCS2+ vuoto 2 µg
DOTAP 7,5 µl (3µl ogni µg di DNA) Fast Green 1X
TECNICHE DI ESPRESSIONE
Reazione cromogenica della β-galattosidasi
Come reporter della localizzazione del messaggero microiniettato, è stato coiniettato l'mRNA della β-galattosidasi nucleare. L'enzima prodotto catalizzerà una reazione cromogenica trasformando il substrato cromogeno Salmon-gal in un precipitato di color rosso-salmone.
1. Per prima cosa, si fissano gli embrioni, raccolti in vials di vetro, stadiati (cioè fatti crescere fino allo stadio desiderato) in MEMFA per 45 minuti.
2. Seguono due lavaggi in PBS 1X.
3. Si immergono gli embrioni nella soluzione di ferri contenente 1/10 del volume finale di salmon-gal (stock al 5% in metanolo) e si attende che la reazione avvenga, lasciando le vials a 37°C e controllando periodicamente la rivelazione. 4. Avvenuta la reazione, gli embrioni vengono nuovamente fissati in MEMFA per
altri 45 minuti e conservati a -20°C in etanolo 100%.
Soluzioni MEMFA MOPS 0,1M pH 7,4 EGTA 2 mM MgSO4 1 mM Formaldeide 37% Soluzione di ferri K3Fe(CN)6 30 mM K3Fe(CN)6H2O 30 mM PBS 1X
PBS 10X
NaCl 80 g
KCl 2 g
NaHPO4 15,56 g
KH2PO4 2 g
Ibridazione in situ su embrioni interi "Whole mount"
Tale tecnica consiste nel permeabilizzare gli embrioni in modo da far ibridizzare una sonda a RNA antisenso marcata con digossigenina con il messaggero senso presente nei tessuti dell'embrione per valutare il pattern di espressione di geni di interesse, sia negli organismi wild-type, sia in quelli trattati sovraesprimendo la JHDM1D. Il primo passaggio è la fissazione degli embrioni in MEMFA (fissa i tessuti e mantiene l'RNA dell'embrione) e la loro successiva conservazione a -20°C in EtOH 100%. Segue poi la procedura di ibridazione, della durata minima di tre giorni.
Procedura di ibridazione.
Primo giorno
Gli embrioni vengono reidratati gradualmente con una serie di passaggi in concentrazioni di etanolo decrescenti, tramite lavaggi di 5 minuti ciascuno, in PBTw a temperatura ambiente (TA):
1. Un lavaggio di 5 minuti a TA su bascula con EtOH 75% in PBTw (PBS 1X +
Tween 20 allo 0,1% e H2O)
2. Un lavaggio di 5 minuti a TA su bascula con EtOH 50% in PBTw 3. Un lavaggio di 5 minuti a TA su bascula con EtOH 25% in PBTw 4. Un lavaggio di 5 minuti a TA su bascula con PBTw
Dopo la reidratazione, viene effettuata la permeabilizzazione degli embrioni con la proteinasi k. Questo faciliterà il passaggio della sonda nell'embrione intero, poiché a TA la proteinasi k, che è massimamente attiva a 37°C, allenta i tessuti connettivi e le matrici:
5. Aggiungere 0,5 ml per vial di proteinasi k (stock da 20μg/ml) diluita 1:2000 in PBTw e lasciare per 5 minuti esatti a TA senza agitazione.
