6
6
S
S
V
V
I
I
L
L
U
U
P
P
P
P
O
O
D
D
I
I
U
U
N
N
A
A
L
L
G
G
O
O
R
R
I
I
T
T
M
M
O
O
P
P
E
E
R
R
I
I
L
L
C
C
A
A
L
L
C
C
O
O
L
L
O
O
D
D
E
E
L
L
L
L
A
A
N
N
U
U
M
M
E
E
R
R
O
O
S
S
I
I
T
T
À
À
C
C
A
A
M
M
P
P
I
I
O
O
N
N
A
A
R
R
I
I
A
A
C
C
O
O
N
N
I
I
L
L
S
S
O
O
F
F
T
T
W
W
A
A
R
R
E
E
R
R
R è un linguaggio di programmazione ad alto livello utilizzato soprattutto in statistica e, più precisamente in ambito biostatistico. R, infatti, fornisce un insieme di funzioni per l’analisi statistica di dati di qualsiasi tipo e un’ampia raccolta di pacchetti indirizzati alla biostatistica.
Nel suo complesso, più che un semplice linguaggio, R può essere considerato un vero e proprio ambiente di sviluppo per la manipolazione, l’analisi e la rappresentazione grafica dei dati.
La scelta di implementare l’algoritmo per il calcolo della numerosità campionaria utilizzando questo software piuttosto che altri, ugualmente validi, è stata dettata principalmente dal fatto che si tratta di un una risorsa completamente gratuita e continuamente aggiornata. L'alto livello della programmazione permette, inoltre, di creare facilmente librerie di funzioni per nuove applicazioni, oltre a quelle, già numerose, disponibili in rete sui siti www.r-project.org e www.bioconductor.org. Bioconductor è un contenitore di pacchetti “open source”, basato sulla piattaforma R, che fornisce validi strumenti per la soluzione di problemi nell’ambito della bioinformatica e dell’analisi dei dati microarray in particolare.
Nonostante l’importanza che una stima della dimensione campionaria assume nella fase preliminare di progettazione di un esperimento microarray, i pacchetti messi a punto dagli sviluppatori di R e Bioconductor per tale scopo hanno un’applicabilità limitata in quanto si riferiscono alle situazioni sperimentali più semplici come il confronto tra due gruppi (dipendenti o indipendenti) oppure il confronto fra più di due classi che differiscono per un unico fattore. Le funzioni integrate in questi pacchetti richiedono inoltre da parte dell’utente non soltanto una sufficiente familiarità con la programmazione informatica, ma anche una buona conoscenza dei metodi statistici di analisi dei dati.
L’algoritmo ideato durante lo sviluppo di questa tesi va pertanto inquadrato nell’ottica di un superamento delle suddette limitazioni.
Prima di illustrare tale algoritmo, nel prossimo paragrafo vengono brevemente introdotte le funzioni principali raccolte in 2 pacchetti, denominati “ssize” e “ssize.fdr” per l’analisi della potenza e il calcolo della numerosità campionaria in esperimenti di “class comparison” condotti con i microarray. In entrambi i pacchetti l’approccio adottato è quello di valutare la potenza e/o la dimensione campionaria gene per gene.
6.1 PACCHETTI DISPONIBILI PER IL CALCOLO DELLA POTENZA E
DELLA NUMEROSITÀ CAMPIONARIA IN UN ESPERIMENTO
MICROARRAY
6.1.1 Il pacchetto “ssize” di Bioconductor
Le funzioni contenute nel pacchetto “ssize” (sviluppato da G. Warnes e disponibile sul sito di Bioconductor, http://bioconductor.org/packages/2.4/bioc/html/ssize.html) consentono di calcolare la potenza, la dimensione campionaria e l’”effect size” nel caso di esperimenti di “class comparison” che prevedono il confronto tra due gruppi di campioni indipendenti.
