CHAPTER 2: Evaluation of new GPR17 ligands
GPR17 is pathologically activated during acute CNS injury, thus contributing to early necrotic death inside the lesion, but also participates in the subsequent remodeling and repair of the lesioned area occurring in the days and weeks after injury. Moreover, GPR17 has emerged as a new timer in oligodendrogenesis. A modulation of GPR17 expression in OPCs in vitro depending upon the presence of GFs and highlight its major role in oligodendrocyte maturation and as danger signal in the presence of high extracellular ATP concentrations has also been demonstrated (Ceruti et al., 2010). Thus, GPR17 ligands could represent a new class of neuroprotective and neuroreparative agents in several types of human neurodegenerative diseases, such as stroke, trauma, and multiple sclerosis.
In this chapter, the characterization of new GPR17 ligands is reported. In the first part of this charter (Chapter 2.1), the activity of new ligands, obtained through structural modification of known agonists or antagonists is reported. The potential drug candidates toward GPR17 have been screened by the application of frontal affinity chromatography-mass spectrometry, along with molecular modeling studies. The aim of this work has been to validate the chromatographic results have been with a reference functional assay ([35S]GTPγS binding assay). Moreover, the receptor nucleotide-binding site was studied by setting up a column where a mutated GPR17 receptor (Arg255Ile) has been immobilized. The behavior of the tested nucleotide derivatives together with in silico studies have been used to gain insights into the structure requirement of GPR17 ligands.
In the second part (Chapter 2.2) I reported the characterization of new compounds with chemical structure different from GPR17 classic ligands, obtained through molecular modeling approach based on multiple templates leading to a "chimeric" GPCR structure. A high-throughput virtual screening exploration of GPR17 binding site with more than 130,000 lead-like compounds, allowed to successfully identify five chemically-diverse compounds. The aim of this project has been the functional and pharmacological validation of the top-scoring chemical structures, to verify if these molecules can act on GPR17.
Experimental section
2.1 Frontal affinity chromatography-mass spectrometry
useful for characterization of new ligands for GPR17
Introduction
Frontal affinity chromatography is a biophysical method involving the continuous infusion of analyte through a column and, in its conventional approach, can be used to measure binding constants of the analyte for the immobilized biomolecule with accuracy and precision. The feasibility of this approach has already been demonstrated for some GPCRs with the development of a series of immobilized membrane-based liquid chromatographic stationary phases obtained from various cell lines and tissues. These phases have been based on the immobilized artificial membrane (IAM) stationary phase developed by Pidgeon and coworkers (Pidgeon et al., 2003) and include membranes containing opioid GPCRs, the β2-adrenergic receptor, and the P2Y1R (Beigi et al., 2003; 2004; Moaddel et al., 2007). The IAM liquid
chromatographic stationary phase is composed of silica particles to which a monolayer of phospholipid analogues, with functional head groups, has been covalently coupled. The specific feature of IAM beads is their resemblance to a hydrophobic environment that allows immobilization of membrane proteins and receptors.
Massolini and coworkers (Temporini et al., 2008) recently have show the development and characterization of a GPR17-based liquid chromatographic stationary phase containing immobilized membranes. The optimized stationary phase has been inserted in standard chromatographic equipment coupled to a mass spectrometer and used in FAC studies for determining the dissociation constants of three ligands that previously have been shown to interact with this receptor. The results indicate that the immobilized receptor retains its ability to bind these ligands, as demonstrated by the consistency of the calculated Kd values with previously reported data where the affinity of these same compounds had been estimated in a completely different assay, the [35S]GTPγS binding assay (Ciana et al., 2006) suggesting that the application of GPR17(+)–IAM to ranking affinity studies may represent a suitable, rapid, and convenient method for the selection of new potential candidates. Subsequently, the
applied to the screening of a small library of compounds toward GPR17 immobilized on a chromatographic support. In addition, the simplicity of the method has allowed to explore the binding mode of the nucleotide site of the receptor by extending this approach to a column where an artificial GPR17 receptor bearing a mutation in a key amino acid for ligand recognition has been immobilized.