Una volta permeabilizzati gli embrioni, questi vanno rifissati con paraformaldeide (PFA) al 4%, un fissativo più blando rispetto al MEMFA:
6. Lavare per due volte gli embrioni con PBTw per 1-2 minuti per rimuovere la proteinasi k.
7. Fissare con 1ml di PFA al 4% in PBS 1X, per 20 minuti esatti, con agitazione occasionale.
Per preparare gli embrioni alla ibridazione vera e propria, abituandoli alla diversa densità della soluzione di ibridazione, una volta lavata via completamente la PFA, gli embrioni vengono ricoperti con una soluzione composta al 50% da miscela di ibridazione (NIH) e al 50% da PBS 1X; comincia la fase di pre-ibridazione:
8. Lavare due volte con 1ml di PBS 1X per vial per 2 minuti.
9. Sostituire con 0,5 ml di 50% NIH – 50% PBS 1X per vial e lasciarlo per 3 minuti
10. Sostituire con 0,5 ml di NIH 100% e lasciare la vial 2-3 ore a 60°C
Alla pre-ibridazione segue l'ibridazione vera e propria, in cui la sonda marcata con digossigenina viene prima denaturata:
11. Denaturare 50 ng di sonda in 500 μl di NIH a 95°C per 2 minuti. Rimettere subito la sonda in ghiaccio per mantenerla denaturata evitando la formazione di eventuali strutture secondarie.
12. Ibridare gli embrioni con la miscela di ibridazione contenente la sonda denaturata, per 12-16 ore a 60°C.
Procedura di ibridazione. Secondo giorno
I trattamenti del secondo giorno di whole mount in situ hybridization servono a rendere più specifico possibile il segnale (aumentando la stringenza) e a preparare gli embrioni alla rivelazione con l'anticorpo che andrà a riconoscere la digossigenina incorporata nella sonda a RNA.
La prima fase prevede lavaggi in soluzioni preriscaldate a 60°C, a forza ionica decrescente per aumentare la stringenza del processo ed eliminare la sonda in eccesso. E' importante che i lavaggi siano fatti alla stessa temperatura di ibridazione, poiché queste sono le condizioni capaci di ridurre al minimo le reazioni di ibridazione non specifica:
1. Togliere l'NIH contenente la sonda e conservarlo a -20°C.
2. Mettere 1 ml per vial di 50% NIH – 50% 2X SSC/0,1% CHAPS e lasciare per 10 minuti a TA.
3. Lavare con 1 ml per vial di 2X SSC/0,1% CHAPS per 30 minuti a 60°C, due volte.
4. Sostiuire con 1 ml per vial di 0,2X SSC/0,1% CHAPS per 30 minuti a 60°C e lavare due volte.
Segue la fase di pre-incubazione. Per aumentare la specificità del segnale, gli embrioni vengono pre-incubati in blocking buffer, una miscela di TBSX, lamb serum e blocking reagent ad alte concentrazioni, poichè quello che di fatto penetrerà nell'embrione sarà una piccola quantità. In aggiunta, il blocking buffer contiene l'EGG extract, un estratto proteico ricavato dall'omogeneizzato di embrioni a diversi stadi, che serve a ridurre ulteriormente il segnale aspecifico:
5. Sostiuire con 0,5 ml di 50% 0,2X SSC/0,1% CHAPS – 50 % di TBS 1X e lasciare per 5 minuti a TA.
6. Sostituire con 0,5 ml di TBSX (TBS 1X e Triton x-100 allo 0,1%) e incubare 5 minuti a TA.
7. Sostituire il TBSX con 0,5 ml di blocking buffer e incubare per 2 ore a 4°C sulla bascula.
Alla fase di pre-incubazione segue la fase di incubazione vera e propria dell'anticorpo anti-dig, che viene preparato diluendolo 1:2500 in blocking buffer e tenuto per 2 ore in questa miscela, a 4°C su bascula:
8. Incubare gli embrioni con 0,5 ml di anticorpi anti-dig diluiti in blocking buffer per 4 ore a TA sulla bascula.
9. Lavare per 30 secondi in TBSX e sostituirlo riempiendo la vial. Lasciare ON a 4°C sulla bascula.
Procedura di ibridazione. Terzo giorno
Nell'ultimo giorno avviene la rivelazione del segnale. Il primo passaggio prevede l'eliminazione dell'anticorpo in eccesso. Poi gli embrioni vengono lavati con il buffer della fosfatasi alcalina (AP-buffer) a cui è aggiunto il tetramisole, un inibitore delle fosfatasi endogene:
1. Fare 4 lavaggi con TBSX riempiendo la vial per 1 ora a TA sulla bascula. 2. Fare 2 lavaggi con 1 ml per vial di AP-buffer , per 5 minuti a TA.