Per determinare la numerosità campionaria si utilizza la funzione denominata ssize() che prevede la specificazione da parte dell’utente dei valori dei seguenti parametri: - vettore delle deviazioni standard gene-specifiche (sd), ricavate da un precedente studio
pilota sullo stesso sistema biologico;
- minimo “effect size” che si vuole rilevare, espresso come logaritmo in base 2 del “fold change”(delta),
- massimo valore del FWER (Family-Wise Error Rate) (sig.level), - potenza desiderata (power).
Fissato il livello di significatività per l’intero esperimento uguale ad α, la funzione ssize() consente di calcolare il livello di significatività corrispondente per il singolo confronto αG utilizzando la correzione di Bonferroni (opzione: alpha.correct =.”Bonferroni”) oppure di definire un unico valore di α senza tenere conto dei confronti multipli (opzione: alpha.correct = “None”).
La sintassi della funzione ssize() è la seguente:
n<-ssize(sd=dev.st, delta=1, sig.level=0.05, power=0.8, alpha.correct = "Bonferroni")
La numerosità campionaria viene calcolata considerando ogni gene separatamente e risolvendo l’equazione (6.1) che coincide con la formula del t-test per campioni indipendenti con varianza combinata o “pooled”:
(6.1) ) 2 | ( ) 2 | ( 1 1 2 /2, 2 2 /2, 2 n T T n T Tn n n n G G σ δ σ δ β = − − α − + − −α − −
dove Td( |θ) è la funzione di distribuzione cumulativa per una distribuzione t non centrale con d gradi di libertà e parametro di non centralità θ.
Il risultato prodotto dalla funzione è un vettore i cui elementi sono i valori dell’ampiezza campionaria calcolati per ogni gene.
L’algoritmo su cui è basata la funzione ssize ( ) prescinde dal tipo di disegno sperimentale adottato, pertanto il dato fornito in uscita è solo indicativo e non mette in evidenza le notevoli differenze che esistono tra i vari tipi di disegno.
La funzione ssize.plot() consente di visualizzare i risultati sotto forma di un diagramma cumulativo in cui sull’asse delle ascisse è riportato il numero n di campioni per classe mentre sull’asse delle ordinate il numero e la proporzione di geni che richiedono una numerosità campionaria maggiore o uguale ad n per poter rilevare la differenza di espressione stabilita con la potenza desiderata.
Fig. 6.1: Grafico per il calcolo della numerosità campionaria generato dalla funzione
Il grafico consente allo sperimentatore di scegliere il giusto compromesso tra la numerosità campionaria richiesta e la potenza che si desidera ottenere. Per esempio, la curva riportata in figura 6.1 mostra che per il 67 % dei geni è necessaria un’ampiezza campionaria (per gruppo) pari a 6 per poter osservare con una potenza dell’80% un “fold-change” uguale a 2.
Grafici analoghi si possono ottenere fissando la numerosità campionaria e facendo variare uno degli altri parametri (la potenza o l’”effect size”), mediante le funzioni power.plot() e delta.plot().
6.1.2 Il pacchetto “ssize.fdr” di R
Il pacchetto “ssize.fdr” (http://cran.r-project.org/web/packages/ssize.fdr/index.html) contiene un insieme di funzioni per il calcolo dell’ampiezza campionaria in esperimenti microarray che coinvolgono uno, due o più gruppi.
La numerosità campionaria viene determinata scegliendo come tasso di errore di I tipo il False Discovery Rate e fissando la proporzione attesa di geni non differenzialmente espressi (Liu e Hwang, 2007). Il risultato viene fornito, oltre che in forma numerica, anche in forma grafica mediante una curva che rappresenta la potenza in funzione della numerosità campionaria.