The compound library (library A) for GPR17 was selected taking into account the chemical structures of three known receptor ligands with different potencies: the antagonists 1 (cangrelor; IC50=0.7 nM) and 2 (MRS 2179; IC50 = 508 nM) and the
agonist 3 (UDP; EC50=1.14 µM) (see also Chapter 1.3). The synthesis of the known
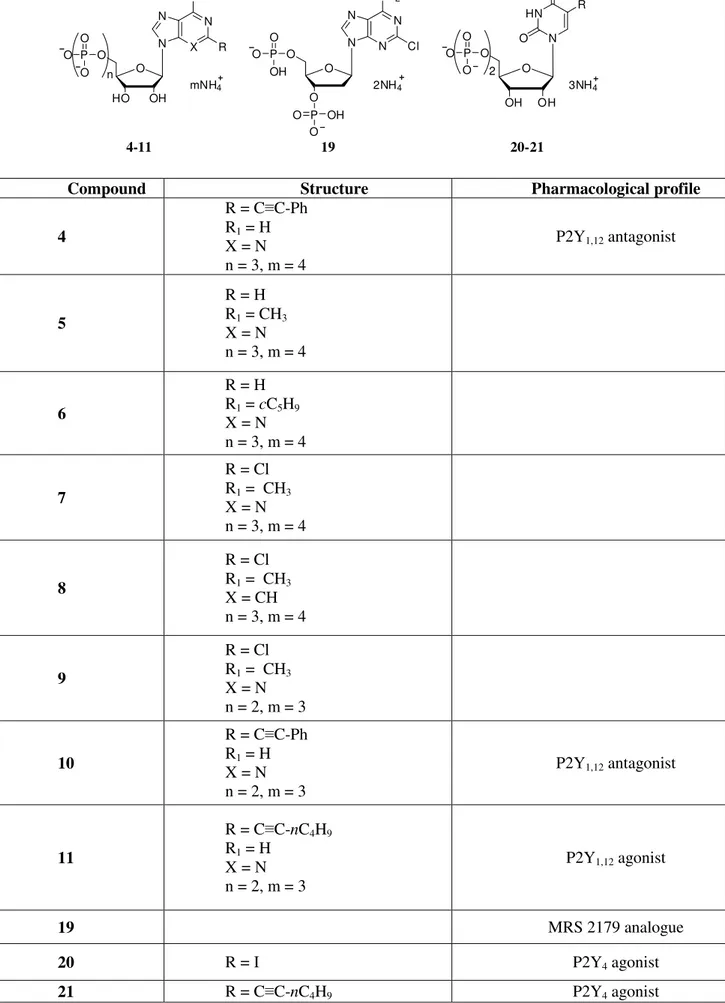
tested compounds was carried out from Department of Chemical Sciences, University of Camerino, Italy. Starting from the cangrelor structure and with the aim of investigating the structural requirements for ligand recognition at the nucleotide receptor active site, a series of ATP derivatives 4-7, substituted at the 2- and/or N6-position, were selected. Furthermore, in order to evaluate the importance of the 3-nitrogen purine ring and the grade of nucleotide phosphorylation, the 2, N6-disubstituted ATP 3-deaza analogue 8 and some diphosphate nucleotides 9-11 were evaluated. Furthermore, compound 12, a bisphosphate derivative analogue to MRS 2179, and the two 5-substituted UDP analogues 13 and 14 were selected. New nucleotides 7, 8, and 14 were prepared by phosphorylation of the corresponding nucleosides. Nucleosides were first phosphorylated to the monophosphates, and from these the corresponding di- and triphosphates were prepared. Chemical structures of the newly synthesized compounds are reported in Table 1.
Table 1. Chemical structures of the newly synthesized compounds selected for FAC-MS studies. N N N N NH2 O O O Cl 19 N X N N NH O OH HO R O P O O O n mNH4 O O H HN N O O OH R O 4-11 20-21 P O O O 2 3NH4 P O O OH P O O OH 2NH4 R1
Compound Structure Pharmacological profile
4 R = C≡C-Ph R1 = H X = N n = 3, m = 4 P2Y1,12 antagonist 5 R = H R1 = CH3 X = N n = 3, m = 4 6 R = H R1 = cC5H9 X = N n = 3, m = 4 7 R = Cl R1 = CH3 X = N n = 3, m = 4 8 R = Cl R1 = CH3 X = CH n = 3, m = 4 9 R = Cl R1 = CH3 X = N n = 2, m = 3 10 R = C≡C-Ph R1 = H X = N n = 2, m = 3 P2Y1,12 antagonist 11 R = C≡C-nC4H9 R1 = H X = N n = 2, m = 3 P2Y1,12 agonist 19 MRS 2179 analogue 20 R = I P2Y4 agonist
To explore the ability of FAC-MS to rank mixtures containing multiple ligands, a column (column GPR17-IAM) was prepared by immobilizing crude membranes of 1321N1 cells expressing wild type GPR17, following the acquisitions from previous studies on the optimization of the GPCR stationary phase where the nonspecific interactions were minimized by analyzing the retention of the three reference compounds on a blank column only containing IAM and an additional column containing membranes from cells transfected with an empty vector. Because our previous studies demonstrated that nonspecific interactions contribute to the total retention time but do not interfere with the specific displacement study, we have reasoned that it was no longer necessary to run a blank column in parallel for every single ranking experiment.
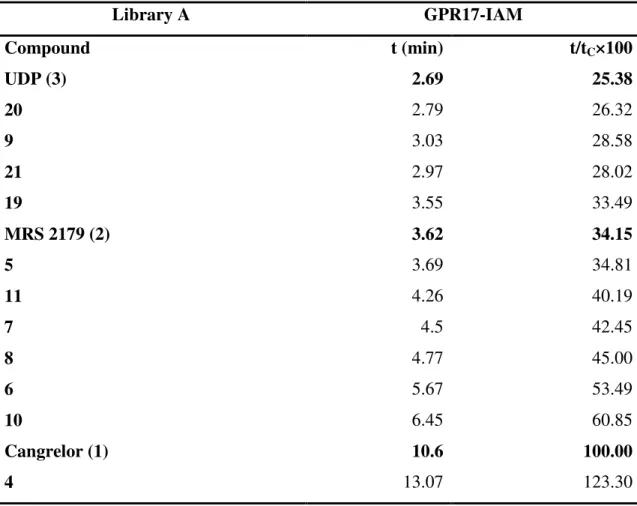
As expected, the elution order of the three reference compounds in the two experiments was consistent with previous results (Temporini et al., 2008) and in agreement with their potency at this receptor (cangrelor>MRS 2179>UDP) (Table 2). Moreover, a very good correspondence between the relative breakthrough % time of the less potent reference compounds (MRS 2179 and UDP) with respect to cangrelor was obtained.
