Materiali
e
2.1. COLTURE CELLULARI
In questo lavoro di tesi sono state utilizzate cellule di astrocitoma umano 1321N1 trasfettate stabilmente con il recettore GPR17 umano (hGPR17). Le cellule sono state mantenute in incubazione alla temperatura di 37°C in un’atmosfera umidificata con il 5% di CO2. Il terreno di
coltura è il Dulbecco’s modified Eagle medium (DMEM), addizionato di L-glutammina 2 mM, penicillina-streptamicina 100 U/mL e siero fetale bovino (FBS) al 10%.
2.2. VALUTAZIONE DELL’ASSOCIAZIONE DELLE β-ARRESTINE CON GPR17
Per analizzare il profilo di reclutamento delle β-arrestine 1 e 2 da parte del GPR17 in seguito a stimolazione con le due classi di ligandi, le cellule 1321N1 hGPR17 sono state stimolate con UDP-glucosio o LTD4. Abbiamo quindi preparato i lisati cellulari e immunoprecipitato il recettore
GPR17, per isolarlo e poter successivamente studiare con analisi Western Blot se fosse associato alle β–arrestine 1 e/o 2.
2.2.1. Preparazione di lisati cellulari di cellule 1321N1 hGPR17
Le cellule 1321N1 vengono trattate per 5 minuti con il solo mezzo di coltura (controllo) o UDP-glucosio 100 μM o LTD4 100 nM. Al termine dei trattamenti, si aspira il mezzo e si lavano le cellule
con 5 mL di PBS (NaH2PO4 9.1 mM, Na2HPO4 1.7 mM, NaCl 150 mM, pH 7.4). Le cellule vengono
quindi staccate dalla piastra meccanicamente con uno scraper, raccolte in falcoon e centrifugate a 1200g per 5 minuti. Si aspira il sovranatante e si sospendono i pellet ottenuti in RIPA contenente inibitori delle proteasi (RIPA: Na-deossicolato 0.5%, IGEPAL 1%, SDS 0.1%, pH 7.4 – inibitori delle proteasi: PMSF 0.6 mM, Aprotinina 0.6 μM, Ortovanadato 1 mM).
I lisati ottenuti vengono raccolti in eppendorf, messi in agitazione per 1 ora a una temperatura di 4°C e infine centrifugati a 13500g per 45 minuti a 4°C. Viene quindi raccolto il sovranatante, che costituisce il lisato, e si esegue il dosaggio proteico.
2.2.2. Dosaggio proteico di Folin
La quantificazione delle proteine totali presenti nei lisati di cellule 1321N1 hGPR17 è stata effettuata tramite il saggio colorimetrico di Folin, che sfrutta la reazione delle proteine con tartrato di rame e reagente di Folin in soluzione alcalina. In questa reazione, la proteina (in particolare gli aminoacidi tirosina e triptofano, in misura minore cistina, cisteina, e istidina) riduce il rame da Cu++ a Cu+ e il rame Cu+ riduce a sua volta il reagente di Folin. La riduzione del
reagente determina la perdita di 1, 2 o 3 atomi di ossigeno, sviluppando una caratteristica colorazione celeste-blu con un picco di assorbanza a 750 nm. In questi esperimenti viene usato il
Kit Bio-Rad, costituito da REAGENTE A (soluzione alcalina di tartrato di rame) e REAGENTE B
(reagente di Folin diluito).
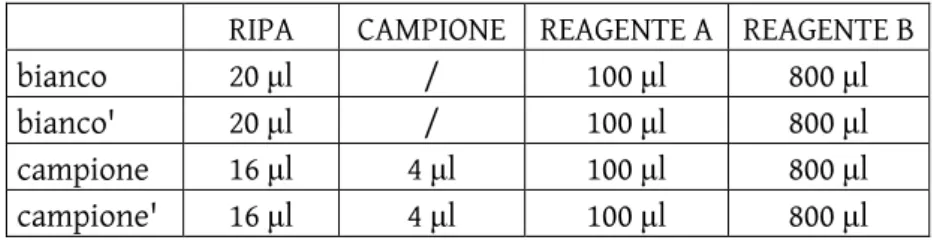
Lo schema di preparazione del dosaggio è mostrato in Tabella 1:
RIPA CAMPIONE REAGENTE A REAGENTE B
bianco 20 μl / 100 μl 800 μl
bianco' 20 μl / 100 μl 800 μl
campione 16 μl 4 μl 100 μl 800 μl
campione' 16 μl 4 μl 100 μl 800 μl
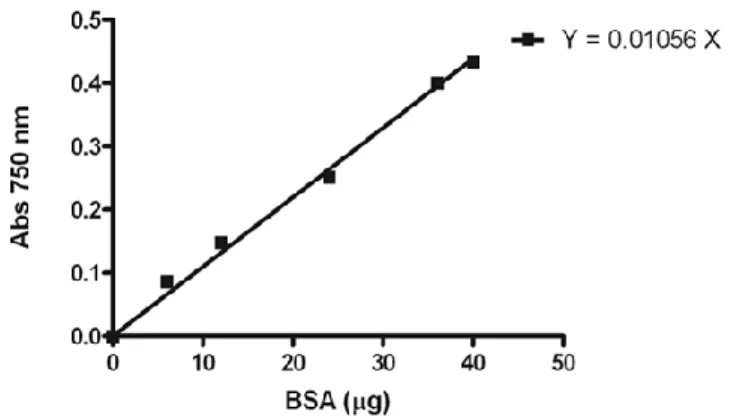
I campioni vengono lasciati in incubazione per 30 minuti ed in seguito si legge l’assorbanza a 750 nm allo spettrofotometro. Dai valori di assorbanza ottenuti, utilizzando il coefficiente della retta di taratura del saggio, possiamo risalire alla concentrazione di proteine presenti nei 4 μl di campione utilizzati. Per costruire la retta di taratura di questo saggio si solubilizzano circa 2 mg
di albumina bovina (BSA) in 1 mL di RIPA per ottenere una soluzione 2 μg/μl e si dosano concentrazioni crescenti di BSA. La retta di taratura che si ottiene è mostrata nella Figura 1.