Per tutte le funzioni incluse nel pacchetto, l’utente deve specificare la potenza desiderata (power), il False Discovery Rate (fdr), la proporzione attesa di geni non differenzialmente espressi (pi0) e il massimo valore della numerosità campionaria (maxN) che può essere utilizzato per svolgere i calcoli. L’utente può fornire in ingresso alla funzione più di un valore per la proporzione attesa di geni non differenzialmente espressi; in tal caso i calcoli per la determinazione dell’ampiezza campionaria vengono ripetuti per ogni valore della proporzione.
Il metodo utilizzato per il calcolo della numerosità campionaria deriva dalla definizione del False Discovery Rate. Se con α si indica il valore del FDR scelto, vale la seguente relazione: (6.2) ) 1 | Pr( ) 0 | Pr( 1 1 0 0 = Γ ∈ = Γ ∈ = − − T H H T π π α α dove:
- π0 è la proporzione attesa di geni non differenzialmente espressi;
- rappresenta la probabilità che il valore assunto dalla statistica test T appartenga alla regione di rifiuto Γ dell’ipotesi nulla, noto che l’ipotesi nulla è vera (H = 0), ossia che il gene considerato non è differenzialmente espresso;
) 0 | Pr(T∈Γ H =
- indica la probabilità che T appartenga alla regione di rifiuto Γ condizionata al fatto che l’ipotesi alternativa è vera, cioè che il gene è differenzialmente espresso.
) 1 | Pr(T∈Γ H =
La statistica test sarà la t di Student quando l’esperimento coinvolge 2 soli gruppi o la F di Fisher quando i gruppi a confronto sono più di 2.
Per ogni valore di n compreso tra 2 e maxN, vengono determinati gli estremi (i valori critici) della regione di rifiuto che soddisfano l’equazione (6.2) e la potenza che si riesce ad ottenere.
In particolare la funzione ssize.twoSamp() consente di calcolare l’ampiezza campionaria per esperimenti microarray in cui il confronto è limitato a due sole classi. In aggiunta ai parametri precedentemente elencati, l’utente deve specificare l’”effect size” (delta) e la deviazione standard (sigma), assunti uguali per tutti i geni. La sintassi della funzione è la seguente:
ssize.twoSamp(delta, sigma, fdr = 0.05, power = 0.8, pi0 = 0.95, maxN = 35, side = "two-sided", cex.title=1.15, cex.legend=1)
L’argomento side con opzioni “two-sided”, “upper”, o “lower” permette di scegliere tra test bilaterale, unilaterale destro e unilaterale sinistro rispettivamente.
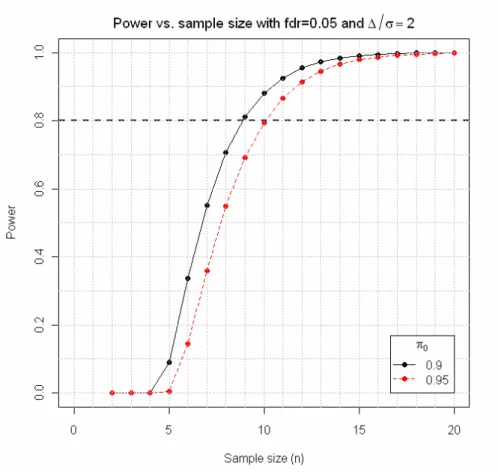
Il grafico creato dalla funzione ssize.twoSamp() per delta = 1, sigma= 0.5 e per due diversi valori della proporzione attesa di geni non differenzialmente espressi è riportato in figura 6.2.
Fig. 6.2: Grafico della potenza in funzione della numerosità campionaria creato dalla
funzione ssize.twoSamp( )
La funzione ssize.twoSampVary() differisce dalla precedente per il fatto che vengono fatte delle ipotesi sulla distribuzione degli “effect-size” e delle varianze tra i geni; in particolare si suppone che i primi seguano una distribuzione Normale mentre le seconde una distribuzione Gamma Inversa. In questo caso l’utente deve specificare anche i valori dei parametri delle 2 distribuzioni.