The binding order of the phosphorylated compounds was consistent with the degree of phosphorylation; hence, in library A the binding order was 3P>2P (compare 4 and 7 with 10 and 9, respectively). The large breakthrough volumes for compounds 4, 10, and 21 indicate that they can be identified as high binding candidates. On the other hand, compounds 5-8 and 11 seem to be medium-high binding ligands, since they were eluted. with breakthrough times between those of cangrelor and MRS 2179. Compound 12 elutes with a breakthrough time close that of MRS 2179. This finding is in agreement with the structure of the two compounds that are very similar bisphosphate analogues. The same consideration can be drawn for the 5-substituted UDP derivatives 13 and 14, which elute close to UDP. These compounds can thus be classified as weak binders toward GPR17, like UDP itself. Interestingly, cangrelor and MRS 2179 are antagonists at GPR17 while UDP acts as an agonist;14 thus, as already demonstrated for many other receptors, also for GPR17, antagonists display higher binding affinity with respect to agonists.
Table 2. Frontal Affinity Chromatography-Mass Spectrometry (FAC-MS) results for Library A screened with immobilized GPR17.
Library A GPR17-IAM Compound t (min) t/tC×100 UDP (3) 2.69 25.38 20 2.79 26.32 9 3.03 28.58 21 2.97 28.02 19 3.55 33.49 MRS 2179 (2) 3.62 34.15 5 3.69 34.81 11 4.26 40.19 7 4.5 42.45 8 4.77 45.00 6 5.67 53.49 10 6.45 60.85 Cangrelor (1) 10.6 100.00 4 13.07 123.30 a
t is the breakthrough time of the ligand with the immobilized GPR17 b
tC represents the breakthrough time of the indicator cangrelor.
As reported in Chapter 1, a 3D molecular model of GPR17 embedded in a solvated phospholipid bilayer and refined by molecular dynamics simulations has been created (Parravicini et al., 2008; 2010). Molecular dynamics simulations suggest that the GPR17 nucleotide binding pocket is similar to that described for the other P2Y receptors, although only one of the three basic residues that have been typically involved in ligand recognition is conserved (Arg255).
On this basis, to assess the actual role of this residue in receptor binding, starting from the previously published model of GPR17, this basic amino acid has been mutated to isoleucine. The mutated receptor in Arg255 has been generated and expressed in 1321N1 cells. To challenge the reliability of the FAC-MS analysis, a column from these cells has been prepared (mutated GPR17-IAM), in parallel with a column loaded with membranes from cells expressing the WT receptor (GPR17-IAM), containing the same number of cells (19.5 millions)/ column.
The same trend has been observed with the exception of the structurally similar compounds 12 and MRS 2179, whose breakthrough times are reversed in mutated GPR17 column (Table 3).
Table. 3. FAC-MS data obtained on wild-type GPR17-IAM and mutated GPR17-IAM.
Compound Wild type GPR17
ta (min) Mutated GPR17 ta (min) UDP 2.69 2.15 20 2.79 2.51 19 3.55 MRS 2179 3.62 3.47 5 3.69 7 4.5 8 4.77 6 5.67 Cangrelor 10.6 13.16 4 13.07 16.07
Columns: wild-type GPR17-IAM (19.5 million cells), mutated GPR17-IAM (19.5 million cells). a t is the breakthrough time of the ligand with the immobilized GPR17 b versus
UDP-glucose 10 µM
The strength of a FAC-MS method is the ability to rank multiple ligands simultaneously, offering the possibility to discriminate between low, medium, and high affinity candidates; however, it identifies compounds simply on the basis of their binding affinities, irrespective of their ability to activate the target. On this basis, the aim of this work has been to verify if the newly synthesized compounds could bind and activate GPR17 and to determine their activity and pharmacological profile towards the human receptor. Moreover, the same experiments have been performed on mutated GPR17, in order to assess the importance of the basic residue Arg255 in GPR17 nucleotide binding pocket.
Methods
Cell culture and transfection. Human 1321N1 cells were cultured as described in Ciana et al., 2006. For [35S]GTPγS, 106 cells were seeded on 75cm2 flasks and transfected, by the calcium phosphate precipitation method, with both the pcDNA3.1 vector containing the construct encoding for the human GPR17 receptor and the empty pcDNA3.1 vector as a control (Ciana et al., 2006).