2.2.3. Immunoprecipitazione del recettore GPR17
Aliquote di lisati vengono trattate con l’anticorpo specifico per la proteina da immunoprecipitare (nel nostro caso anticorpo anti-GPR17, 5 μg/mg proteine). I lisati vengono quindi lasciati in agitazione tutta la notte a 4°C, dopodiché si aggiungono 80 μl di Proteina A Sepharose in ogni campione, per far precipitare il complesso antigene-anticorpo. Si mette in agitazione per almeno 2 ore a 4°C, si centrifuga a 13000g per 1 minuto a 4°C, si aspira il sovranatante e si lavano i pellet con RIPA contenente inibitori delle proteasi. A questo punto aggiungiamo 20 μl di soluzione di Laemmli (Tris HCl 0.25 M, pH 6.7-7.4, SDS 5%, β-mercaptoetanolo 10%, glicerolo 20%, blu di bromofenolo 400 mg/L) con DTT 200 mM in ogni campione e si fanno scaldare i campioni a 100°C per 5 minuti, per favorire la denaturazione della Proteina A Sepharose e il suo distacco dal complesso anticorpo-recettore. Le proteine vengono quindi separate mediante elettroforesi in condizioni denaturanti (SDS-PAGE). La corsa elettroforetica delle proteine è condotta su un gel di poliacrilammide, in presenza di sodio dodecil solfato (SDS), un composto in grado di legarsi alle proteine e denaturarle, distruggendone legami a idrogeno e ponti disolfuro e promuovendo
così la perdita della struttura secondaria o terziaria delle proteine stesse. Questa denaturazione è indispensabile per separare tra loro i complessi proteina-SDS in funzione della sola massa, annullando le cariche degli aminoacidi, che influirebbero sulla migrazione. I complessi ottenuti sono tutti carichi negativamente e migrano verso il polo positivo del campo elettrico applicato con una velocità che risulta essere, esclusivamente, inversamente proporzionale al log10 del loro
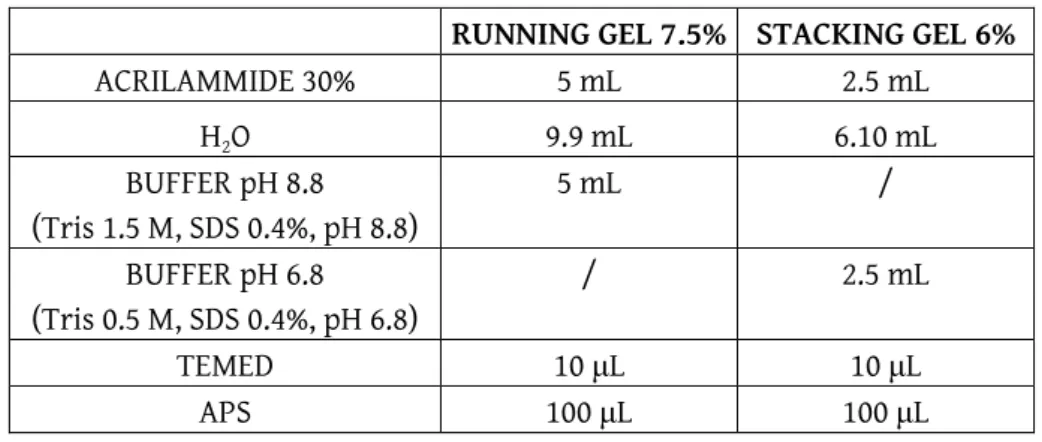
peso molecolare. I gel utilizzati per l’elettroforesi SDS-PAGE sono costituiti da: RUNNING GEL (permette la separazione delle proteine dei vari campioni in base al loro peso molecolare); STACKING GEL (permette di posizionare e concentrare i campioni negli appositi pozzetti, affinché questi possano iniziare la corsa elettroforetica dallo stesso punto).
La preparazione dei gel è riportata nella Tabella 2.
RUNNING GEL 7.5% STACKING GEL 6%
ACRILAMMIDE 30% 5 mL 2.5 mL H2O 9.9 mL 6.10 mL BUFFER pH 8.8 5 mL / (Tris 1.5 M, SDS 0.4%, pH 8.8) BUFFER pH 6.8 / 2.5 mL (Tris 0.5 M, SDS 0.4%, pH 6.8) TEMED 10 μL 10 μL APS 100 μL 100 μL
Le due miscele vengono agitate e degassate per circa 15 minuti per evitare che l’ossigeno atmosferico formi radicali e impedisca l’innesco del processo di polimerizzazione del gel. Solo al momento dell’uso vengono aggiunti APS (Ammoniopersolfato 10%) e TEMED (Tetrametil-etilen-diammina). APS è un agente ossidante, impiegato come fonte di radicali liberi, per iniziare la polimerizzazione dell’acrilamide e TEMED, una volta attivato da APS, funziona da catalizzatore e rende reattiva l’acrilamide, inducendo una polimerizzazione radicalica. Una volta avvenuta la polimerizzazione del Running gel si carica lo Stacking gel, al cui interno si inserisce un pettine per formare i pozzetti in cui verranno caricati i campioni. Avvenuta la polimerizzazione del gel, gli
immunoprecipitati di GPR17, sospesi in Laemmli contenente DTT 200 mM, vengono caricati nei pozzetti e si imposta la corsa elettroforetica a 17 mA per circa 90 minuti, mantenendo una temperatura mediamente fredda (Figura 2).