Quando le classi a confronto sono più di 2 si utilizza la funzione ssize.F(). La filosofia su cui si basa questa funzione è quella dei modelli lineari, pertanto in questo caso l’utente deve definire la matrice del disegno, il vettore dei parametri ed eventualmente la matrice o il vettore dei contrasti che stabilisce quali sono i confronti di interesse.
Anche quando l’esperimento coinvolge più gruppi, l’utente può scegliere se considerare la deviazione standard e l’”effect size” come parametri fissi, uguali per tutti i geni (funzione ssize.F())o come variabili random che seguono una distribuzione assegnata per la quale occorre specificare gli opportuni parametri (funzione ssize.Fvary()).
6.2 SVILUPPO DELL’ALGORITMO
L’algoritmo per il calcolo della numerosità campionaria sviluppato in questo lavoro di tesi si basa essenzialmente sulla teoria dei modelli lineari di analisi dei dati, ampiamente illustrata nel capitolo 4.
Di seguito vengono descritte, in logica successione, le considerazioni che hanno portato alla formulazione dell’algoritmo.
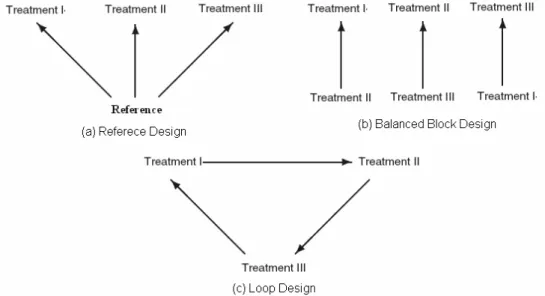
Una delle prerogative degli esperimenti microarray rispetto ad altri tipi di indagine sperimentale (per esempio gli studi epidemiologici) è rappresentata dalla varietà di disegni sperimentali che è possibile utilizzare per raggiungere lo stesso obiettivo. Limitando l’attenzione agli esperimenti di “class comparison”, nel capitolo 2 sono stati descritti i disegni sperimentali che vengono impiegati con maggiore frequenza, ossia il “reference design”, il ”balanced block design” e il “loop design”. Stabilito il numero di classi/trattamenti/varietà coinvolti nell’esperimento, per ogni disegno è possibile definire uno schema di base, senza considerare alcuna forma di replicazione. Sia s il numero di array necessari per realizzare tale disegno. Si consideri, per esempio, un esperimento microarray in cui si vogliono confrontare tre trattamenti. Gli schemi di base per il “reference design”, il ”balanced block design” e il “loop design” sono rappresentati nella figura 6.3, dalla quale si evince che s = 3 in tutti e tre i casi con la differenza che nello schema di base del ”balanced block design” bisogna considerare 2 repliche biologiche per ciascun trattamento, mentre nel “loop design” sono necessarie 2 repliche tecniche per classe.
Il quesito al quale si vuole dare risposta è quanti set di s array, e quindi quante repliche, bisogna collezionare per rilevare una data differenza di espressione con la potenza desiderata.
Per ciascun gene g, il disegno sperimentale può essere tradotto in un modello lineare (che, per semplicità, si è supposto ad effetti fissi), uguale per ogni set i, con i=1,2,…n, e dato da:
(6.3) Ygi = Xβg +εgi dove:
- β è il vettore dei parametri per il gene g, di dimensione (p x 1) dove p è il numero di g parametri considerati nel modello,
- è il vettore delle osservazioni (espresse sotto forma di “log-ratio”) relative al gene g e al set i,
gi
Y
- X è la matrice del disegno e - ε è il termine di errore. gi
Dato un disegno sperimentale, la matrice del disegno non è unica ma dipende dalla parametrizzazione scelta e dagli effetti che si vogliono includere nel modello. Nello sviluppo dell’algoritmo si è considerato il modello più semplice in cui è presente solo l’effetto del trattamento che è anche il fattore di interesse. Questa scelta è stata operata sulla base del fatto che altri fattori come, per esempio, la variabilità biologica fra campioni appartenenti alla stessa classe, sono assegnati dall’utente e non devono essere valutati dalla funzione. Per includere la stima di altri effetti, oltre all’effetto del trattamento, e non dare una valutazione a priori della loro variabilità bisognerebbe utilizzare un modello lineare differente, basato sui logaritmi delle intensità piuttosto che sui “log-ratio”, in linea con quanto previsto dai modelli ANOVA. Questo comporta però la necessità di un numero molto maggiore di campioni per poter garantire un numero di gradi di libertà sufficiente per stimare le diverse componenti della varianza.
