CHAPTER 1
TSPO LIGANDS
3
1.1 INTRODUCTION
Over the last 40 years, benzodiazepines (Bz) have been recognized as valuable therapeutic molecules and their widespread use in the clinic is based upon potent anxiolytic, anticonvulsant, hypnotic, muscle-relaxant and sedative properties. The central benzodiazepine receptor (CBR) that is restricted to the central nervous system (CNS) mediates these actions. Besides CBR, an additional binding site for benzodiazepine Diazepam was identified in 1977 in peripheral tissues, and it was named “Peripheral-type benzodiazepine receptor (PBR)”.1
In 2006, Papadopoulos and co-workers2 renamed the PBR to Translocator Protein (18KDa) with the abbreviation “TSPO”. The renaming of this protein was thought to represent more accurately recent findings regarding its structure, subcellular roles and putative functions. Further review of the current nomenclature highlights the following key reasons and rationales that support this name change for the PBR.
(i) The term „benzodiazepine‟ is inaccurate because many ligands of other structures, such as cholesterol and protoporphyrin IX, also bind to the PBR. In addition, not all benzodiazepines bind to the PBR, so that including „benzodiazepine‟ in the nomenclature is potentially misleading and inaccurate. (ii) The term „peripheral-type‟ does not reflect the real tissue distribution of the PBR because it is also expressed in glial and ependymal cells, which are located in the CNS.
(iii) The PBR protein itself is not a receptor in the traditional sense. Cellular localization is usually, even thought not always, mitochondrial. Although some data indicate that extra mitochondrial localization of the PBR could result from mutations, there are no data indicating common functions of the nuclear and mitochondrial fractions.
(iv) The term „binding site‟ does not define function with adequate specificity; binding per se does not reflect the importance of the protein or receptor, and it is too general term.
(v) Although „receptor‟ functionality is appealing in the context of cell signalling, it might not be an optimal descriptor given the diverse nature of
4
candidate physiological and pharmacological ligands and the currently incomplete understanding of the potential role of the PBR protein in the transduction of cell signals.
There are three main Structure Activity Relationships (SAR) for the TSPO: (i) cholesterol binding and transport; (ii) protein import; and (iii) porphyrin binding and transport. Given all of these considerations, the expert panel reached a consensus to recommend renaming the PBR to TSPO.2
The TSPO is an evolutionarily well-conserved 18 kDa protein consisting of 169 amino-acids,3 which is mainly located at the contact sites between the outer and inner mitochondrial membranes,4 although it is also expressed at low levels in other subcellular compartments, such as plasma membranes and nuclear fraction of cells.2 In mitochondria, the three-dimensional structure of TSPO, which is highly hydrophobic and rich in tryptophan, is characterized by five α-helices spanning through the phospholipids bilayer of the mitochondrial membrane. Photolabelling studies indicated that this receptor is strictly associated in a trimeric complex with the 32 kDa voltage-dependent anion channel (VDAC) and the 30 kDa adenine nucleotide translocase (ANT), to constitute the mitochondrial permeability transition pore (MPTP). Figure 1
Figure 1. Translocator Protein.2
To date, four cytosolic TSPO interacting proteins have been identified, the function of two of them being unknown: PRAX-1, which interacts with the C-terminal end of TSPO and p10, a 10 kDa protein which was coimmunoprecipitated with the TSPO, but whose identity has not been unravelled.1 The two other TSPO partners are the steroidogenic acute regulatory
5 protein, StAR, which binds cholesterol and promotes mitochondrial cholesterol transfer5 and PKA-associated protein 7, PAP7, which regulates steroidogenesis as shown in antisense and overexpression studies.1,6
Varieties of endogenous molecules with different chemical structures that bind to TSPO have been identified. Among these, the main endogenous ligands include protoporphyrins (protoporphyrin IX, mesoporphyrin IX, deuteroporphyrin IX, hemin), phospholipase A2 (PLA2), diazepam binding inhibitor (DBI), and its biologically active derivatives.7 Cholesterol is also considered an endogenous nanomolar affinity ligand for TSPO.
TSPO is widely expressed throughout the body, with higher levels in steroid producing tissues, but also in other peripheral tissues including liver, heart, kidney, lung, immune system. In the CNS, TSPO is mainly located in glial and ependymal cells.2
TSPO is involved in a variety of biological processes including cholesterol transport, steroidogenesis, calcium homeostasis, lipid metabolism, mitochondrial oxidation, cell growth and differentiation, apoptosis induction, and regulation of immune functions.8,9
1.1.1 TSPO and Regulation of Steroidogenesis
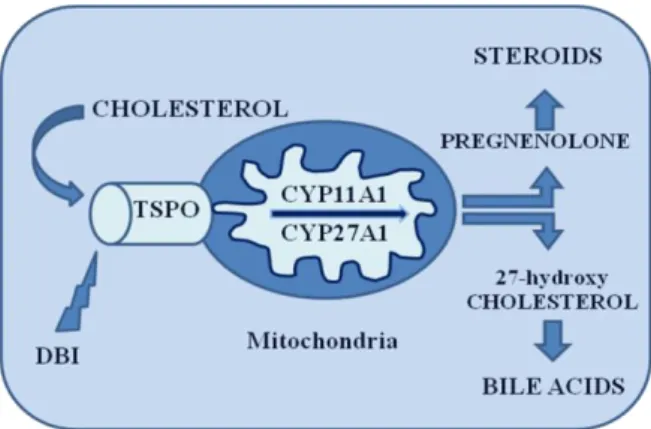
Biosynthesis of tissue specific steroids typically involves the conversion of cholesterol into pregnenolone, which occurs through cholesterol side chain cleavage by cytochrome P450 enzyme (P450scc) and auxiliary electron transferring proteins, localized on the inner mitochondrial membrane. Cholesterol transport from the outer to the inner mitochondrial membrane is the rate-determining step in steroid and bile acid syntheses.10,11 Widespread studies on the location and function of TSPO have found its role to be primarily involved cholesterol transport through mitochondrial membranes and thus steroid synthesis. Cholesterol transport from the outer to inner mitochondrial membrane is activated by specific ligand binding to TSPO, after which cholesterol undergoes metabolism and begins the steroidogenesis cascade.2 Cholesterol may interact with a receptor binding site on the c-terminus of the TSPO12, triggering its transport from outer to inner mitochondrial membranes.11,13 In the inner
6
mitochondrial membrane cholesterol is converted to pregnenolone by the C27 cholesterol side chain cleavage cytochrome P450 enzyme (P450scc), catalyzing a series of reactions involving the formation of the 22R-hydroxycholesterol and 20,22R-hydroxycholesterol intermediates followed by cleavage of the bond between C20 and C22. Pregnenolone then leaves the mitochondrion to undergo enzymatic transformation in the endoplasmic reticulum that will give rise to the final steroid products.14 Figure 2
Figure 2. Cholesterol metabolism in hepatic and steroidogenic cells. Cholesterol binds to TSPO
and its transported up on binding of endozepine also called DBI. In hepatic cells, cholesterol is hydroxylated by cytochrome P450 (CYP27A1) to give 27-hydroxycholesterol, whereas in steroidogenic cells, cholesterol is cleavage by another cytochrome P450 (CYP11A1) that cleave side chain to give pregnenolone.