2.2.4. Western Blot su immunoprecipitati di cellule 1321N1 hGPR17
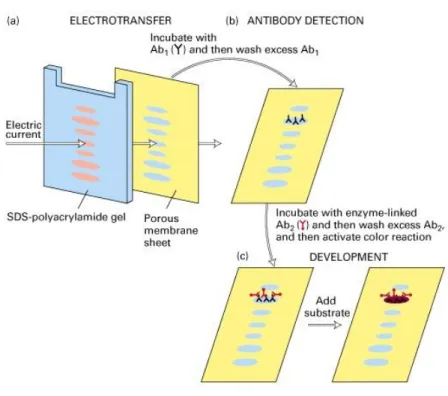
Una volta separate le proteine immunoprecipitate con l'anticorpo anti-GPR17 mediante elettroforesi, abbiamo valutato la co-immunoprecipitazione delle β-arrestina 1 e 2 mediante marcatura di quest'ultime proteine con la tecnica del Western Blot. Le proteine seaparate sul gel di poliacrilamide vengono trasferite su una membrana di polivinilidenfluoruro (PVDF), precedentemente attivata in metanolo, mediante elettroblotting. Il trasferimento viene fatto creando un “sandwich” (Figura 3).
I due sandwich vengono inseriti nella camera di trasferimento, riempita con il tampone di Transfert freddo (glicina 0.19 M, Tris 25 mM, SDS 3.5 mM, MeOH 200 mL, H2O mQ q.b. a 1L) e si
imposta un voltaggio costante di 120 V per 45 minuti perpendicolare al gel, provocando così la migrazione delle proteine dal gel al foglio di PVDF. La camera viene messa in ghiaccio, per mantenere bassa la temperatura durante il trasferimento. Terminato il trasferimento, si apre il
sandwich e si mette in una vaschetta la membrana di PVDF, in modo che sia completamente
immersa in Milk [TBS (Tris-HCl 10 mM, NaCl 150 mM, pH 8); Milk 5%; Tween-20 0.05%], mantenendola in agitazione sul basculante per almeno 1 ora a temperatura ambiente. Questo passaggio risulta fondamentale, dal momento che il Milk ha la funzione di bloccare tutti i siti di legame aspecifici presenti sul foglio. Dopodiché la membrana viene lasciata in incubazione con l’anticorpoprimario che riconosce la proteina di interesse tutta la notte a 4°C in agitazione. In questi esperimenti abbiamo utilizzato gli anticorpi diretti contro la β–arrestina 1 (Santa Cruz
Biotechnology, diluito 1:100) e la β–arrestina 2 (Abcam, diluito 1:500). La mattina seguente si lava il
foglio di PVDF tre volte con TBS-Tween-20 (Tris-HCl 10 mM; NaCl 150 mM, pH 8; Tween-20 0.05%) e si lascia in incubazione con l’anticorpo secondario diluito in Milk, per almeno due ore. In questi esperimenti abbiamo utilizzato un anticorpo secondario anti-goat IgG coniugato alla perossidasi, diluito 1:20000. Si effettuano quindi tre lavaggi da 5-10 minuti con TBS-Tween-20 e un lavaggio da 5-10 minuti con TBS. A questo punto si può procedere allo sviluppo della lastra. Per prima cosa, si recupera il foglio di PVDF con una pinzetta, lasciandolo sgocciolare su carta assorbente, e
si ripone in una vaschetta pulita e asciutta, sulla quale versare i reagenti luminescenti E.C.L. (in rapporto 1:1, Perkin Elmer) per circa 1 minuto. Si asciuga la membrana e si posiziona tra due pellicole trasparenti, per poi metterla a contatto con la lastra fotografica per un intervallo di tempo variabile di 2-5 minuti. L’anticorpo secondario, che riconosce l’anticorpo primario precedentemente legato, è marcato con la perossidasi di rafano, che in presenza di perossido di idrogeno ossida il luminolo, dando luogo al fenomeno della chemioluminescenza, in grado di impressionare una lastra fotografica esposta al PVDF.
Uno schema riassuntivo dei passaggi sopra elencati è riportato in Figura 4.
2.3. VALUTAZIONE DELLA FOSFORILAZIONE DELLE ERK 1/2 MEDIATA DA
AGONISTI DEL GPR17
Per valutare l’effetto degli agonisti del GPR17 sul pathway di segnale delle ERK 1/2, abbiamo studiato la cinetica di fosforilazione di queste MAP-chinasi, utilizzando un saggio ELISA di tipo
indiretto su cellule 1321N1 hGPR17, stimolate per diversi tempi (1- 60 minuti) con UDP-glucosio o
LTD4.