Le matrici del disegno relative allo schema di base per i 3 tipi di disegno sperimentale, con 2, 3 e 4 classi a confronto sono integrate nel codice della funzione. Per un esperimento microarray con 2 classi a confronto, la matrice del disegno è data da:
(6.4) ⎟⎟ nel caso del “reference design”, ⎠ ⎞ ⎜⎜ ⎝ ⎛ − − = 1 1 1 0 0 1 X
(6.5) ⎟⎟ nel caso del “loop design” e del ”balanced block design”. ⎠ ⎞ ⎜⎜ ⎝ ⎛ − = 1 1 X 1 −1
Sebbene la matrice del disegno sia la stessa per il “loop design” e per il ”balanced block design”, bisogna tenere presente che nel primo caso le repliche considerate per ciascuna classe sono repliche tecniche (il “loop design” con 2 classi è di fatto un “dye-swap”), mentre nel secondo caso si tratta di repliche biologiche.
L’obiettivo di uno studio di “class comparison” consiste nello stabilire se un dato gene risulta differenzialmente espresso tra 2 o più gruppi. Questo problema si traduce di fatto nel confronto tra i valori medi di espressione di quel gene relativi a ciascun gruppo. Le ipotesi da verificare si possono pertanto esprimere in termini di una combinazione lineare dei parametri del modello, attraverso la definizione di una matrice o di un vettore dei contrasti C.
Con k classi, il numero di confronti indipendenti è pari a (k – 1), per definire i confronti di interesse si è allora costruita una delle possibili matrici dei contrasti C, di dimensione p x (k-1).
Con riferimento ad un esperimento microarray effettuato su due gruppi di campioni, il vettore dei contrasti definito all’interno dell’algoritmo è dato da:
(6.6) C =
(
1 −1 0)
T nel caso del “reference design,(
)
T(6.7) C = 1 −1 nel caso del “loop design” e del ”balanced block design”.
Le ipotesi nulla ed alternativa possono essere così formulate: H0: CTβg = 0
H1: CTβg ≠ 0, in particolare H1: CTβg = δg, dove δg è l’entità dell’”effect size” che si vorrebbe rilevare. δg sarà rispettivamente uno scalare o un vettore a seconda che i confronti di interesse sono uno o più di uno.
La statistica test è data dal rapporto: (6.8) ) ( / ) ( ) ( ) ( / ) ( ] / ) ( [ ) ( 1 1 n d X Y X Y C rg C n C X X C C F g g T g g g T T T T g T n β β β β − − = − − dove:
- rg(C) è il rango della matrice dei contrasti e
- d(n) è una funzione del parametro incognito n, che dipende dal tipo di disegno.
Sotto l’ipotesi nulla, Fn segue una distribuzione F di Fisher con gradi di libertà υ1 = rg(C) e υ2 = d(n). Sotto l’ipotesi alternativa, Fn segue invece una distribuzione F non centrale con gli stessi gradi di libertà e con parametro di non centralità λ dato da:
(6.9) ( ) 1( g) T g T g T C C β β λ = Σ − dove:
- Σg =σg2C(XTX)−1C/n è la varianza dei confronti di interesse, ossia Σg =Var(CTβg), e
- σg2 è la varianza dei dati relativi al gene g. Realisticamente 2 e δ
g
σ g variano da gene a gene, tuttavia per ricavare una stima della numerosità campionaria nella fase preliminare di progettazione dell’esperimento si può
supporre che tutti i geni abbiano lo stesso set di parametri σ2 e δ. In tal caso, la potenza media calcolata su tutti i geni, cioè su tutti i confronti, coincide con la potenza fissata per ogni gene.