L'aggiunta del tetramisole permette di inibire le fosfatasi endogene, ma non la fosfatasi alcalina legata all'anticorpo poiché è mutagenizzata. L'ultimo passaggio prevede l'aggiunta del substrato della fosfatasi alcalina, il BM Purple, così chiamato per la caratteristica colorazione blu-violacea prodotta dalla sua trasformazione:
3. Mettere 500 μl di BM-Purple per vial; ricoprire con carta stagnola e lasciare in rivelazione a TA, a 14 o a 4°C, sulla bascula, controllando di tanto in tanto. 4. Fermare la reazione lavando con TBSX per 10 minuti.
Depigmentazione (bleaching)
Per distinguere meglio la marcatura delle strutture embrionali, si opera il "bleaching" degli embrioni stessi, ossia la depigmentazione con acqua ossigenata. Tale operazione viene evitata se gli embrioni sono albini e quindi già privi di pigmento:
1. Lavare gli embrioni con EtOH al 70% per 10 minuti.
2. Lavare gli embrioni con una soluzione EtOH 50% e SSC 0,5X.
3. Lasciare gli embrioni per 1-2 ore sotto illuminazione diretta di una lampada, nella soluzione depigmentante:
SSC 0,5X H2O2 1%
Formammide5%
4. Lavare gli embrioni per 5 minuti con EtOH 70% 5. Conservare gli embrioni in EtOH 100% a -20°C
Fissazione degli embrioni a stadio 42
1. Trasferire gli embrioni a stadio 42 dalla piastra Petri in un tubo da batteri contenente paraformaldeide (PFA) al 4% in PBS 1X e lasciarli fissare per 1 ora e mezza a TA sulla bascula in agitazione. La PFA è in grado di legarsi con legami covalenti a numerosi gruppi chimici contenenti un atomo di idrogeno reattivo e alle concentrazioni a cui è usata e al pH di 7,3-7,4 reagisce soprattutto con anelli aromatici e con gruppi aminici (-NH2 ). una volta legatasi per addizione ad una proteina, la formaldeide può reagire con un idrogeno reattivo di una seconda molecola formando ponti intermolecolari.
2. Sostituire con saccarosio al 20% in PBS 1X e lasciare in agitazione sulla bascula a TA finché gli embrioni non affondano, oppure a 4°C ON (over night). Il saccarosio crioproteggerà gli embrioni per la loro conservazione a -80°C e per il taglio a -20°C.
3. Trasferire gli embrioni in appositi stampi di plastica e ricoprirli di O.C.T.TM.
4. Orientare gli embrioni nel senso del taglio.
5. Effettuare un congelamento rapido adagiando i blocchetti in una vaschetta contenente etanolo a -80°C per 15 minuti circa.
6. Conservare a -80°C fino al momento del taglio, facendo trascorrere almeno una notte.
7. Lasciare i blocchetti a -20°C per circa 30 minuti prima di procedere col taglio.
Questo passaggio è necessario per ammorbidire la resina O.C.T.TM.
8. Tagliare al criostato sezioni di 12 µm di spessore.
9. Fare aderire le sezioni al vetrino avvicinandolo parallelamente ad esse. I vetrini sono polarizzati e l'adesione avviene elettrostaticamente.
10. Lasciare asciugare i vetrini a TA per circa un'ora. Poi fare due lavaggi in PBS1X di 5 minuti ciascuno per rimuovere l’O.C.T.
11. Far gocciolare 1ml di Hoechst diluito 0,1:1000 con il PBS1X. Incubare per 20 minuti circa.
12. Lavare 3 volte con PBS 1X 15 minuti.