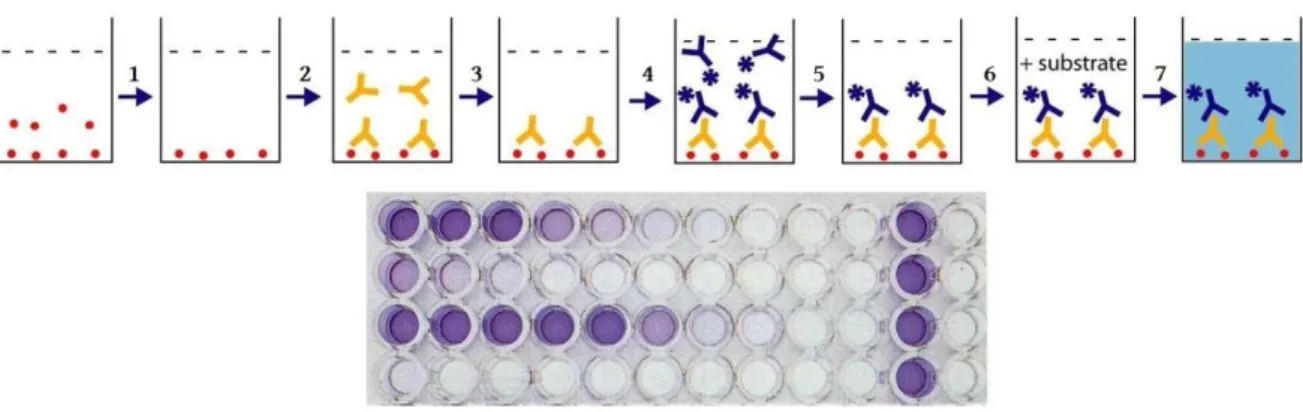
Il saggio ELISA, acronimo inglese per Enzyme-Linked ImmunoSorbent Assay (Saggio
Immuno-Assorbente legato ad un Enzima), è un metodo di analisi immunologica, utile per rilevare la presenza
di una sostanza attraverso l’impiego di due anticorpi, uno dei quali è collegato ad un enzima che catalizza una reazione il cui prodotto è visibile. Il saggio ELISA di tipo indiretto è caratterizzato dalle seguenti fasi:
1. Si fissa al supporto il campione biologico del quale vogliamo verificare la presenza dell’antigene di interesse;
2. Si inserisce l’anticorpo primario diretto contro l’antigene di interesse e si lascia in incubazione per favorire la formazione del complesso antigene-anticorpo. A questo punto, si effettuano dei lavaggi per rimuovere l’eccesso di anticorpo (che non si è legato); 3. Si inserisce l’anticorpo secondario diretto contro l’anticorpo primario precedentemente utilizzato. L’anticorpo secondario è collegato ad un enzima capace di catalizzare una reazione, il cui prodotto sia rilevabile. In genere viene utilizzato come enzima la
perossidasi del rafano, che catalizza la reazione: H2O2 + substrato ridotto 2 H2O +
substrato ossidato. Dopo un periodo di incubazione per favorire il legame dell’anticorpo secondario al primario contro cui è diretto, si effettuano dei lavaggi per rimuovere l’anticorpo in eccesso.
Dopo il lavaggio dell’eccesso dell'anticorpo secondario collegato all’enzima, si aggiunge la
di colore, si blocca la reazione con una Stop Solution e si legge l’assorbanza alla lunghezza d’onda di riferimento.
I vari step del saggio utilizzato per valutare la fosforilazione delle ERK descritto sono riportati di seguito (Figura 5):
Per la valutazione della fosforilazione delle ERK, è stato seguito il protocollo del Kit Elisa (Active
Motif), che prevede le seguenti fasi:
1. Seminare cellule 1321N1 hGPR17 in multiwell da 96 pozzetti e trattare con UDP-glucosio o LTD4 per tempi diversi;
2. Fissare le cellule con 100 μl di Fixing Buffer, costituito da formaldeide al 4% in PBS 1X; 3. Incubare 20 minuti a temperatura ambiente;
4. Rimuovere Fixing Buffer e fare tre lavaggi da 5 minuti con 200 μl di Wash Buffer sotto leggera agitazione;
5. Rimuovere Wash Buffer e aggiungere 100 μl di Quenching Buffer; 6. Incubare 20 minuti a temperatura ambiente;
7. Rimuovere Quenching Buffer e fare due lavaggi da 5 minuti con 200 μl di Wash Buffer; 8. Rimuovere Wash Buffer e aggiungere 100 μl di Antibody Blocking Buffer;
Figura 5: Schematizzazione delle fasi di un saggio ELISA di tipo indiretto. Sia dopo l’incubazione con l’anticorpo
primario (2) che dopo l’incubazione con l’anticorpo secondario collegato all’enzima (4), si effettuano dei lavaggi (3) e (5) per rimuovere l’eccesso di anticorpo. Dopo l’inserimento del substrato della reazione catalizzata dall’enzima (6), si osserva una variazione di colore dovuta alla formazione del prodotto (7). Come si vede nella parte inferiore della
9. Incubare 1 ora a temperatura ambiente;
10. Rimuovere Antibody Blocking Buffer e fare due lavaggi da 5 minuti con 200 μl di Wash Buffer; 11. Rimuovere Wash Buffer e aggiungere 40 μl di Anticorpo I p-ERK (diluito in Antibody Diluition
Buffer in rapporto 1:250) o, per il controllo, 40 μl di Antibody Diluition Buffer e coprire;
12. Incubare a 4°C overnight;
13. Rimuovere Anticorpo I p-ERK diluito e fare tre lavaggi da 5 minuti con 200 μl di Wash Buffer; 14. Rimuovere Wash Buffer e aggiungere 100 μl di Anticorpo II (diluito in Antibody Diluition
Buffer in rapporto 1:200);
15. Incubare 1 ora a temperatura ambiente;
16. Durante l’incubazione trasferire in un secondo contenitore la Developing Solution fornita dal kit (fotosensibile) e lasciare a temperatura ambiente per almeno 1 ora;
17. Rimuovere Anticorpo II diluito e fare tre lavaggi da 5 minuti con 200 μl di Wash Buffer e due lavaggi da 5 minuti con 200 μl di PBS 1X;
18. Rimuovere PBS 1X e aggiungere 100 μl di Developing Solution;
19. Incubare da 2 a 20 minuti a temperatura ambiente al riparo dalla luce (durante l’incubazione monitorare il viraggio della colorazione da trasparente a blu non troppo intenso, facendo attenzione a non sovrasviluppare);
20. Aggiungere 100 μl di Stop Solution in ogni pozzetto allo stesso momento (viraggio dal blu al giallo) e leggere assorbanza allo spettrofotometro a 450 nm.