Per calcolare la numerosità campionaria si sfrutta la definizione di potenza statistica che rappresenta la probabilità di rifiutare l’ipotesi nulla quando questa è falsa. In simboli: (6.10) 1−β =Pr( _ 0 : β =0| β =δ). T T C C H rifiutare
Questo equivale a risolvere l’equazione:
(6.11) 1−β =Pr( (υ1,υ2,λ)> | β =δ)
T crit
n F C
F
nell’incognita n, dove Fcrit = F(υ1,υ2,λ=0,α) è il valore di soglia della regione di rifiuto dell’ipotesi nulla, che dipende dal livello di significatività α scelto.
Noto il valore di n, si ricavano facilmente il numero di array e il numero di repliche biologiche per ognuno dei 3 disegni sperimentali considerati.
6.2.1 Guida all’utilizzo della funzione “sample_size”
Dalla descrizione delle funzioni per il calcolo dell’ampiezza campionaria integrate nei vari pacchetti bioinformatici emergono due importanti limiti legati al loro impiego. Innanzitutto esse o prescindono dal tipo di disegno utilizzato o propongono una casistica spesso insufficiente di disegni sperimentali, ma, soprattutto, possono essere utilizzate soltanto dagli “addetti ai lavori” poiché presuppongono che l’utente abbia, da un lato, una buona familiarità con il linguaggio di programmazione (R, nella fattispecie), dall’altro una conoscenza approfondita dei metodi statistici di analisi dei dati. Per esempio, per utilizzare la funzione ssize.F() del pacchetto “ssize.fdr”, l’utente deve specificare la struttura della matrice del disegno, il vettore o la matrice dei contrasti e tanti altri parametri il cui significato è in generale noto agli esperti di statistica, ma non alle altre figure professionali con formazione diversa che generalmente compongono i gruppi di ricerca che lavorano con i microarray.
Alla luce di queste considerazioni, la funzione “sample_size” è stata costruita in modo da limitare l’immissione da parte dell’utente soltanto di quei parametri statistici come il livello di significatività, la potenza o la deviazione standard, il cui significato è noto a chi, con qualunque formazione, lavori con i microarray.
Si descrivono di seguito i passaggi che l’utente deve compiere per utilizzare la funzione “sample_size” e ottenere il dato in uscita.
Nel sistema operativo Windows per lanciare il software R è sufficiente fare doppio click sull'icona corrispondente o avviarlo dal menu programmi come un qualsiasi altro programma.
Una volta che R è stato lanciato, si apre la schermata di avvio: tutte le istruzioni sono eseguibili dalla linea di comando dell'ambiente caratterizzata dal prompt “ >”.
Per poter utilizzare la funzione, l’utente deve innanzitutto specificare la directory di lavoro, ovvero la cartella dove è stato salvato il file contenente il codice della funzione e dove intende salvare i file relativi a quella sessione di lavoro. A tal fine basta digitare il comando: > setwd (dir)
il cui argomento è il percorso per raggiungere la directory di lavoro corrente, a cui è stato assegnato il nome “working dir “, come illustrato nella figura 6.4.
Fig. 6.4: Definizione della directory di lavoro
Il passo successivo consiste nel caricare la funzione nell’area di lavoro, assegnando il file contenente il codice della funzione ad una variabile locale mediante il comando:
> myf<- edit (file=”nome_file”) come mostrato in figura 6.5.