1.1.2 TSPO and apoptosis
The apoptotic cascade leading to cell death has been well characterized and in this process the dissipation of the mitochondrial transmembrane potential caused by the opening of the MPTP represent a critical initiating event. The resulting increase volume in the mitochondrial matrix leads to the disruption of the outer membrane integrity and the release of intermembrane proteins from the mitochondria. These proteins include cytocrome c and the apoptosis inducing factor, AIF. The flavoprotein AIF translocates to the nucleus where it induces nuclear chromatin condensation and a large scale DNA fragmentation. In the cytosol, the cytochrome c interacts with Apaf-1 (apoptosis activating factor 1) and caspase 9, leading to the activation of caspase 9, which in turn cleaves pro-caspase 3 to yield active pro-caspase 3. This pro-caspase activates a range of enzymes critical for inducing the structural rearrangement of the nucleus, cytosckeleton and
7 plasma membrane that are characteristic of apoptosis.1 Apoptosis is defined as the transient opening of MPTP, as reclosure of the pore ensures this transient process does not result in necrosis and ATP levels are ultimately maintained inside the cell.11 TSPO is an endogenous modulator of this process but the exact mechanism has not yet been definitively established. TSPO regulation may act at different levels and some proposed mechanisms include the modulation of the MPTP or the direct interaction with pro- or anti-apoptotic molecules.1
In 2007 Li and co-workers15 showed that the specific TSPO ligand PK 1119516 induces mitochondrial release of cytochrome c and ultimately induced mitochondrial uncoupling. PK 11195 also facilitates the induction of apoptosis, reverses Bcl-2-mediated inhibition of apoptosis, and facilitates TNF-R-induced necrosis, producing a multicomponent effect on the induction of cell death. Outline, it is important to note that the proapoptotic effects of PK 11195 are significant only at concentrations 1000-fold higher that those required for specific binding to TSPO.11
1.1.3 TSPO and Immunomodulation
The presence of TSPO in a wide range of immunomodulatory cells such as microglia, blood monocytes, lymphocytes, and leukocytes implies its involvement in immune response. The mechanism through which this occurs is largely unknown. Macrophages express high levels of TSPO binding sites, and in mouse studies, TSPO ligands, specifically benzodiazepines, inhibit the capacity of macrophages to produce ROS and inflammatory cytokines such as IL-1, TNFR, and IL-6. Furthermore, TSPO is involved in the regulation of phagocyte oxidative metabolism, a process that is normally required for inducing effective elimination of foreign antigens. This immunosuppressive function of some TSPO ligands suggests an important role in host defense mechanisms and inflammatory response.
In the health CNS, TSPO is minimally expressed on microglial cells. Upon the injection of excitotoxic compounds, there occurs a dose-dependent increase in the level of TSPO, an up-regulation that is correlated closely with microglial activation. Inflammatory mechanisms initiated by microglia are implicated as part
8
of the primary and secondary mechanisms of inflammatory neurodegenerative diseases such as Alzheimer‟s disease (AD),17
whereby the activation of microglia initiates an inflammatory response that may exacerbate neuronal damage. The inflammation that occurs in the brain during such neurodegenerative diseases is thought to involve TSPO through its increased presence in activated microglia, thus presenting the possibility for the use of specific TSPO ligands to prevent or limit neuroinflammation. However, the involvement of activated microglia in different CNS diseases differs with respect to its role in disease progression and severity. By use of TSPO as a marker for activated microglia, it is possible to determine exactly what role neuroinflammation plays in specific CNS disease states, opening doors for treatment or inhibition of disease progression.11
1.1.4 The TSPO Altered Expression
TSPO basal expression is up-regulated in a number of human pathologies, including a variety of tumours and neuropathologies, such as gliomas and neurodegenerative disorders (Huntington‟s and Alzheimer‟s diseases), as well as various forms of brain injury and inflammation.18,19,20 Furthermore, changes in TSPO receptor levels were found in anxiety and mood disorders.7,21,22 Recent evidences suggest that TSPO on glial cells may regulate the biosynthesis of neurosteroids, leading to the hypothesis of its potential role as key determinant for the treatment of neuropathological conditions.5,11
1.1.5 TSPO in Cancer
The recent literature on TSPO includes many reports dealing with TSPO and cancer. The rationale behind the potential application of TSPO-targeted therapies in cancer is based on two main features: i) alteration of TSPO expression in tumour cells and ii) TSPO-dependent apoptosis modulations. Considering TSPO expression, some of highest densities of TSPO are observed in neoplastic tissues and cell lines. Ovarian, hepatic and colon carcinomas, adenocarcinoma, and glioma all show increased TSPO densities relative to untransformed tissues. Even higher levels of TSPO density were observed in more rapidly proliferating breast cancer cells and more aggressive breast cancer phenotypes. In further, an
9 important relationship between TSPO expression, proliferation rate and tumour aggressivity was recently documented by Hardwich and co-workers.23 These data suggest that monitoring TSPO expression may be relevant in the clinic and that the protein could be used as a diagnostic and/or prognostic marker. Some TSPO ligands may have potential as antitumor molecules which would be related to their antiproliferative and pro-apoptotic properties. Consistent with this, FGIN-1-2724 and PK 1119516 were shown to have antitumoral activities and to induce apoptosis and cell cycle arrest in colorectal, or esophageal cancer cell lines and cancer primary cell cultures. These observations indicate that TSPO is an attractive target in cancer therapy.1
1.1.6 TSPO in Inflammation and Auto-immune Diseases
TSPO is also involved in the regulation of immune responses. In the immune system, TSPO is widely expressed: in the thymus, where the protein is restricted to the medulla, in the white pulp of the spleen, in the lymph nodes, in the Peyer patches of the intestine and in all human peripheral blood leukocyte subsets. As observed for tumoral cell lines, changes in TSPO expression have already been associated with several inflammatory conditions: PK 11195 binding is up-regulated in experimental autoimmune encephalitis, motor neuron axotomy, sciatic nerve degeneration and regeneration.25 TSPO ligands exhibit anti-inflammatory properties. They modulate monocyte chemotaxis and humoral responses such as macrophage production of reactive oxygen intermediates, TNF, or IL-1 and IL-6.26 Both Ro 5-4864 and PK 11195 were show to limit the severity and progression of different inflammatory processes. For instance, they were highly potent at inhibiting inflammatory sings in two mouse models of acute inflammation, where a treatment with carrageenan induced a paw oedema formation and pleurisy.27 The potential therapeutic benefit of TSPO ligands is not restricted to arthritis treatment and may be enlarged to other auto-immune pathologies. The protective effect of Ro 5-4864, PK 11195 and SSR 180575 was also demonstrated in a spontaneous inflammatory skin pathology developed in an
10
targeting TSPO may have beneficial effect in the treatment of inflammatory disorders and auto-immune pathologies.1
1.1.7 TSPO in Viral Infection
TSPO and TSPO ligands were shown to modulate responses to viral infection. Apoptosis blockade is a strategy adopted by the virus to circumvent the normal protective host response against viral infection. The interaction between ML11, a virulence factor produced by the Myxoma poxvirus, and TSPO resulted in the inhibition of the dissipation of the transmembrane mitochondrial potential and the blockade of the mitochondrial release of cytochrome c.29 This latter study was important as it demonstrated, for the first time, the direct interaction of TSPO with an anti-apoptotic protein. The demonstration that a pathogenic virus targets TSPO to inhibit apoptosis offers new prospects for the dissection of TSPO function but also for the definition of original anti-viral strategies.1
1.1.8 TSPO in Neurodegenerative Diseases
A dramatic increase in TSPO density has been observed following experimental injuries of the CNS and in pathological situations. In most lesions, the increase in TSPO density was associated with the inflammatory reaction of the nervous system. These conditions included inflammation,30 metabolic stress,31 traumatic, or ischemic chemically-induced brain injury in animal models.32 Similar changes in TSPO density are observed in acute and chronic neurodegenerative states in humans. For example, temporal cortex obtained from patients with Alzheimer‟s disease show an increase in TSPO density. A highly significant increase in TSPO density was also observed in the putamen and a moderate but significant increase in the frontal cortex of patients suffering from Huntington‟s disease.33
Finally, Vowinckel and co-workers observed a correlation between [3H]PK 11195 binding and inflammation both in vitro and in vivo in multiple sclerosis and experimental autoimmune encephalomyelitis.34 Given these modulations, labelled PK 11195 and positron emission tomography (PET) have been used for in vivo imaging of neuroinflammation in a variety of brain diseases and at different disease stages to i) detect in patients inflammatory changes and ii)
11 monitor the progression of neuroinflammation as a marker of the disease activity. Consistent with these modulations and considering both the steroidogenic potential of TSPO ligands and the antiapoptotic property of the protein, it has been hypothesized that the modulation of TSPO functions could play neurotrophic and neoroprotective roles during neuronal damage. TSPO ligands might promote neuronal survival by acting at different levels, through the regulation of apoptosis which favoured the survival of glial cells and/or the production of mediators such as neurosteroids, cytokines or other neurotrophic factors that support nerve survival. For example in a same study the ligand SSR 180575 and Ro 5-4864 promoted neuronal survival and repair following axotomy.1,35
1.1.9 TSPO in Anxiety
In the brain, neurosteroids such as allopregnanolone and pregnenolone, acting as positive modulators of -aminobutyric type A (GABAA) receptors, exert anxiolytic activity.