Successivamente si esegue la colorazione delle cellule con Crystal Violet, per verificare la concentrazione cellulare in ciascun pozzetto e poter quindi attribuire univocamente la diversità di fosforilazione delle ERK, rilevata con il saggio sovraesposto, all’effetto della stimolazione con UDP-g e LTD4, e non ad un diverso numero di cellule presenti. Il Crystal Violet è un colorante
organico sintetico costituito dal cloridrato di un derivato della fucsina. È usato in microscopia per evidenziare le cellule facendole apparire viola. Il protocollo seguito per la colorazione con
1. Dopo la lettura a 450 nm, fare due lavaggi da 5 minuti con 200 μl di Wash Buffer e due lavaggi da 5 minuti con 200 μl di PBS 1X;
2. Tamponare la piastra con carta assorbente per rimuovere l’eccesso di liquido dai pozzetti e lasciare all’aria per 5 minuti a temperatura ambiente;
3. Aggiungere 100 μl di Crystal Violet e incubare per 30 minuti a temperatura ambiente; 4. Fare tre lavaggi da 5 minuti con 200 μl di PBS 1X;
5. Rimuovere PBS 1X e aggiungere 100μl di SDS 1%;
6. Incubare per 1 ora a temperatura ambiente sotto leggera agitazione; 7. Leggere assorbanza allo spettrofotometro a 595 nm.
2.3.1. Valutazione del contributo del pathway proteina G
i-dipendente alla
fosforilazione delle ERK 1/2 mediata da agonisti del GPR17
Per determinare il contributo del pathway Gi-dipendente all’attivazione delle MAP-chinasi da
parte degli agonisti del GPR17, le cellule sono state pretrattate con l’inibitore della proteina Gαi, la
tossina della pertosse (PTX) (200 ng/mL), per 16 ore prima della stimolazione con gli agonisti al recettore. Successivamente abbiamo stimolato le cellule con UDP-glucosio e LTD4 a tempi diversi
(da 1 a 30 minuti) e valutato la fosforilazione delle ERK con il saggio ELISA di tipo indiretto precedentemente esposto.
2.3.2. Valutazione del contributo del pathway β-arrestina-dipendente alla
fosforilazione delle ERK 1/2 mediata da agonisti del GPR17
Per determinare il contributo delle β-arrestine all’attivazione delle ERK mediata da ligandi del GPR17, abbiamo usato la tecnica dell’RNA Interference per ridurre l’espressione della β-arrestina 1 e/o della β-arrestina 2 endogene in cellule 1321N1 hGPR17. Successivamente abbiamo stimolato
le cellule con UDP-glucosio e LTD4 e valutato la fosforilazione delle ERK tramite il saggio ELISA di
tipo indiretto utilizzato negli esperimenti precedenti.
2.3.2.1. Spingimento delle β–arrestine 1 e 2 tramite Small Interfering RNA
(siRNA)
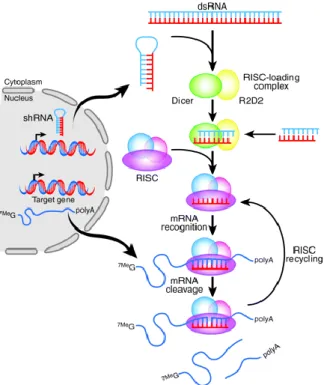
I siRNA, Small Interfering RNA, sono frammenti di circa 25 nucleotidi coinvolti nel silenziamento dell’espressione di specifici geni. I siRNA vengono introdotti artificialmente nella cellula attraverso metodi di trasfezione e, in linea teorica, qualsiasi gene a sequenza nota può essere scelto come bersaglio di un siRNA. Il meccanismo di silenziamento tramite siRNA è mostrato in
Figura 6.
Il protocollo di trasfezione siRNA utilizzato è quello della Santa Cruz Biotechnology, che prevede:
Giorno 1: seminare in multiwell da 24 pozzetti circa 30000 cellule in 2 mL di mezzo di
coltura con siero ma senza antibiotici selezionanti; Giorno 2: preparare le soluzioni A e B.
SOLUZIONE A: 0.625 μg di siRNA duplex in 100 μL di mezzo di trasfezione ogni 4 pozzetti; SOLUZIONE B: 5 μl di reagente di trasfezione in 100 μL di mezzo di trasfezione ogni 4 pozzetti.
Si uniscono le due soluzioni e si lasciano incubare per 45 minuti a temperatura ambiente. A questo punto, la soluzione A+B ottenuta viene aggiunta alle cellule per 5-7 ore. Si aggiunge poi ad ogni pozzetto il mezzo di coltura 2X (doppio siero e antibiotici) e si mettono in incubazione le cellule per 18-24 ore. Il giorno seguente si aspira la soluzione e si sostituisce con il mezzo di coltura completo normale 1X. L’esecuzione del saggio viene effettuata solo dopo 48 ore dalla rimozione del siRNA dalle colture cellulari.
Per verificare che i siRNA utilizzati fossero realmente specifici per le β-arrestine 1 e 2, abbiamo estratto l’mRNA da cellule 1321N1 hGPR17, precedentemente trattate con siRNA β-arrestina 1 o
siRNA β-arrestina 2, e da cellule trattate con siRNA random, come controllo. Abbiamo quindi
retro-trascritto gli specifici mRNA ed eseguito l’analisi PCR, utilizzando primer specifici per la β-arrestina 1, la β-β-arrestina 2 e la β-actina umane.