Fig. 6.5: Caricamento della funzione nell’area di lavoro
A questo punto non resta che eseguire una chiamata alla funzione, digitando il suo nome seguito da una coppia di parentesi, dal momento che essa non prevede la specifica di alcun argomento:
> sample_size( )
Sullo schermo vengono quindi visualizzate una serie di domande attraverso le quali l’utente è invitato ad introdurre il valore scelto per un dato parametro o a scegliere fra due o più opzioni alternative. In particolare l’utente deve immettere, nell’ordine, i seguenti parametri:
1. il numero di classi/varietà/trattamenti coinvolti nell’esperimento. Le scelte possibili considerate sono tre: 2, 3 o 4 classi, che corrispondono anche alle situazioni più frequenti nella realtà;
2. il livello di significatività α del test: l’utente può decidere se utilizzare il valore predefinito, pari a 0.001 (vedi fig. 6.6), oppure lasciare all’algoritmo il calcolo di α fornendo il numero di geni presenti sull’array e il numero di falsi positivi che l’utente è disposto a tollerare (vedi fig. 6.7);
Fig. 6.6: Sequenza delle domande che si presenta all’utente nel caso in cui decide di utilizzare
Fig. 6.7: Sequenza delle domande che si presenta all’utente nel caso in cui decide di calcolare
il livello di significatività α sulla base del numero di ipotesi da verificare 3. la potenza,
4. una stima della deviazione standard dei dati, ricavata dalla letteratura o da uno studio pilota;
5. l’entità dell’”effect size” che desidera rilevare, espresso in termini di “fold-change”; 6. il valore del rapporto tra la varianza biologica e quella tecnica che, sulla base dei dati di
letteratura, è tipicamente compreso tra 2 e 10.
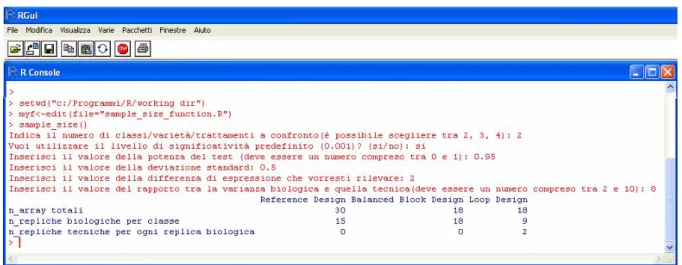
Il risultato della funzione viene visualizzato sotto forma di una matrice i cui elementi sono il numero totale di array e il numero di repliche biologiche per gruppo richieste dalle 3 tipologie di disegno sperimentale prese in considerazione. La figura 6.8 mostra l’uscita prodotta dalla funzione “sample_size” per i seguenti valori dei parametri:
- numero di classi/varietà/trattamenti a confronto = 2 - livello di significatività α = 0.001
- potenza 1 - β = 0.9
- deviazione standard σ = 0.5
- Fold-Change = 2 (che corrisponde a δ =log2(Fold-Change) = 1) - rapporto tra varianza biologica e varianza tecnica = 4.
Fig. 6.8: Visualizzazione del risultato prodotto dalla funzione sample_size( )
A parità dei valori dei parametri in ingresso, il numero di array necessari per il “reference design” è 28, contro i 16 array richiesti nel caso si voglia utilizzare un ”balanced block design” o un “loop design”. Le repliche biologiche che occorre collezionare per ogni gruppo sono invece 14 per il “reference design”, 16 per il ”balanced block design”, 8 per il “loop design”.