[35S]GTPγγγγS binding assay. Control and transfected cells were homogenized in 5 mM Tris–HCl and 2 mM EDTA (pH 7.4) and centrifuged at 48 000 g for 15 min at 4 °C. The resulting pellets (plasma membranes) were washed in 50 mM Tris–HCl and 10 mM MgCl2 (pH 7.4) and stored at -80 °C until used. Nucleotide-stimulated [35S]GTPγS
binding in membranes of cells expressing the human receptor was performed as described previously (Fumagalli et al., 2004; Ciana et al., 2006). The pharmacological profile of the new ligands (agonists or antagonists) toward the GPR17 receptor was evaluated by assessing the effect of different ligand concentrations to modulate GPR17 receptor-G protein coupling. In parallel, in order to investigate if compound-mediated effects were really ascribed to the interaction with GPR17 binding site, GTPγS binding was also performed in 1321N1 cells transfected with the empty vector.
Statistical analysis. For the analysis and graphic presentation of [35S]GTPγS binding data, a nonlinear multipurpose curve fitting computer program (Graph-Pad Prism) was used. All data are presented as the mean ± SEM of three different experiments. Data were tested for statistical significance with the paired Student’s t test or by analysis of variance (one-way ANOVA), as appropriate. When significant differences were observed, the Newman-Keuls multiple comparison test (one-way ANOVA) was performed. A value of P < 0.05 was considered significant.
Results and Discussion
Functional Assay on wild type GPR17. In order to evaluate the functional activity of the new compounds on GPR17, the derivatives were tested in a well established GPCRassay, the [35S]GTPγS binding, based on the ability of agonists to increase the binding of radioactive GTP to the activated GPCR. Antagonist activity has been assessed by the ability to counteract the increase of [35S]GTPγS binding induced by the agonist UDP-glucose. The GPR17 antagonists cangrelor, MRS2179, and the agonist UDP were also assayed in parallel as reference compounds. The [35S]GTPγS binding results in comparison with FAC-MS data are reported in Table 4.
The obtained potency values of the tested ligands versus wild-type GPR17 closely reflect the elution order of the analytes in the GPR17 wild-type column. A strong correlation was found by comparing these results, expressed as halfmaximal response concentrations (EC50) or half-maximal inhibition (IC50) values for agonists and
antagonists, respectively, with the data obtained with the GPR17 column by FAC-MS, thus confirming the validity of our approach in the ranking of new potential ligands for GPR17. The ligands 13, 6, 8, 7, and 4 were able to increase [35S]GTPγS binding, with potency values in the micromolar and subnanomolar range, showing an agonist profile (Figure 1, panel a). In particular, the ATP analogue 4, bearing a lipophilic steric hindered substituent in the 2-position, was a very potent agonist of GPR17 with an EC50
of 36 pM, in agreement with the fact that this is the compound with the longer breakthrough time from the GPR17-column. Also, a lipophilic substituent in the N6-position of ATP favored, although to a less extent, the interaction with the GPR17 receptor. In fact, comparison of potency of 6 (EC50=1.4 nM), bearing a N6-cyclopentyl
group, and 7 (EC50=11 nM), bearing a smaller methyl group, demonstrated clearly the
contribution of the more lipophilic substituent. With regard to the purine ring, we can confirm that the presence of a nitrogen in the 3-position is not essential (see above), since its isosteric replacement by a CH induced even a 7-fold increase of activity as shown by comparing 7 (EC50 = 11 nM) and 8 (EC50=1.7 nM).
Compound 12 did not induce any significant increase in [35S]GTPγS binding; conversely, it was able to counteract [35S]GTPγS binding stimulation induced by the purinergic agonist UDP-glucose (used at 10 µM), with an antagonist profile (Figure 1, panel b) and an affinity constant (IC50) in the nanomolar range, comparable to that
UDP derivative 13 showed an activity comparable to that of UDP itself (EC50=0.945
versus 1.140 µM, respectively).
Fig. 1 [35S]GTPγS experiments on wild-type GPR17: (a) dose-response curves of some compounds of library A in 1321N1 cells expressing wild-type GPR17; (b) antagonistic effect of 12 on UDP-glucose stimulation of wild-type GPR17 in the [35S]GTPγS binding, where each point is the mean (SEM of three independent experiments run in triplicate.
Structure-Affinity Relationships (SARs). The obtained results have been used to draw preliminary binding SAR speculations at the hit discovery stage that would reveal ways of designing potent and selective GPR17 ligands. The data suggest that lipophilic substituents on the C-2 and on the N6 amino group of purine ring can lead to a tight interaction with the hydrophobic residues in the binding pocket. In fact, compounds that strongly bind to the receptor bear lipophilic groups in the 2-position like the phenylalkynyl chain or halogens (see 4, 10, and 21) and/or an alkyl/cycloalkyl substituent on the N6 amino group (see 6, 7,and 8). The replacement of the phenyl group with a n-butyl moiety brings about a reduction of binding affinity to GPR17 (compare 10 and 11). On the other hand, it could be that the presence of a nitrogen in 3-position is not essential, since its isosteric replacement with a CH does not significantly change the breakthrough time (compare 7 and 8). It is clear that the nature and the position of substituents on the purine ring modulate the receptor binding affinity, the presence of rigid lipophilic groups in 2-position being the most favourable for the interaction of these compounds with the receptor.