2.3.2.2. Tecnica di estrazione del mRNA e retrotrascrizione in cDNA
Seguendo il protocollo del Kit Qiagen, l’estrazione del mRNA prevede le seguenti fasi:
1. Aggiungere al pellet 350 μl di RTL + 10 μl/mL di β-mercaptoetanolo e un’eguale volume di EtOH 70%;
2. Agitare su vortex per un minuto;
3. Trasferire il campione nella colonnina del kit (per un volume non superiore a 700 μl, altrimenti ripetere il passaggio due o più volte);
4. Centrifugare 15 secondi a 10000g;
5. Scartare il residuo dal tubo, aggiungere 700 μl di RW1 e centrifugare a 10000 rpm per 15
secondi;
6. Cambiare tubo raccoglitore, aggiungere 500 μl di RPE e centrifugare per 15 secondi a 10000g;
7. Scartare il residuo dal tubo, aggiungere 500 μl di RPE e centrifugare per 2 minuti a 10000g;
8. Trasferire la colonnina in eppendorf da 1.5 mL, aggiungere H2O RNasi free e centrifugare a 10.000g per 1 minuto; si ottiene così il PRIMO ELUATO;
9. Ripetere il passaggio 7 ed 8 per ottenere il SECONDO ELUATO.
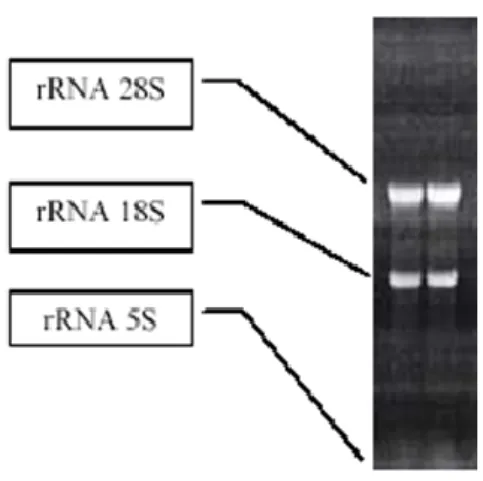
L’RNA estratto deve essere conservato a -20°C. Viene inoltre effettuata una corsa su gel di agarosio all’1% (1 μl di RNA, 1 μl di colorante 6X, 4 μl di H2O) per verificare l’integrità del
prodotto. Essendo gli RNA ribosomiali (rRNA) gli RNA più abbondanti in una cellula, la valutazione della corsa viene effettuata controllando la presenza di due bande nette corrispondenti alle subunità 28S e 18S dell’rRNA. Quando l’rRNA si degrada, le bande 18S e 28S non sono più presenti (Figura 7).
Una volta completata l’estrazione, si procede alla retrotrascrizione del mRNA in cDNA, seguendo il protocollo del Kit Biorad, composto da: 4 μl di buffer; 1 μl d enzima retrotrascrittasi; 3-7 μl di RNA estratto; H2O q.b. a 20 μl. I campioni vengono sottoposti ad un ciclo di:
1. 25°C per 5 minuti;
2. 42°C per 30 minuti (retrotrascrizione);
3. 85°C per 5 minuti (disattivazione dell’enzima); 4. 4°C per infinito.
2.3.2.3. Tecnica di amplificazione genica: PCR
In biologia molecolare, la PCR, dall’acronimo inglese Polymerase Chain Reaction (reazione a catena della polimerasi), è una tecnica di amplificazione genica, che permette di ottenere un elevato numero di copie di DNA, a partire da una singola molecola di acido nucleico o da un frammento di DNA di cui si conosce la sequenza nucleotidica iniziale e finale. In questo modo, possiamo preparare in vitro, in modo molto rapido, la quantità di materiale genetico richiesta.
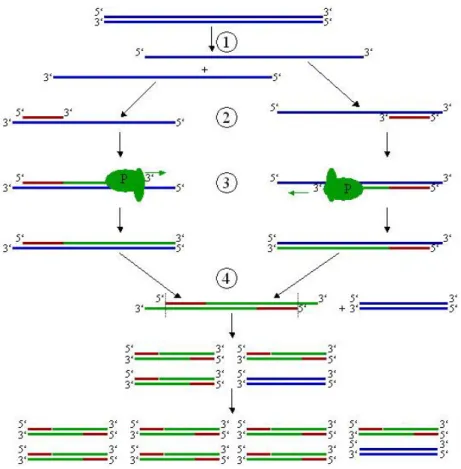
Questo processo di amplificazione si basa sulla naturale reazione di sintesi di un nuovo filamento di DNA, da parte della DNA-polimerasi (Figura 8). Tuttavia, alle elevate temperature raggiunte dalla PCR (96-99°C), la DNA-polimerasi umana si degrada e quindi non può essere usata per l’esecuzione di questa tecnica. Al suo posto si impiega la Taq-polimerasi, una polimerasi più resistente ottenuta dal batterio termofilo Thermophilus aquaticus. Nella PCR si realizza una sequenza di numerosi cicli, in ognuno dei quali viene duplicato il DNA sintetizzato nelle fasi precedenti, ottenendo così una “reazione a catena della polimerasi”.
La reazione a catena della polimerasi condotta in laboratorio utilizza una miscela di soluzione di DNA, desossiribonucleotidi trifosfato, ioni magnesio, primer e Taq-polimerasi. Le fasi della PCR sono schematizzate in Figura 9:
1. DENATURAZIONE o Melting (94-99°C); a queste temperature si promuove la separazione dei due filamenti della molecola stampo, rendendo così accessibile la regione da amplificare.
2. APPAIAMENTO o Annealing (40-55°C); a queste temperature si favorisce il legame dei
primer alle sequenze complementari dei filamenti denaturati, dando così inizio alla
sintesi del DNA.
Figura 8: Meccanismo di amplificazione genomica. 1. Denaturazione; 2. Annealing; 3. Allungamento (P =
Polimerasi); 4. Primo ciclo completo, i due filamenti di DNA risultanti costituiscono il DNA stampo per il ciclo successivo, raddoppiando così la quantità di DNA duplicato per ogni ciclo.
3. ALLUNGAMENTO o Extension (65-72°C); a queste temperature si ottimizza la capacità della Taq-polimerasi di promuovere l’allungamento dei primer, utilizzando come stampo il singolo filamento di DNA.
Il ciclo di denaturazione - appaiamento - estensione viene ripetuto in genere 30-40 volte, senza mai superare i 50 cicli totali. Questo è dovuto al fatto che i primi prodotti apprezzabili di PCR si formano a partire dal terzo ciclo e si accumulano con andamento esponenziale, fino al raggiungimento di un plateau; inoltre, un numero eccessivo di cicli rischia di estendere l’amplificazione anche a materiale indesiderato, contaminante.