Per verificare la correttezza dei risultati prodotti dalla funzione si è fatto riferimento ai dati riportati in un articolo di Dobbin e collaboratori (Dobbin et al.,2003). In questo articolo vengono riportati il numero di array e il numero di repliche biologiche richiesti in un esperimento di “class comparison” con il quale si vogliono identificare quali geni risultano differenzialmente espressi tra due classi. In particolare viene fatto un confronto tra il “reference design” e il ”balanced block design”, per i quali, fissati i valori dei parametri (α = 0.001, 1 – β = 0.95, δ = 1, σ = 0.5, varianza biologica/varianza tecnica = 8) si trova che il numero di array totali è rispettivamente pari a 30 e 17, da cui segue che il numero di repliche biologiche per classe è 15 per il “reference design” e 17 per il ”balanced block design”. In realtà con 17 array il disegno a blocchi non è bilanciato rispetto ai fluorocromi. Per il bilanciamento è necessario un numero pari di array, quindi sale a 18 il numero di array e di repliche biologiche richiesti da un ”balanced block design”.
Utilizzando la funzione con i parametri forniti dall’articolo si ottengono gli stessi risultati, come dimostra la figura 6.9.
Fig. 6.9: Risultati prodotti dalla funzione sample_size( ) sulla base dei dati forniti dal lavoro
di Dobbin e collaboratori.
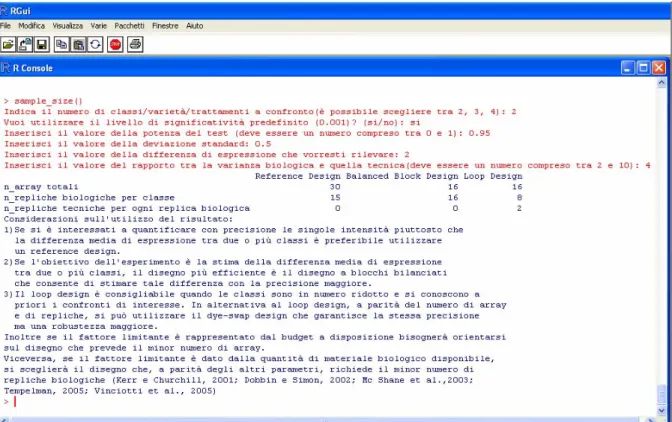
A partire dai risultati forniti dalla funzione, l’utente deve poi decidere quale disegno sperimentale adottare. Tale scelta è subordinata a quello che è riconosciuto come fattore limitante nell’esperimento; in particolare se il vincolo è rappresentato dal budget a disposizione bisognerà orientarsi sul disegno che prevede il minor numero di array. Viceversa, se il fattore limitante è dato dalla quantità di materiale biologico disponibile, si sceglierà il disegno che, a parità degli altri parametri, richiede il minor numero di repliche biologiche.
Un’altra considerazione che deve guidare lo sperimentatore in questa fase di pianificazione dell’esperimento è l’obiettivo dello studio. In particolare se si è interessati a quantificare con precisione le singole intensità piuttosto che la differenza media di espressione tra le classi, è preferibile utilizzare un “reference design”.
Se, al contrario, l’obiettivo dell’esperimento è la stima della differenza media di espressione tra due o più classi, il disegno più efficiente è il ”balanced block design” che consente di stimare tale differenza con la precisione maggiore (ovvero con la varianza più piccola) in quanto si basa sul confronto diretto tra le classi (Mc Shane et al., 2003). Tale disegno diventa tuttavia molto complesso quando i gruppi a confronto sono più di 2 e lo sperimentatore è interessato a confrontare ciascun gruppo con tutti gli altri.
Infine il “loop design” è consigliabile quando le classi sono in numero ridotto e si conoscono a priori i confronti di interesse. In alternativa si può utilizzare il “dye-swap design” che, a parità di precisione, ha l’indiscutibile vantaggio di essere molto più robusto (Kerr e Churchill, 2001; Dobbin e Simon, 2002; Tempelman, 2005; Vinciotti et al., 2005).
Per agevolare l’utente nella scelta del disegno più adatto ai suoi scopi, le considerazioni fatte sopra vengono visualizzate in forma ridotta sullo schermo, come mostrato in figura 6.10.
Fig. 6.10: Visualizzazione delle considerazioni prodotte dalla funzione sample_size( ) per