Specific ligands targeting TSPO increase neurosteroid production and for this reason have been suggested to play an important role in anxiety modulation. Unlike benzodiazepines (Bzs), which represent the most common anti-anxiety drugs administered around the world, selective TSPO ligands have shown anxiolytic effects in animal models without any of the side effects associated with Bzs. Therefore, specific TSPO ligands that are able to promote neurosteroidogenesis may represent the future of therapeutic treatment of anxiety disorders. Furthermore, TSPO expression levels are altered in several different psychiatric disorders in which anxiety is the main symptom.7 Anxiety is a normal reaction to stress, which helps people deal with daily difficulties and cope with them. When anxiety is excessive and disabling, it may become pathological and fall under the classification of an anxiety disorder according to the diagnostic manual definition.36 Anxiety disorders are the most common and frequent mental disorders, affecting a significant percentage of the world's population (8-13%), and often overlapping with mood disorders and drug abuse.37 Anxiety disorders markedly affect the living conditions of patients and their relatives, jeopardizing work and family setting. In general, anxiety disorders are treated with medication,
12
specific types of psychotherapy, or both. Treatment choices depend on the problem and the person‟s preference. The diagnosis is difficult and complex because of the variety of possible causes and symptoms, often arising from highly personalized experiences. Anxiety disorders are classified as panic disorder (PD), obsessive-compulsive disorder (OCD), post-traumatic stress disorder (PTSD), generalized anxiety disorder (GAD) and social and specific phobias. A combination of pharmacological and genetic manipulation studies has provided forceful evidences that the GABAergic system is strongly linked to the development of anxiety behaviours. The GABAA receptor is one of two ligand-gated ion channels responsible for mediating the effects of GABA, the major inhibitory neurotransmitter in the brain. In addition to the GABA binding site, the GABAA receptor complex appears to have distinct allosteric binding sites for benzodiazepines, barbiturates, ethanol, inhaled anaesthetics, furosemide, kavalactones, neuroactive steroids, and picrotoxin. The receptor is a multimeric transmembrane receptor consisting of five subunits (the most common type in the brain is a pentamer comprising two α, two β, and a γ subunit (α2β2γ)), arranged around a central pore.38 Once bound by endogenous GABA, the protein receptor changes conformation within the membrane, opening the pore to allow chloride ions to pass down the electrochemical gradient. Activation of GABAA receptors tends to stabilize the resting potential. In that condition, the excitatory neurotransmitters cannot depolarize the neuron generating the action potential, so the net effect is typically inhibitory, reducing the activity of the neuron. The BZs are a class of psychoactive drugs with anxiolytic, sedative, hypnotic, anticonvulsant, and muscle relaxant properties. BZs bind at the interface of the
and subunits on the GABAA receptor (BZR site). The long-term use of BZs can cause physical dependence with risk of withdrawal symptoms and rebound syndrome. However, BZs are the most commonly used medications to treat states of anxiety, and are ever prescribed for short term relief of severe anxiety disorders. Common medications are lorazepam, clonazepam, alprazolam, and diazepam. Once bound to the BZR, the BZ ligand locks the BZR into a conformation in which the GABA neurotransmitter has a much higher affinity for
13 its receptor. This increases the frequency of opening of the associated chloride ion channel, thereby hyperpolarizing the membrane of the associated neuron.7
Neurosteroids synthesised de novo in the brain, modulate neuronal functioning and could play an important pathophysiological role. Interestingly, a variety of neurological and psychiatric disorders, such as anxiety, depression, and schizophrenia, have been associated with both abnormal levels of neurosteroids and perturbations of neurotransmission. Some of the endogenous neurosteroid GABAA modulators are allopregnanolone (3α,5α-tetrahydroprogesterone, ALLO), pregnenolone (PREG), and tetrahydrodeoxycorticosterone (THDOC). In addition, dehydroepiandrosterone sulphate (DHEAS), the most abundant steroid found in the body, exhibits a bimodal effect on GABAA receptors: it demonstrates positive allosteric modulation at low nanomolar concentrations, and negative allosteric modulation at high micromolar concentrations. The anxiolytic properties of DHEAS may also occur since it is metabolised to androsterone and androstanediol, which are positive modulators of the GABAA receptor.
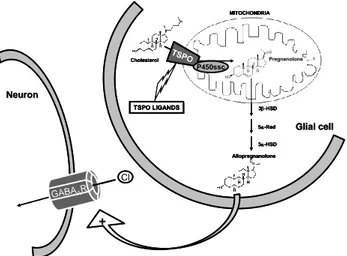
It has been recognized that neurosteroids are synthesized from cholesterol in the CNS by a cascade of enzymatic processes, controlled by both P450 and non-P450 cytochrome enzymes.7,39 The first step in the neurosteroid synthesis is cholesterol conversion to pregnenolone, which occurs in mitochondria and is facilitated by TSPO. This step is the rate-limiting reaction of neurosteroid synthesis and appears to regulate the neurosteroid levels in the brain. The binding of TSPO ligands induces cholesterol transport and steroid formation, leading to the formation of active neurosteroids (ALLO), which positively modulate the activity of the GABAA receptor potentiating GABAergic transmission and thus explaining the anxiolytic activity, as primarily shown in animal models of anxiety.39 Figure 3
14 Cl GABAAR + Neuron Neuron Allopregnanolone 3-HSD 5-Red 3-HSD Glial
Glial cellcell Cholesterol TSPO P450ssc Pregnenolone MITOCHONDRIA TSPO LIGANDS Cl GABAAR + Neuron Neuron Allopregnanolone 3-HSD 5-Red 3-HSD Glial
Glial cellcell
Allopregnanolone 3-HSD 5-Red 3-HSD
Glial
Glial cellcell Cholesterol TSPO P450ssc Pregnenolone MITOCHONDRIA TSPO LIGANDS Cholesterol TSPO P450ssc Pregnenolone MITOCHONDRIA TSPO TSPO P450ssc Pregnenolone MITOCHONDRIA TSPO LIGANDS
Figure 3. Role of TSPO ligands in neurosteroid-mediated allosteric modulation of the GABAA receptor (TSPO, Translocator Protein; P450ssc, Cholesterol side-chain cleavage cytochrome P-450 complex; 3β-HSD, 3-hydroxysteroid dehydrogenase; 5-Red, 5-Reductase; 3-HSD, 3 -hydroxysteroid dehydrogenase; GABAA R, GABAA receptor).