PRIMER: oligonucleotidi sintetici che funzionano da innesco per l’inizio della sintesi del nuovo
filamento di DNA. Secondo la loro complementarietà vengono distinti in reverse (complementari al filamento in direzione 5’-3’) e forward (complementari al filamento in direzione 3’-5’). La scelta dei primer è un aspetto critico, importante per la buona riuscita della PCR. Infatti, questi oligonucleotidi determinano alcune caratteristiche fondamentali dell’intero processo:
Devono ibridarsi in maniera efficiente e specifica alla sequenza di interesse; A seconda dello scopo della PCR la loro tipologia può cambiare;
La lunghezza può variare da 20 a 30 paia basi e sarebbe opportuno che questa non fosse mai inferiore a 16 paia basi, per non compromettere la specificità del processo;
Durante la preparazione della PCR, la distanza tra i due primer è piuttosto flessibile. Può andare da 100 a 10000 paia basi, anche se l'efficienza di amplificazione diminuisce superate le 3000 paia basi;
Controllare che nel resto del genoma non vi siano sequenze omologhe;
Possono essere disegnati tenendo conto del contenuto di CG (tra il 45-50%) e della presenza di sequenze complementari o ripetute invertite, per evitare la formazione di dimeri o strutture a forcina tra primer.
MAGNESIO: influenza l'attività della polimerasi e l'ibridizzazione dei primer e aumenta la
temperatura nella fase di denaturazione. Vista la sua grande rilevanza, si deve controllare che nella miscela di reazione non sia presente un'eccessiva quantità di agenti chelanti, come EDTA, o di gruppi carichi negativamente, come gruppi fosfato, poiché entrambi possono sequestrare il magnesio e non renderlo più disponibile al processo di amplificazione.
NUCLEOTIDI: vengono utilizzati in genere alla concentrazione di 200 μM. Valori di
concentrazione eccessivamente più alti riducono l'efficienza della reazione, poiché i gruppi fosfato, carichi negativamente, si possono legare al magnesio rendendolo meno disponibile. Infatti, a concentrazioni superiori a 200 μM, la percentuale d'errore della polimerasi aumenta notevolmente.
ENZIMA: la Taq-polimerasi, data la sua caratteristica di enzima altamente termostabile, ha
consentito il raggiungimento di temperature di Annealing e di allungamento più elevate, permettendo di migliorare la specificità della reazione di PCR. Inoltre, la Taq-polimerasi garantisce un’amplificazione di frammenti con lunghezza superiore a 400 pb fino un massimo di 10 Kb e presenta un basso tasso di errore, mostrando un’attività esonucleasica solo in direzione 5’-3’ e non in direzione 3’-5’.
Nel nostro lavoro abbiamo allestito il saggio di PCR utilizzando, per un volume totale di 25 µL:
polimerasi (5 U/μl) 0.2 μl (0.04 U/μl in prova) MgCl2 (25 mM) 1.5 μl (1.5 mM in prova)
dNTP (10 mM) 0.5 μl (200 μM in prova) Buffer 5X 5 μl (1X in prova)
Primer 0.75 μl FOR (1 µM in prova) 0.75 μl REV (1 µM in prova)
cDNA 4 μl
H2O 12.3 μl
Lo schema di PCR utilizzato è il seguente:
1. 94°C per 5 minuti (attivazione della Taq polimerasi); 2. 95°C per 1 minuto (denaturazione dei due filamenti); 3. 55°C per 30 minuti (Annealing dei primer);
4. 72°C per 1 minuto (estensione dei filamenti); 5. 72°C per 7 minuti termina allungamenti incompleti.
I primer utilizzati sono riportati nella Tabella 3:
REV FOR
β-ACTINA CTCGTTAATGTCACGCAC CGTCTTCCCCTCCATCG
β -ARRESTINA 1 GTCAGTGGCTTCTTGTCCTC GGTCCTGGTGGATCCTGAGT β -ARRESTINA 2 GTCTTCGTGCTTGAGTTGCC TGAGACAGTATGCCGACATC
Al termine della PCR si effettua una corsa su gel di agarosio al 2% con 10 μl di bromuro di etidio. In questo modo possiamo verificare, nelle cellule utilizzate come controllo, la presenza del gene
Per 40 cicli totali
di interesse, e, nel caso del trattamento con siRNA, la diminuzione rispetto al controllo. La valutazione delle bande nell’analisi dell’RNA viene fatta prendendo sempre come termine di paragone tra i due campioni l’espressione della β-actina, gene espresso in modo ubiquitario (cosiddetto gene housekeeping).
2.4. VALUTAZIONE DELLA TRASLOCAZIONE DELLE ERK FOSFORILATE DAL
CITOPLASMA AL NUCLEO
Per valutare i livelli di ERK fosforilate nel nucleo e nel citoplasma delle cellule 1321N1 hGPR17 dopo stimolo con gli agonisti del GPR17, le cellule sono state trattate con solo mezzo (controllo) o UDP-glucosio 100 μM o LTD4 100 nM a tempi diversi (2.5-5-30-60 minuti). Al termine dei
trattamenti, si aspira il mezzo e si lavano le cellule con 5 mL di PBS. Le cellule vengono quindi staccate dalla piastra meccanicamente con uno scraper, raccolte in falcoon e centrifugate a 1200g per 5 minuti. Si aspira il sovranatante, che corrisponderà alla frazione citosolica, e si procede all’estrazione nucleare, che prevede i seguenti passaggi:
1. Sospendere i pellet ottenuti in tampone ipotonico freddo (HEPES 20 mM, NaF 5 mM, Na2MoO4 10 μM ed EDTA 0.1 mM, pH 7.5);
2. Incubare 15 minuti in ghiaccio, quindi aggiungere Nonidet 0.5%; 3. Vortexare per 10 secondi e centrifugare per 30 secondi a 13000g a 4°C;
4. Raccogliere in eppendorf il sovranatante (che costituisce la frazione citosolica), in modo da separarlo dal pellet (che costituisce la frazione nucleare);
5. Sospendere ognuna delle frazioni in 20 μl di RIPA contenente inibitori delle proteasi e mettere in agitazione per 30 minuti a una temperatura di 4°C;
6. Determinare la concentrazione proteica con il saggio proteico di Folin precedentemente riportato;
A questo punto le proteine presenti nei campioni possono essere separate mediante elettroforesi e analizzate attraverso la tecnica del Western Blot precedentemente illustrata. Per la rilevazione delle ERK, si utilizza un anticorpo primario diretto contro le p-ERK.