Functional assay on mutated GPR17. To further confirm the data reported above, some analytes from library A (i.e., 4 and 13, demonstrating the highest and the lowest affinity toward wild-type GPR17, respectively) together with the three standard compounds (UDP, MRS 2179, and cangrelor) were also tested in parallel on wild-type and mutated GPR17 in the [35S]GTPγS functional binding assay. All the tested compounds maintained the same pharmacological profile (agonist/antagonist) at both the wild-type (Fig. 1a and 1b) and the mutated GPR17 receptor (Fig. 2a and 2b). No significant changes in receptor affinity (i.e., EC50) were demonstrated for all tested
agonists, but a trend toward a reduced affinity was observed for UDP and compound 13, whereas a trend toward an increased affinity was evident for compound 4 (Table 4). In contrast, the antagonists cangrelor and MRS 2179 showed a significant increase in affinity toward the mutated vs the wild type receptor (P<0.05; see Table 4).
Data from [35S]GTPγS functional experiments paralleled those obtained in affinity chromatography studies. In fact, a decrease in the breakthrough time was observed for those compounds showing a trend toward a reduced affinity for the mutated receptor (e.g., UDP and 13; Table 5), whereas a higher breakthrough time was observed for cangrelor and compound 4, showing an increased affinity toward the mutated receptor (Table 4). Among all the tested compounds, despite a significant
decrease in the IC50 value toward the mutated receptor, no significant shift in the
retention time was observed for MRS 2179 (Table 4).
1321N1 hGPR17 mutated
-14.5 -12.0 -9.5 -7.0 -4.5 90 100 110 120 130 140 150 160 3 4 20a
log[M] G T Pγγγγ S b in d in g (% o v e r b a s a l v a lu e )1321N1 hGPR17 mutated
-13.5 -12.0 -10.5 -9.0 -7.5 -6.0 -4.5 90 100 110 120 130 140 1 2b
log[M] G T Pγγγγ S b in d in g (% o v e r b a s a l v a lu e )Fig. 2 [35S]GTPγS experiments on mutated GPR17: (a) dose-response curves of some compounds of library A in 1321N1 cells expressing mutated GPR17; (b) antagonistic effect of 12 on UDP-glucose stimulation of mutated GPR17 in the [35S]GTPγS binding, where each point is the mean (SEM of three independent experiments run in triplicate.
Table 4. FAC-MS data in comparison with [35S]GTPγS binding results obtained on wild-type GPR17-IAM and mutated GPR17-IAM.
GPR17-wild type Mutated-GPR17
Cpd ta (min) EC50 IC50b ta (min) EC50 IC50b UDP 2.69 1.14 ± 0.2 µM 2.15 3.0 ± 0.3 µM 20 2.79 945 ± 48 nM 2.51 1.87 ± 0.20µM 19 3.55 582 ± 57 nM MRS2179 3.62 508 ± 29 nM 3.47 227 ± 22 nM 5 3.69 112 ± 7 nM 7 4.5 11 ± 1 nM 8 4.77 1.7 ± 0.1 nM 6 5.67 1.4 ± 0.1 nM Cangrelor 10.6 0.7 ± 0.02 nM 13.16 0.15 ± 0.01 nM 4 13.07 36 ± 3 pM 16.07 25.4 ± 1.0 pM
Columns: GPR17-IAM (19.5 million cells), mutated GPR17-IAM (19.5 million cells) a
t is the breakthrough time of the ligand with the immobilized GPR17 b versus
UDP-glucose 10 µM
Comparison between wild type and mutated GPR17. The trend of reduced affinity showed by UDP towards the mutated GPR17 has been confirmed from computational assay. Simulations of the forced unbinding of UDP from the wild type (WT) and the mutant (R255I) receptor models have been compared: the detected significant difference in energy peak intensities suggests that the mutation actually affects the binding of UDP. Moreover, to allow a theoretical interpretation of results and to get more information regarding the molecular interactions of the most interesting
ligands with GPR17, in silico docking studies using a virtual library of the synthetic nucleotide derivative ligands tested in vitro.
Two of the most representative ligand candidates (MRS 2179 and 4, a bisphosphate and a triphosphate compound, respectively) were chosen. Docking results showed that, while Arg255 is involved in GPR17 binding to MRS 2179, another residue (Arg87) seems to be primarily involved in binding of the receptor to the phosphate chain of 4. These different docking poses could account for the different effect of theArg255 mutation on the behavior of bisphosphate and triphosphate ligands on the GPR17 column. Specifically, it can be hypothesized that, different from bisphosphate derivatives like MRS 2179, for triphosphate derivatives such as 4, mutation of Arg255 is not enough to reduce the compound affinity in the GTPγS binding assay, as the longer phosphate chain can interact with other arginine residues (e.g., Arg87) through electrostatic interactions. Moreover, it could be hypothesized that triphosphate 4, bearing a lipophilic phenylethynyl chain, can be better accommodated into the more lipophilic pocket of mutated GPR17, thus leading to an increased breakthrough time. This effect is less evident for diphosphate derivatives where the loss of electrostatic interaction, due to the mutation, is preponderant with respect to the increased lipophilicity.