This evidence has suggested TSPO involvement in the secretion of neurosteroids, whose levels have been reported to be changed in several diseases and to be implicated in the pathogenic mechanisms of anxiety.7,40 Indeed, the important involvement of TSPO in anxiety in humans has been extensively demonstrated by numerous studies in which TSPO expression levels have been reported to be changed. Specifically, TSPO density is up-regulated in acute stress conditions and down-regulated in chronic or repeated stress. A decrease of TSPO density has been shown in blood cells (platelets or lymphocytes) of patients affected by different psychiatric disorders, mainly characterized by anxiety, such as generalized anxiety disorder, generalized social phobia, post-traumatic stress disorder, panic disorder, chronic obsessive–compulsive disorder and also in suicidal patients,41 as well as in healthy subjects with high trait anxiety levels. We have demonstrated that TSPO expression was significantly decreased in lymphocytes of patients with post-traumatic stress disorder,42 and in platelets of patients with panic disorder or major depressive disorder associated with severe separation anxiety symptoms.7
1.1.10 Tspo Synthetic Ligands
Numerous synthetic class of compounds are known to bind the TSPO with varying selectivity and affinity.9 Figure 4
15
Benzodiazepines (Diazepam, Ro 5-4864)
Isoquinoline Carboxamides (PK 11195)
Benzothiazepines (THIA-66, THIA-67, THIA-4i)
Benzoxazepines (Oxa-17f, Oxa-17j)
Imidazopyridines (Alpidem)
Phenoxyphenyl-Acetamide Derivatives (DAA 1106)
Pyrazolopyrimidines (DPA-713)
Indolacetamide Derivatives (FGIN-1, SSR180575)
Indol-3-ylglyoxylamides (IND-18)
1.1.10.1 Benzodiazepine
The classical benzodiazepine, Diazepam, has long been used as an anxiolytic and also as an effective anticonvulsant. Its ability to enhance the affinity of GABAA for its receptor by binding to the modulatory site has been well characterized. In 1977, Braestrup and Squires performed a study concerning the binding properties of [3H]Diazepam, identifying an alternative, saturable and specific binding site located on rat brain membranes, as well as on mitochondrial fractions from rat kidney, liver and lung.43 Starting from this discovery, Bz-binding sites have been divided into central and peripheral types. Diazepam binds with nearly similar nanomolar affinity to both receptors, but other ligands show selectivity. Actually, in the same study, the clinically inactive benzodiazepine Ro 5-4864 (4'-clorodiazepam), showed high affinity for this peripheral site, now TSPO, and was unable to displace [3H]Diazepam from the central binding site. The main disadvantage of Ro 5-4864 is its species dependence, which implied markedly different results between rats and humans, partially limiting its usefulness as a tool to study TSPO.7
16 N N CH3 CH3 O Cl CH3 PK 11195 N N O Cl CH3 Cl Ro 5-4864 N N O Cl CH3 Diazepam S N OR X R'
THIA-66: R=COCH3, R'=H, X=OCH3
THIA-67: R=SO2CH3, R'=H, X=OCH3
THIA-4i: R=CON(Et)2, R'=4-Cl, X=H O N R X OXA-17f: R=OCON(Et)2, X=H OXA-17j: R=OCON(Et)2, X=CH3 N N CON(C3H7)2 Cl Cl Alpidem O CH3 N O H3CO F OCH3 DAA 1106 N N H3C N N CH3 CH3 OCH3 O DPA 713 CH3 N H O N X R1 R2 R3 FGIN-1 FGIN-1-27: R1=R2= n-C6H13; R3=F N H N N O N CH3 CH3 O SSR 180575 N H R4 N O O R1 R2 R3 IND-18: R1=R2= n-C6H13, R3=F, R4=H R5 Figure 4 1.1.10.2 Isoquinoline Carboxamide
The Isoquinoline Carboxamide PK 11195, was the first non-benzodiazepine type compound identified to bind the TSPO with high potency,
Figure 4.
A study by Versijpt et al. demonstrated that PK 11195 reduced the amount of inflammatory mediators, such as TNFα and nitric oxide (NO), released by activated microglia cells.44 This indicates that TSPO ligands may be able to
17 altered biochemical processes in the brain and hence may have therapeutic benefits in certain neurological disorders. PK 11195 has been used extensively in characterising the TSPO in both normal and pathological states. It has not only provided useful information on the molecular identity of TSPO but has also showed promise as a potential therapeutic due to its anti-inflammatory action and its ability to increase the amount of neurosteroids in the CNS. However, the use of PK 11195 is severely limited by its far from ideal in vivo properties including its high lipophilicity.9
1.1.10.3 Benzothiazepines
The Benzothiazepines ligands including THIA-66 and THIA-67, (Figure
4) displayed nonomolar affinity and notable selectivity for TSPO binding sites;
however they were less potent than PK 11195. To date, THIA-4i has been reported as the most potent TSPO ligand from this class with an affinity comparable to that of the gold standard TSPO ligand PK 11195. Although these ligands might be useful probes for in vitro studies, further evaluation is required to elucidate their in vivo pharmacological profile.9
1.1.10.4 Benzoxazepines
The Benzoxazepines (Figure 4) were synthesised based on a pyrrolobenzoxazepine skeleton. Ligands from this class were evaluated in binding studies for their ability to displace [3H]PK 11195 from the receptor site. A number of these compounds displayed Ki values in the subnanomolar range. OXA-17f and OXA-17j were reported as the most potent TSPO ligands from this series with Ki values of 0.26 and 0.36 nM, respectively compared to Ki value of 0.78 nM for PK 11195 in the same assay. All of these benzodiazepine-like compounds were shown to stimulate neurosteroids production with comparable potency to PK 11195 and Ro 5-4864 in mouse Y-1 adrenocortical cell line.9
1.1.10.5 Imidazopyrdines and Phenoxyphenyl-Acetamides
The progenitor of the class of Imidazopyridines has to be considered Alpidem
(6-chloro-2-(4-chloro-phenyl)-N,N-dipropylimidazo[1,2-a]pyridine-3-18
acetamide) (Figure 4), known to bind both TSPO and BZR with nanomolar affinity (Ki 0.5-7 nM and 1-28 nM, respectively). Alpidem possesses anxiolytic activity with a profile that is substantially different from that of BZs. Alpidem was releasedin 1991 (Ananxyl®, Sanofi-Aventis) as a highly active anxiolytic agent and it was generally prescribed to patients with moderate to severe anxiety, particularly when these patients exhibited either sensitivity or resistance to benzodiazepine therapy. Alpidem was withdrawn from the market in most of the world following reports of severe liver damage caused by Ananxyl.7
In 1997, Trapani‟s research group reported a wide SAR study on alpidem, developing a series of 2-phenylimidazo[1,2-a]pyridine derivatives (CB) that showed various degrees of affinity and selectivity for TSPO with respect to BZR, dependent on the value of n and the nature of the various substituents R1, R2, X, Y, Z.45 Figure 5 N N CONR1R2 Z Y X 1 2 3 4 5 6 7 8 n CB
Figure 5. General formula of 2-phenylimidazo[1,2-a]pyridine derivatives CB.