2.5. VALUTAZIONE DELLA FOSFORILAZIONE DELLE CREB MEDIATA DA
AGONISTI DEL GPR17
Per valutare i livelli di fosforilazione delle CREB in seguito a stimolazione del recettore GPR17, abbiamo utilizzato un saggio ELISA di tipo indiretto, secondo il protocollo del Kit Phospho-CREB
Activation Assay (Active Motif).
L’estrazione nucleare viene effettuata su cellule a confluenza in piastre da 75 cm2
(approssimativamente 0.15 μg di proteine nucleari per 9x106 cellule). Le fasi sono le seguenti:
1. Seminare le cellule 1321N1 hGPR17 e trattare con UDP-glucosio o LTD4 per tempi diversi;
2. Lavare le cellule con 10 μl di PBS/PIB freddo (0.5 mL PIB in 10 mL PBS 1X – PIB, Phosphate
Inibitors Buffer è costituito da NaF 125 mM, β-glicerolfosfato 250 mM, p-nitrofenilfosfato
250 mM, NaVO3 25 mM, H2O q.b. a 10 mL);
3. Aggiungere 10 μl di PBS/PIB e scrapare le cellule;
4. Trasferire le cellule in eppendorf e centrifugare a 300g per 5 minuti a 4°C;
5. Sospendere il pellet in 1 μl di HB Buffer freddo (HEPSE 20 mM, NaF 5 mM, Na2MoO4 10 μM
ed EDTA 0.1 mM, pH 7.5);
6. Trasferire le cellule in eppendorf da 1.5 mL e incubare 15 minuti in ghiaccio; 3. Aggiungere Nonidet 0.5%;
7. Vortexare per 10 secondi e centrifugare 30 secondi a 4°C in microcentrifuga;
8. Rimuovere il sovranatante che costituisce la frazione citoplasmatica (che può essere conservata a -80°C);
9. Sospendere la frazione nucleare in 50 μl di Complete Lysis Buffer (Lysis Buffer AM1 fornito dal kit, con aggiunta di 1 μl di DTT 1 M fornito dal kit e 10 μl di cocktail di inibitori delle
proteasi per 1 mL di Lysis Buffer AM1);
10. Incubare in ghiaccio sul basculante per 30 minuti;
11. Centrifugare 10 minuti a 14000g e 4°C: il sovranatante ottenuto è l’estratto nucleare (che può essere conservato a -80°C);
12. Determinare la concentrazione proteica con il saggio proteico di Folin precedentemente riportato.
La procedura di analisi della fosforilazione delle CREB con il saggio ELISA di tipo indiretto è costituita dalle seguenti fasi:
1. Aggiungere 30 μl di Complete Binding Buffer (1 μl DTT 1 M, 10 μl Herring Sperm DNA 1 μg/μl per μl di Binding Buffer AM1) ad ogni pozzetto;
2. Aggiungere: nei bianchi 20 μl di Complete Lysis Buffer; nei campioni 2-20 μg di estratto nucleare dei campioni diluito in 20 μl di Complete Lysis Buffer;
3. Sigillare con il coperchio adesivo fornito dal kit e incubare per 3 ore a temperatura ambiente sotto leggera agitazione;
4. Fare tre lavaggi con 200 μl di Wash Buffer 1X, tamponando ad ogni lavaggio dopo aver svuotato i pozzetti;
5. Aggiungere 100 μl di Anticorpo Phospho-CREB (Ser133 diluito 1:500 con Antibody Binding
Buffer);
6. Sigillare con il coperchio adesivo e incubare per 1 ora a temperatura ambiente senza agitazione;
7. Fare tre lavaggi con 200 μl di Wash Buffer 1X;
8. Aggiungere 100 μl di Anticorpo secondario-HRP (diluito 1:1000 con Antibody Binding Buffer); 9. Sigillare con il coperchio adesivo e incubare per 1 ora a temperatura ambiente senza
10. Aggiungere la Developing Solution fornita dal kit (fotosensibile) a temperatura ambiente; 11. Fare quattro lavaggi con 200 μl di Wash Buffer 1X;
12. Aggiungere 100 μl di Developing Solution;
13. Incubare da 5 a 15 minuti a temperatura ambiente al riparo dalla luce;
10. Aggiungere 100 μl di Stop Solution in ogni pozzetto allo stesso momento (in presenza di acido il colore vira dal blu al giallo);
11. Leggere l’assorbanza dei campioni allo spettrofotometro a 450 nm entro 5 minuti.
2.6. ANALISI DEI DATI
Per l’analisi dei dati e per le presentazioni grafiche è stato utilizzato il programma Graph-Pad
Prism 5 (Graph-Pad). La densità ottica delle bande immunoreattive ottenute dalle analisi Western Blot è stata misurata impiegando il programma Image J.
Tutti i dati rappresentano la media ± le deviazioni standard di tre distinti esperimenti. Per verificare una significativa divergenza dei dati ottenuti, in cellule di controllo e in cellule trattate, abbiamo utilizzato il test statistico Student t-test. Un valore di P<0.05 è stato considerato significativo.