Unlike other affinity-based screening strategies, which are not specific enough for chemical identification and/or suffer from low throughput, FAC-MS methodology is highly specific and can be used for the discovery of classes of small molecule ligands (agonists and antagonists) in a single fast screening campaign. The strength of the FAC-MS approach has been validated using a reference functional assay ([35S]GTPγS assay), and the data allowed the drawing of preliminary structure-activity relationships.
However, the design and synthesis of selective GPR17 agonist/ antagonist ligands would greatly benefit from the knowledge of receptor 3D structure and from the definition of its ligand binding mode.
Anyway, the obtained data validate the FAC-MS methodology and address the design and the synthesis of new refined compounds acting as new potent and selective ligands.
2.2 An integrated pipeline for the identification of novel GPR17 ligands
Introduction
GPR17 was selected to apply an up-to-date multidisciplinary pipeline sequentially consisting of: (i) the in silico “chimeric” modeling of this GPCR based on the use of multiple templates for the building procedure of receptor’s loops; (ii) the in silico screening of virtual chemical libraries containing a large variety of molecules, including very diverse chemical structures that are totally unrelated to those already known to interact with either the P2YR or CysLTRs.
Each specific part of any target GPCR was modeled on the structure of the closest available template. The resulting “chimeric” receptor was then used to probe hundreds of thousands of molecules through computational methods; the most interesting compounds were then synthesized and validated through established experimental assays.
In the past, in silico models have been obtained for several pharmacological targets, including GPCRs, through comparative modeling based on the structure of bRh. These models have been successfully exploited for the rational design of new drugs (Katritch et al., 2010). In the case of GPR17, the availability of several recently crystallized proteins of the same receptor family (the human adenosine A2A receptor
(Jaakola et al., 2008), the human β2-adrenergic receptor (Rosenbaum et al., 2007;
Hanson et al., 2008), the turkey β1-adrenergic receptor (Warne et al., 2008)) allowed to
select different templates for the discrete modeling of distinct parts of the receptor. The docking procedure was carried out in two steps, screening and refinement. The 5 ligands whose poses scored at the lowest energy values were selected for the in vitro experiments. The 5 top scoring compounds belong to different chemical classes, suggesting that the application of this method allows to identify putative lead compounds that are not necessarily related to the already known ligands.
Although these molecules have very different chemical structures from each other, they are nevertheless united by their ability to interact with a common and well-defined region of amino acids belonging to the binding site of the GPR17 receptor, as shown by data of Protein Ligand Interaction Fingerprints (PLIF) Annex 1. These ligands have, therefore, structural and conformational features that allow them to bind the receptor, regardless of their structure and / or class of chemistry. This is a particularly important new feature of our approach, since none of the currently available
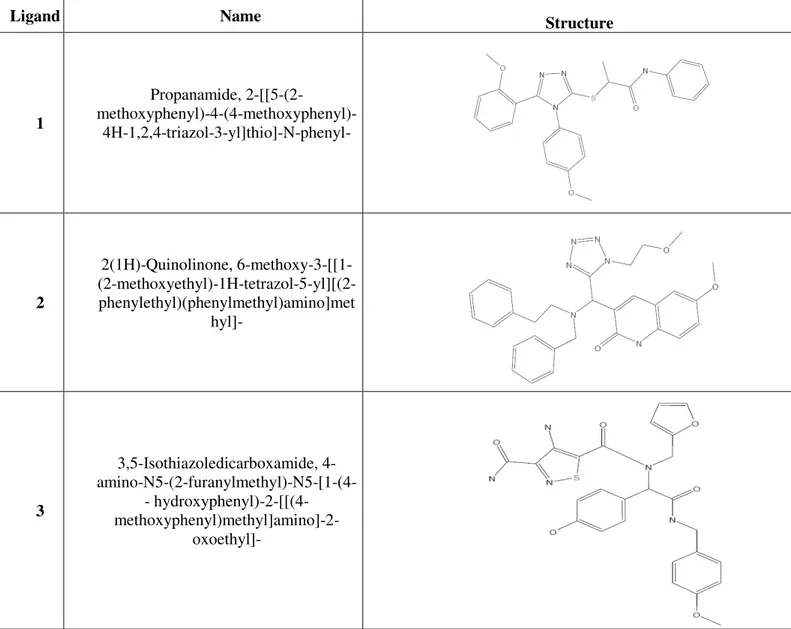
GPR17 ligands, which are chemically related to ligands for already known P2Y and CysLTRs, is indeed specific for GPR17. The chemical structures and names of the five best scoring GPR17 ligands are reported in Table 5.
On this basis, the aim of this work has been to verify if the five candidate molecules, belonging to very different chemical classes, were able to modulate GPR17 activity.