Three lead compounds selected from this series, propyl-[2-(4-chlorophenyl)-6,8-dichloroimidazo[1,2-a]pyridin-3-ylacetamide CB34, N,N-di-n-propyl-[2-phenyl-6,8-dichloroimidazo[1,2-a]pyridin-3-ylacetamide CB50, and
N,N-di-n-propyl-[2-phenyl-6-bromo-8-methylimidazo[1,2-a]pyridin-3ylacetamide
CB54 were investigated for their ability to stimulate central and peripheral steroidogenesis and to produce anticonflict action in rats.46 Figure 6
N N CON(n-C3H7)2 Cl Cl Cl N N CON(n-C3H7)2 Cl Cl N N CON(n-C3H7)2 CH3 Br CB34 CB50 CB54
19 In in vitro experiments performed on cerebral cortex membrane, all three compounds potently inhibited [3H]PK 11195 binding in the order CB34 (IC50 1.03 nM) > CB54 (IC50 1.54 nM) > CB50 (IC50 3.04 nM), without substantially affecting [3H]flunitrazepam binding to BZRs. Intraperitoneal administration of CB compounds in rats resulted in significant dose-dependently increased concentrations of neuroactive steroids, such as pregnenolone, progesterone, allopregnanolone, and allotetrahydrodeoxycorticosterone (THDOC) in plasma and brain. CB34 also increased the brain concentration of neuroactive steroids in adrenalectomized-orchiectomized rats, although to a lesser extent than in sham-operated animals. The effect of these ligands on neurosteroid concentration was attributable to increased steroidogenesis in the brain, rather than to an increased supply from peripheral sources. In this view, CB34 appeared to be a promising compound, which, acting as TSPO agonist, increased neuroactive steroid concentration, and proved beneficial for the treatment of stress and anxiety related diseases.7
In 2004, the research group of Nakazato and colleagues from the Taisho Pharmaceutical and Co. described a novel class of selective TSPO ligands derived from a structural simplification of the bicyclic structure of the benzodiazepine Ro 5-4864, which is the opening of the diazepine ring. The resulting Phenoxyphenyl-Acetamide derivatives with general formulae DAA, were synthesized and evaluated for their affinity and selectivity for TSPO in an extensive SAR study.47
Figure 7 N O H3C Cl Cl O R1 N X1 Y Ar1 Ar2 O O CH3 N X1 CH3 O Ro 5-4864 DAA-I DAA
20
The authors focused their attention on chemical modification of Ar1, Ar2, R1, X1, and Y, obtaining a number of compounds with remarkable affinity and selectivity, so that to define the most important structural features for this class of compounds in binding to TSPO.
Two phenoxyphenylacetamide derivatives, N-(2,5-dimethoxybenzyl)-N-(5-fluoro-2-phenoxyphenyl)acetamide (DAA 1106,) and
N-(4-chloro-2-phenoxyphenyl)-N-(2-isopropoxybenzyl)acetamide (DAA 1097, 7-099) (Figure
8), have been extensively characterized with respect to their receptor binding and
behavioural profile, as well as to their effect on steroidogenesis.
O CH3 N O Cl O H3C CH3 O CH3 N O H3CO F OCH3 DAA 1106 DAA 1097
Figure 8. Structure of DAA 1106 and DAA 1097 derivatives.
Both DAA 1106 and DAA 1097 inhibited [3H]PK 11195 and [3H]Ro 5-4864 binding to crude mitochondrial preparations of rat whole brain with IC50 values in the subnanomolar range (0.92 and 0.28 nM, and 0.64 and 0.21 nM, respectively).48 These compounds were also highly selective, as they did not inhibit the binding of BzR ligand [3H]flunitrazepam (IC50 > 10,000 nM) in the same preparation, and showed weak or negligible affinity (IC50 values are approximately 10,000 nM) for 58 other receptors, including those of related neurotransmitters, ion channels, uptake/transporter and second messengers.
In a separate study the same research group performed an exhaustive characterization of the binding of DAA 1106 in the mitochondrial fractions of rat brain by means of its [3H]radiolabelled analogue. The binding of [3H]DAA 1106 was saturable, with a dissociation constant (Kd) determined from a Scatchard plot
21 analysis of 1.2 nM. The binding was inhibited by several TSPO ligands in an order similar to that observed for [3H]PK 11195, with DAA 1106 being the most potent inhibitor. In addition, [3H]DAA 1106 was highly TSPO selective, as its binding was not affected by several neurotransmitter-related compounds, including adrenoceptor, GABA, dopamine, 5-hydroxytriptamine (5-HT), acetylcholine, histamine, glutamate and BZR ligands even at a concentration of 10 μM. When tested on mitochondrial fractions of the rhesus monkey cerebral cortex, [3H]DAA 1106 showed a high affinity for TSPO (Kd 0.426 nM), with its binding being potently inhibited by DAA 1106 and PK 11195, but not by Ro 5-4864, even at 1 μM. The results indicated that this ligand was not species dependent, similar to PK 11195 and contrary to Ro 5-4864. Autoradiography and biochemical studies on rat brain showed that the highest binding of [3H]DAA 1106 is localized in the olfactory bulb and ventricular structures related to the secretion of cerebrospinal fluid (choroid plexus), followed by the cerebellum and cerebral cortex.7 In in vivo studies, DAA 1106 and DAA 1097 showed potent anxiolytic-like properties in laboratory animals, in both the light/dark exploration test in mice and the elevated plus-maze test in rats.46
Even though these two phenoxyphenylacetamides have a similar structure, potently bind to TSPO and exert anxiolytic effects, their effects on steroidogenesis appeared to be opposite. Culty and colleagues 49 examined the effects of these two ligands on steroidogenic response using MA-10 Leydig tumour cells, C6-2B glioma cells, and rat brain mitochondria. It has been observed that DAA 1097 activated steroidogenesis in all the three preparations in a similar fashion to that described for PK 11195, and more efficiently on brain than Leydig cells. Surprisingly, DAA 1106 did not activate steroidogenesis, despite its high affinity for TSPO.
It has been suggested that the DAA 1097 and DAA 1106 binding sites on TSPO share a common domain with that of PK 11195, but also appear to contain additional motif(s) that do not interact efficiently with PK 11195 site. The different effect on steroidogenesis has been rationalized by hypothesizing that the binding of DAA 1097 induce changes in the receptor similar to that triggered by PK 11195, allowing steroidogenesis activation. On the contrary, the binding of
22
DAA 1097 leads to conformational changes that do not permit or antagonize TSPO steroidogenic function. It has been thus concluded that DAA 1097 appears to be an agonist and DAA 1106 may act as a competitive antagonist.7, 47
1.1.10.6 Pyrazolopyrimidines
The Pyrazolopyrimidines (Figure 4) are bioisteres of the imidazopyridine derivatives and hence are structurally related to alpidem. Binding assays, using [3H]PK 11195 as the radioligand and membranes from rat kidney tissue as receptor source, showed that a subset of ligands from this pyrazolopyrimidine series displayed high binding affinity (Ki) for the TSPO ranging from 0.8-6.1 nM. Some of these compounds were shown to stimulate steroid biosynthesis in C6 glioma rat cells with a few capable of increasing pregnenolone synthesis with similar potency to Ro 5-4864 and PK 11195.50
1.1.10.7 Indoleacetamide Derivatives
The Indoleacetamide Derivatives were found to enhance steroidogenesis and to be highly selective for the TSPO. The lead compound from this class is known as N,N-di-n-hexyl-2-(4-fluorophenyl)indole-3-acetamide (FGIN-1-2724
Figure 4). FGIN-1-27 has a high affinity (Ki 5.0 nM, displacement study using [3H]PK 11195) and is able to penetrate the blood brain barrier. FGIN-1-27 has also been shown to induce sedation and ataxia at micromolar intravenous doses. It was hypothesised that the pharmacological actions of this compound are most likely due to its indirect action on GABAA receptors via the stimulation of neurosteroid production.9
SAR studies allowed the authors to identify the structural features of FGIN-1 derivatives essential for high affinity: (i) the presence of two alkyl groups on the amide nitrogen; (ii) the length of such alkyl chains, with the N,N-dihexyl groups conferring an optimum of affinity, twenty fold higher than the corresponding N,N-dimethyl analogues; (iii) halogenation of the pendant 2-aryl groups and the indole benzofused ring (X and R3 = Cl, F). Broad screening studies revealed that these FGIN-1 derivatives were highly TSPO selective, as they failed to bind with any significant affinity (Ki > 1000 nM) to other receptor systems,
23 including the GABAA, GABAB, glycine, glutamate, dopamine, serotonin, opiate, cholecystokinin, beta adrenergic, cannabinoid, and sigma receptors. Compounds exhibiting the highest TSPO binding affinity were selected to evaluate their ability to stimulate pregnenolone formation from the mitochondria of C6-2B glioma cells.7
Another high affinity TSPO ligand structurally related to this class of compounds, 7-chloro-N,N-5-trimethyl-4-oxo-3-phenyl-3,5-dihydro-4H-pyridazino[4,5 b]indole-1-acetamide (SSR 180575 Figure 4), has been shown to promote neuronal survival and repair. SSR 180575 displays nanomolar affinity, four times greater than that of Ro 5-4864, for both the rat and human TSPO. Moreover, SSR 180575 increased pregnenolone accumulation in the brain implying that the therapeutic effects may be mediated through its ability to stimulate steroidogenesis.9
1.1.10.8 N,N-dialkyl-2-phenylindol-3-ylglyoxylamides
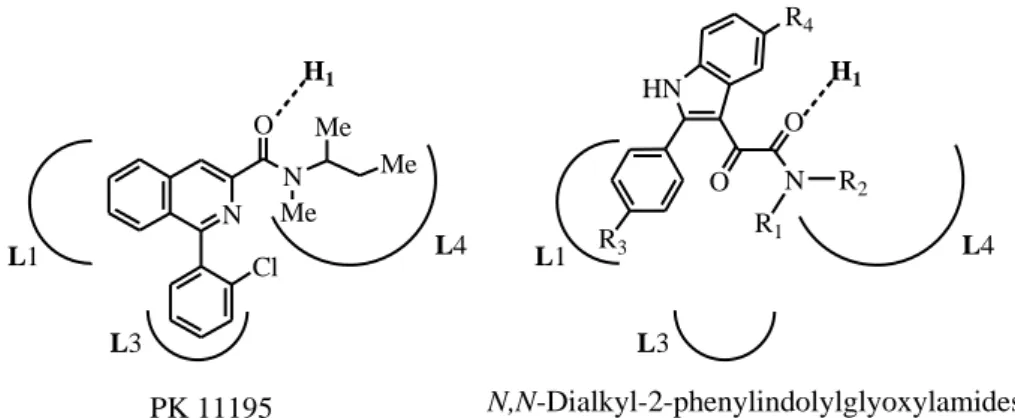
Recently, a novel series of highly potent and selective TSPO ligands, which represent conformationally constrained analogues of FGIN-1 derivatives, was developed in the laboratory where this PhD has been done, and are known as the N,N-dialkyl-2-phenylindol-3-ylglyoxylamides I, (Figure 9).7,51 Within this class, SAR studies were rationalized in light of the recently reported pharmacophore/receptor model made up of three lipophilic pockets (L1, L3, and L4) and an H-bond (H1) donor group, Figure 9.
N O N Me Me Me HN O N R1 R2 R3 R4 O PK 11195 L3 Cl N,N-Dialkyl-2-phenylindolylglyoxylamides I H1 L1 L4 H1 L1 L4 L3
Figure 9. PK11195 and N,N-dialkyl-2-phenylindolylglyoxylamides in the pharmacophore/receptor
24
Most of the new compounds exhibited a high affinity for TSPO, with Ki values in the nanomolar/subnanomolar range. Table 1 The binding affinity of the indolylglyoxylamides Ia-Iaa at the TSPO was determined in rat kidney membranes by competition experiments against [3H]PK 11195. The TSPO/BZR selectivity of Ia-Iaa was evaluated by binding studies using membranes from rat brain tissues and [3H]flumazenil as radioligand.51 Most of the compounds Ia-Iaa showed a high affinity for TSPO in the nanomolar to subnanomolar range and a high selectivity for TSPO over BZR because they inhibited the binding of [3H]flumazenil at rat brain membranes to an extent varying from 0% to 16% at a fixed 10 μM concentration. Table 1 Among the unsubstituted indolylglyoxylamides Ia-Il (R3 = R4 = H), the least effective were the N-monosubstituted derivatives Ia and Ib probably because they cannot occupy both the L3 and L4 lipophilic pockets. Thus, we did not further examine this type of substitution. The N,N-disubstituted derivatives Ic-Ii exhibited nanomolar affinity because they fill both the L3 and L4 sites. Compound Ig, bearing two n-hexyl groups, was the most potent one, with a Ki of 1.4 nM. A progressive enhancement of affinity was generally observed by increasing the length of the linear N-alkyl groups (Ic-Ig), due to the progressively better filling of both the L3 and L4 lipophilic pockets, with If being the only exception. In any case, the affinity data of Ic-Ii suggest that interactions at the L3 and L4 sites are governed primarily by lipophilicity and that steric factors may also play some role. Also, in the subseries of the cyclic amides Ij-Il, the affinity was enhanced with increasing dimensions of the cycle. However, Ij and Ik showed a considerable drop in affinity with respect to the linear derivatives Ic and Id probably because of their incapability of optimally filling both the L3 and L4 sites, which are separated in the binding site, while the more lipophilic and flexible Il displayed an appreciable affinity (Ki 33 nM), again confirming the role of lipophilicity in the interaction of these ligands with the receptor site. The three most potent derivatives Id, Ie, and Ig were selected as leads for further affinity optimization efforts. Insertion of the electron-donating lipophilic methyl group in the 4'-position of the 2-phenyl ring (R3) left the affinity nearly unchanged because Is-Iu were equipotent with the parent compounds Id, Ie and Ig. By introducing an electron-withdrawing substituent