Table 5. Structures of the five best scoring GPR17 ligands
Ligand Name Structure
1 Propanamide, 2-[[5-(2- methoxyphenyl)-4-(4-methoxyphenyl)-4H-1,2,4-triazol-3-yl]thio]-N-phenyl- 2 2(1H)-Quinolinone, 6-methoxy-3-[[1- (2-methoxyethyl)-1H-tetrazol-5-yl][(2-phenylethyl)(phenylmethyl)amino]met hyl]- 3 3,5-Isothiazoledicarboxamide, 4- amino-N5-(2-furanylmethyl)-N5-[1-(4-- hydroxyphenyl)amino-N5-(2-furanylmethyl)-N5-[1-(4--2amino-N5-(2-furanylmethyl)-N5-[1-(4--[[(4amino-N5-(2-furanylmethyl)-N5-[1-(4-- hydroxyphenyl)-2-[[(4- methoxyphenyl)methyl]amino]-2-oxoethyl]-
Ligand Name Structure 4 1H-Benzotriazole-1-acetamide, N-[1- (4-fluorophenyl)-2-oxo-2- [[(tetrahydro-2- furanyl)methyl]amino]ethyl]-N-(4-methoxyphenyl)- 5 1-Piperidinecarboxamide, 4-[3-[(2- chlorophenyl)methyl]-6,7-dihydro-7-oxo- 3H-1,2,3-triazolo[4,5-d]pyrimidin-5-yl]-N-[4-(trifluoromethyl)phenyl]-
Methods
See precedent section.
Results and Discussion
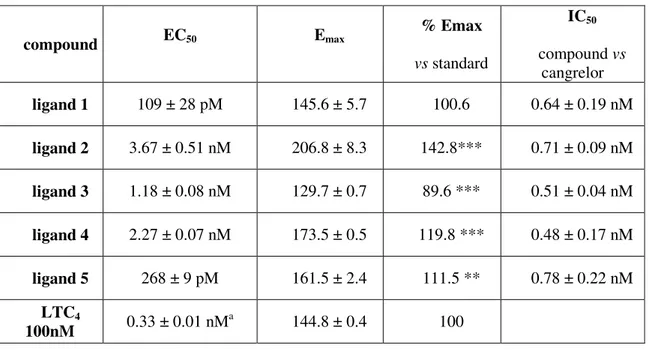
In order to verify the reliability of the in silico results and to evaluate the functional activity on GPR17 of the selected compounds, the 5 candidate molecules were tested in a well established GPCR assay, the [35S]GTPγS binding, based on the ability of agonists to increase the binding of radioactive GTP to the activated GPCR. Antagonist activity was instead assessed through the ability to counteract the increase of [35S]GTPγS binding induced by the model agonist LTC4. [35S]GTPγS binding results
are reported Table 6. All the 5 tested molecules showed a very high potency across a narrow range of concentrations: from sub-nanomolar to nanomolar. Differences in ranking between molecular docking and data from [35S]GTPγS binding assay might be due to different causes, the major being that computed pKi values could directly compare only with results from classical receptor binding experiments. In this respect, it has to be emphasized that the [35S]GTPγS assay utilized here not only verifies the
ability of the tested compounds to bind to GPR17, but also measures their intrinsic activity (α), classifying them as agonists, partial agonists, or antagonists. It is therefore a functional assay that cannot be directly compared with the in silico molecular modeling method.
All the tested compounds, except ligand 5, showed an efficacy comparable to, or even higher than, that of the reference compound LTC4. Only ligand 5 showed an
efficacy (89.6 %) significantly lower that that of the LTC4, suggesting that it could act
as a partial agonist. Figure 3 panel a shows the dose-response curves of the 5 tested compounds in the [35S]GTPγS assay performed on 1321N1 cells expressing GPR17. LTC4 was unable to induce any response in cell transfected only with the control vector
pcDNA3.1 (black line), confirming that the increase of [35S]GTPγS binding is mediated by the presence of GPR17.
The antagonistic effect of cangrelor was tested on GPR17 activation induced by Asinex compounds. All these compounds are antagonized by cangrelor, a P2Y12/13R,
and GPR17 antagonist, which is able to bind to the GPR17 uracil nucleotide-binding site (Ciana et al., 2006).
Table 6. Pharmacological parameters for the tested compounds
compound EC50 Emax % Emax vs standard IC50 compound vs cangrelor ligand 1 109 ± 28 pM 145.6 ± 5.7 100.6 0.64 ± 0.19 nM ligand 2 3.67 ± 0.51 nM 206.8 ± 8.3 142.8*** 0.71 ± 0.09 nM ligand 3 1.18 ± 0.08 nM 129.7 ± 0.7 89.6 *** 0.51 ± 0.04 nM ligand 4 2.27 ± 0.07 nM 173.5 ± 0.5 119.8 *** 0.48 ± 0.17 nM ligand 5 268 ± 9 pM 161.5 ± 2.4 111.5 ** 0.78 ± 0.22 nM LTC4 100nM 0.33 ± 0.01 nM a 144.8 ± 0.4 100 **p<0.01, *** p<0.001 vs LTC4 set to 100% a Ciana et al., 2006
Fig.3 [35S]GTPγS binding assays on GPR17. panel a: Concentration-response curves for the five best-scoring compounds selected through the in silico screening procedure. Membrane aliquots obtained from 1321N1 cells transfected with empty vector (control cells) or pcDNA3.1-hGPR17 were incubated with different Asinex compound concentrations. panel b: Effect of the P2YR antagonist cangrelor on agonist-stimulated [35S]GTPγS binding. Membranes from pcDNA3.1-hGPR17-transfected cells were preincubated for 10 min with graded cangrelor (0.01nM–1µM) concentrations and then stimulated with a selected concentration for each Asinex ligand (10 fold over the EC50 value). All data are expressed as percentages of basal [35S]GTPγS binding (set to 100%) and are the mean ± SEM of 3 different experiments, each performed in duplicate.