25 such as chlorine or fluorine into the 4'-position, we obtained Ip- Ir or Im-Io, respectively, which exhibited a gain in affinity of 2.5-fold to 7.5-fold. Among them, the N,N-di-n-hexyl derivative Io stood out as the most potent of all the ligands, with a Ki of 0.37 nM, similar to that of alpidem. These results suggest that the phenyl ring of 2-phenylindoles might be involved in a π-stacking interaction with an electron-rich aromatic ring within the L1 pocket and that this interaction is reinforced by 4' electron-withdrawing substituents such as the halogens. In the 4'-phenyl-substituted indoles, potency correlated with the N-alkyl length: n-hexyl > n-butyl > n-propyl. This affinity trend was reversed when a chlorine was introduced in the 5-position of the indole nucleus: the longer the side chains, the lower the potency. Among the 5-chloro-4'-phenyl-unsubstituted derivatives Iv-Ix, the highest potency was exhibited by Iv (Ki 2.8 nM), which bears two n-propyl groups on the amide nitrogen. Insertion of a chlorine in both the 5 and 4' positions improved potency with respect to the unsubstituted derivatives when the amide chain featured n-propyl or n-butyl groups (compare Iy and Iz vs Id and Ie); otherwise, it was unfavorable (compare Iaa vs Ig, both bearing n-hexyl groups). In other words, the effects on affinity of a chlorine in the 5 and 4' positions are not additive but depend on the nature of the N,N-dialkyl chains. It is tempting to speculate that the length of the 5,4'-dichloro-2-phenylindole scaffold restricts the mobility of the whole ligand within the binding site. Under these steric constraints, the L3 and L4 pockets are optimally filled by the smaller n-propyl or n-butyl, compared with the bulkier n-hexyl chains. To sum up, the lack of additive effects of substituents NR1R2, R3, and R4 is somehow related to a simultaneous increase in their dimensions, which makes each single interaction at the three lipophilic sites less effective.5
26
Table 1. Binding dates of 2-phenylindol-3-ylglyoxylamides derivatives Ia-aa.[51] N R4 N O O R1 R2 H R3 N R1 R2 R3 R4 TSPO a Ki (nM) CBRb Ia (CH2)2CH3 H H H 81580 3293381 Ib (CH2)3CH3 H H H 116799 12% Ic CH2CH3 CH2CH3 H H 43.04 - Id (CH2)2CH3 (CH2)2CH3 H H 12.21.0 16% Ie (CH2)3CH3 (CH2)3CH3 H H 7.500.7 3% If (CH2)4CH3 (CH2)4CH3 H H 16.02.0 3% Ig (CH2)5CH3 (CH2)5CH3 H H 1.400.2 10% Ih CH(CH3)2 CH(CH3)2 H H 1039.0 - Ii CH(CH3)CH2C H3 CH(CH3)CH2C H3 H H 17.02.0 5% Ij -(CH2)4 H H 2400125 4% Ik -(CH2)5- H H 66530 3% Il -(CH2)6- H H 33.03.0 3% Im (CH2)2CH3 (CH2)2CH3 F H 4.28±0.32 4.3% In (CH2)3CH3 (CH2)3CH3 F H 2.400.81 0% Io (CH2)5CH3 (CH2)5CH3 F H 0.370.13 0% Ip (CH2)2CH3 (CH2)2CH3 Cl H 4.650.52 14% Iq (CH2)3CH3 (CH2)3CH3 Cl H 1.000.27 0% Ir (CH2)5CH3 (CH2)5CH3 Cl H 0.550.19 4.2% Is (CH2)2CH3 (CH2)2CH3 CH3 H 5.500.98 It (CH2)3CH3 (CH2)3CH3 CH3 H 3.800.91 Iu (CH2)5CH3 (CH2)5CH3 CH3 H 1.600.13 Iv (CH2)2CH3 (CH2)2CH3 H Cl 2.800,3 Iw (CH2)3CH3 (CH2)3CH3 H Cl 4.910.4 Ix (CH2)5CH3 (CH2)5CH3 H Cl 58.46 3% Iy (CH2)2CH3 (CH2)2CH3 Cl Cl 0.620.06 Iz (CH2)3CH3 (CH2)3CH3 Cl Cl 1.90.2 Iaa (CH2)5CH3 (CH2)5CH3 Cl Cl 5.80.6 PK 11195 9.30.5 Ro 5-4864 233.1 Alpidem 0.5-7 1-28 a
The concentration of tested compounds that inhibited [3H]PK 11195 binding to rat kidney mitochondrial membranes (IC50) by 50% was determined with six concentrations of the displacers, each performed in
triplicate. Ki values are the mean ± SEM of three determinations. b The inhibition percent of [3H]flumazenil
specific binding at 10 µM of the compound are the mean ± SEM of five determinations. Ki values are the
27 Following these interesting results, a further series of N,N-disubstituted indol-3-ylglyoxylamides, of general formula II, bearing different combinations of substituents R1-R5 were subsequently synthesized and biologically evaluated.52 Some of these compounds bear linear alkyl chains on the amide nitrogen (R1 = R2 = n-propyl, n-butyl, n-hexyl) and modifications to the basic 2-phenylindol-3-ylglyoxylamide scaffold, consisting of the introduction of small groups in the para position of the 2-phenyl ring (R3 = F, NO2, CF3), the 5- (R4 = F, Cl, NO2, OCH3) and 7-position (R5 = Cl, CH3) of the indole ring. Figure 10
N H R5 R4 O R2 N R1 O R3 II Figure 10
A series of N-ethyl-N-benzyl substituted indoles was designed taking as a reference AC-5216 a TSPO ligand that displays antianxiety activity without the undesirable effects caused by typical benzodiazepines.53 Figure 11
N N N N O CH3 N CH3 O AC-5216 N H R5 R4 O N O R3 II Figure 11
We also investigated a subset of asymmetrical N,N-disubstituted indoles to probe the L3 and L4 lipophilic pockets of the TSPO binding site (R1 = methyl, ethyl; R2 = ethyl, n-butyl, n-pentyl; R3 = H, Cl; R4 = H, Cl).
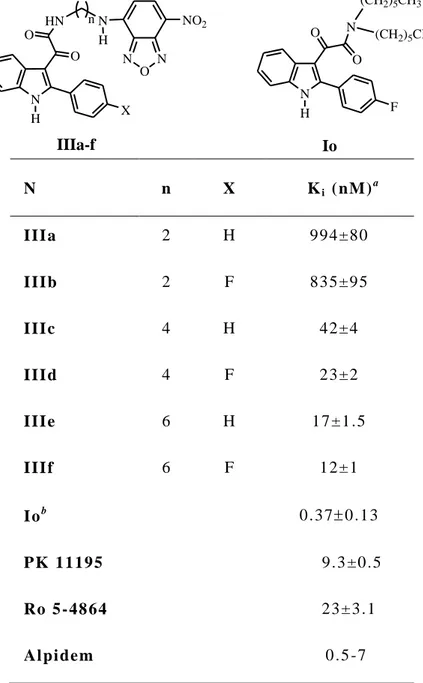
The binding affinities of compounds IIa-IIff, IIgg-IIzz, and IIaaa-IIppp, expressed as Ki values, are given in Tables 2, 3, and 4, respectively, together with the Ki values of the standard TSPO ligands PK 11195, Ro 5-4864, and alpidem.
28
The binding data of some of the previously investigated indole derivatives51 are included for comparison at the bottom of Table 2 (Id, e, g) and Table 3 (Id, y, p,
v). Tables 2-3-4 also include the neurosteroidogenic activity of most of the newly
synthesized compounds, expressed as percent value vs control of the increase in pregnenolone production.
In series IIa-IIff, the amide nitrogen is symmetrically disubstituted (R1 = R2) with n-propyl, n-butyl, and n-hexyl chains. These compounds generally showed a high affinity for TSPO in the nanomolar/subnanomolar range. With the exception of derivatives Iii and IIz, the highest potency was achieved with R1 = R2 = n-hexyl. Introduction of a 4′-NO2 group on the 2-phenyl ring gave the most potent indolylglyoxylamides within this subset: IIb and IIc with a Ki of 0.27 and 0.23 nM, respectively. A 4′-CF3 group was likewise beneficial for potency (compare IId-IIf with Id, e, g), although to a lesser extent with respect to the 4′-NO2 group. These data confirmed the favourable influence on affinity exerted by an electron-withdrawing R3 substituent. This effect was ascribed by Primofiore et al. to a putative π-stacking interaction between the 2-phenyl and an electron-rich aromatic ring within the L1 pocket.51 The affinity was lowered by a NO2 or a OCH3 group at the 5-position of the indole nucleus (R4) (compare IIg-IIn with Id,
e, g) or, in contrast, enhanced by a F in the same position (compare IIo-IIq with Id, e, g).