In order to evaluate if the selected ligands bind to the same site on GPR17, we built dose response curves for the most potent compound in presence of different concentrations of cangrelor (Fig. 4, panel a). The curves were shifted to the right (increased EC50), but no effect was observed on the efficacy. This result suggests that
cangrelor is a competitive antagonist for ligand 1, and that these two molecules compete for the same binding site on GPR17.
The same experiment was repeated with montelukast, an antagonist of the CysLT1R (Fig. 4, panel b). It has already been demonstrated that montelukasts can act
as inhibitors not only of CysLT1R, but also of GPR17 (Ciana et al., 2006). However, the
dose-response curves for ligand 1, obtained in the presence of montelukast, showed a reduction of the efficacy of ligand 1, suggesting a non-competitive antagonism, probably exerted through an orthosteric mechanism on a binding site different from that bound by uracil nucleotides. The evidence that purinergic and cysLT ligands do not share the same binding site on GPR17 had indeed already arisen from previous data (Ciana et al., 2006; Parravicini et al., 2008; 2010).
Fig. 4. Inhibition of agonist binding by purinergic or cysLT GPR17 antagonists in the [35S]GTPγS binding assay. panel a: Antagonistic activity of cangrelor on the stimulation of [35S]GTPγS binding induced by ligand 1. Membranes from GPR17-expressing 1321N1 cells were incubated with ligand 1 (0.01-10nM) in the absence or presence of graded antagonist concentrations as indicated. Binding of [35S]GTPγS to G proteins was then quantified. panel b: Antagonistic activity of montelukast on the stimulation of [35S]GTPγS binding induced by ligand 1. Membranes from GPR17-expressing 1321N1 cells were incubated with ligand 1 (0.01-10nM) in the absence or presence of graded antagonist concentrations as indicated. Binding of [35S]GTPγS to G proteins was then quantified. All data are expressed as percentages of basal [35S]GTPγS binding (set to 100%) and are the mean ± SEM of 3 different experiments, each performed in duplicate.
Conclusions
The screening of 130,000 lead-like and non-targeted compounds allowed us to select a set of putative GPR17 ligands belonging to highly diverse chemical classes. The in vitro evaluation of a subset of the 5 top-scoring compounds through a well-standardized functional assay confirmed the efficiency of the proposed screening pipeline (modeling + docking) for the identification of new GPR17-targeting scaffolds. These ligands are the first examples of molecules acting on GPR17 that have not been developed/used as CysLT1R or P2YR modulators. The molecular pharmacology
experiments suggested that the selected compounds can bind to the GPR17 uracil binding site and can be competitively antagonized by cangrelor. Inhibition of ligand 1 maximal effect (efficacy) induced by montelukast suggests a non-competitive antagonism, which likely involves a binding site different from the site of interaction of uracil and cangrelor. Among the 5 tested ligands, we identified 4 full agonists, with a better potency than the reference ligand (LTC4), and a partial agonist. Considering the
dual role of GPR17 in promoting both damage and repair depending on specific times after the injury (Lecca et al., 2008), the availability of a partial agonist could be very advantageous from a therapeutic point of view. In fact, during the acute phase of an ischemic event, when nucleotides and cysLTs are massively released at the inflammation site, the activity of GPR17 can be finely tuned by a partial agonist by blocking the detrimental effects mediated by excessive and dysregulated receptor activation. On the other hand, by keeping the receptor partially activated during the subsequent ischemic phases, when nucleotides and cysLTs levels decrease, a partial agonist would not repress the repair and trophic actions induced by these endogenous molecules. The positive results obtained through these molecular modeling and docking approaches suggest that this strategy could be extended to other GPCRs to better understand their molecular recognition mechanisms and identify new lead compounds.
![Fig. 1 [ 35 S]GTPγS experiments on wild-type GPR17: (a) dose-response curves of some compounds of library A in 1321N1 cells expressing wild-type GPR17; (b) antagonistic effect of 12 on UDP-glucose stimulation of wild-type GPR17 in the [ 35 S]GTP](https://thumb-eu.123doks.com/thumbv2/123dokorg/7535609.107553/10.892.231.823.171.944/gtpγs-experiments-response-compounds-library-expressing-antagonistic-stimulation.webp)
![Table 4. FAC-MS data in comparison with [ 35 S]GTPγS binding results obtained on wild-type GPR17-IAM and mutated GPR17-IAM](https://thumb-eu.123doks.com/thumbv2/123dokorg/7535609.107553/13.892.123.846.162.735/table-fac-comparison-gtpγs-binding-results-obtained-mutated.webp)
![Fig. 4. Inhibition of agonist binding by purinergic or cysLT GPR17 antagonists in the [ 35 S]GTPγS binding assay](https://thumb-eu.123doks.com/thumbv2/123dokorg/7535609.107553/21.892.222.709.109.723/inhibition-agonist-binding-purinergic-cyslt-antagonists-gtpγs-binding.webp)