These data suggest that R4 has to be electron-withdrawing and also very small for optimal binding to TSPO, a combination of properties featured only by fluorine. Substitutions at the 4′- and 5-positions were merged to yield IIr-IIt (R3 = R4 = F) and IIu-IIz (R3 = F, R4 = Cl). Two halogens in both the 4′- and 5-positions did not increase affinity in an additive manner, all the 4′,5-dihalo derivatives being almost as potent or slightly less potent than the 5-fluoro derivatives IIo-IIq. Insertion of an electron withdrawing (Cl) or an electron-donating (CH3) lipophilic group at the 7-position of the indole nucleus (R5) did not produce any gain in affinity (compare IIaa-IIff with their unsubstituted counterparts Id, e, g).51
Compounds IIa-IIz displayed fair to high steroidogenic activity, with IIz performing best (135% increase in pregnenolone production under conditions in
29 which the activities of PK 11195 and Ro 5-4864 were 48% and 41%, respectively). Consequently, IIz was selected for in vivo studies. It is noted that TSPO affinity did not generally correlate with the steroidogenic activity. On the other hand, the nature of the 7-substituent was decisive in the pregnenolone biosynthesis assay, as the 7-chloro derivatives IIaa-IIcc were devoid of any activity, whereas the 7-methyl derivatives IIdd-IIff were good stimulators of pregnenolone production. The precise molecular determinants of the above differences in activity are unknown. At the moment, we can only speculate about an electronic-based triggering effect exerted by R5 on the translocation function of the TSPO.
Further attempts to optimize potency focused on asymmetrically N,N-substituted indoles (R1 ≠ R2). A first subset of N-benzyl-N-ethyl derivatives
(IIgg-IIzz, Table 3) revealed that this combination of R1 and R2 led to affinities roughly similar to those exhibited by N,N-di-n-butyl derivatives. Such a conclusion can be drawn by comparing the Ki values of IIgg-IIzz with our previously reported symmetrically N,N-substituted indoles.51 Thus, it is likely that the aromatic moiety of the N-benzyl chain is accommodated within the lipophilic pocket L3, which also hosts the phenyl ring of PK 11195, in agreement with our pharmacophore/topological model depicted in Figure 9,52 giving rise to the same type of hydrophobic interactions established by an aliphatic moiety of similar size. In this subset, the effects of R3-R5 on the affinity were similar to those observed in the series of symmetrical N,N-dialkyl derivatives IIa-IIff listed in Table 2. Thus,
IIoo (R3 = NO2, R4 = R5 = H) was the most potent (Ki 0.55 nM), whereas IIrr (R3 = H, R4 = 5-OCH3, R5 = H) was the least potent (Ki 69.5 nM).
The highest levels of steroidogenic activity were displayed by IIii, IImm, and IIoo. Among these three compounds, IImm was tested for its in vivo anxiolytic properties.
Table 4 lists the binding data of asymmetrical N,N-dialkyl derivatives IIaaa-IIppp, designed to evaluate whether differences in the lengths of R1 and R2 could optimize their fit into the lipophilic pockets L3 and L4. For this purpose, the unsubstituted and the 4′,5-dichloro, the 4′-chloro, and the 5-chloro substitution patterns were chosen as representative within the previously investigated
N,N-30
dialkylindoles (see for comparison Id, y, p, v in Table 4). Four combinations of R1 and R2 were selected: methy/ethyl (IIaaa and IIbbb), methyl/n-butyl (IIccc,
IIfff, IIiii, IInnn), methyl/n-pentyl (IIddd, IIggg, IIlll, IIooo), and ethyl/n-butyl
(IIeee, IIhhh, IImmm, IIppp). The 4′,5-dichloro derivatives IIfff (R1 = CH3, R2 = (CH2)3CH3, Ki 0.15 nM) and IIggg (R1 = CH3, R2 = (CH2)4CH3, Ki 0.18 nM) turned out to be the most potent indolylglyoxylamides so far described by us.51 These results indicate that the L3 and L4 pockets differ in their dimensions, and that R1 and R2 should be likewise different in their size. The 4′-chloro and 5-chloro derivatives (IIiii-IIppp) displayed Ki values between those shown by their unsubstituted counterparts (IIccc-IIeee) and by the 4′,5-dichloro derivatives (IIfff-IIhhh). Taken together, these data suggest that each chlorine favourably contributes to the affinity to a similar extent.
Most of the compounds were evaluated for their ability to induce pregnenolone biosynthesis in rat C6 glioma cells, in Table 4 showing a low/medium percentage increase in pregnenolone production vs control, despite their high affinities, thus confirming the lack of correlation between potency and steroidogenic activity.
31
Table 2. Receptor Binding Affinity of Compounds IIa-IIff for TSPO and Their
Stimulatory Effects on Pregnenolone Biosynthesis.
N H R5 R4 O N O R3 R1 R2 Compd R1 = R2 R3 R4 R5 Ki (nM) a increase in pregnenolone production vs control (%)b IIa (CH2)2CH3 NO2 H H 0.95 0.1 70 7 IIb (CH2)3CH3 NO2 H H 0.27 0.07 64 2 IIc (CH2)5CH3 NO2 H H 0.23 0.1 88 7 IId (CH2)2CH3 CF3 H H 1.69 0.2 66 7 IIe (CH2)3CH3 CF3 H H 1.16 0.1 60 6 IIf (CH2)5CH3 CF3 H H 1.0 0.1 85 5 IIg (CH2)2CH3 H NO2 H 20.2 2.02 91 2 IIh (CH2)3CH3 H NO2 H 21.6 2.15 102 10 IIi (CH2)5CH3 H NO2 H 30.3 9.15 115 10 IIl (CH2)2CH3 H OCH3 H 328 45 45 2 IIm (CH2)3CH3 H OCH3 H 65.2 3.4 36 3 IIn (CH2)5CH3 H OCH3 H 35.5 8.7 87 7 IIo (CH2)2CH3 H F H 2.67 0.48 66 7 IIp (CH2)3CH3 H F H 4.00 0.15 88 9 IIq (CH2)5CH3 H F H 0.37 0.12 36 4 IIr (CH2)2CH3 F F H 6.73 1.39 17 1 IIs (CH2)3CH3 F F H 4.36 0.05 91 8 IIt (CH2)5CH3 F F H 0.95 0.1 40 2 IIu (CH2)2CH3 F Cl H 2.83 0.08 42 4 IIv (CH2)3CH3 F Cl H 3.05 0.45 37 4 IIz (CH2)5CH3 F Cl H 7.75 1.55 135 4 IIaa (CH2)2CH3 H H Cl 14.0 1.5 0
32 IIbb (CH2)3CH3 H H Cl 3.40 0.3 0 IIcc (CH2)5CH3 H H Cl 2.4 0.3 0 IIdd (CH2)2CH3 H H CH3 25.0 3.0 129 13 IIee (CH2)3CH3 H H CH3 6.0 0.6 71 8 IIf (CH2)5CH3 H H CH3 1.90 0.1 63 5 Idc (CH2)2CH3 H H H 12.2 1.0 8 1 Iec (CH2)3CH3 H H H 7.5 0.7 23 3 Igc (CH2)5CH3 H H H 1.40 0.2 30 3 PK 11195 9.3 0.5 48 5 Ro 5-4864 23 3.1 41 4 alpidem 0.5―7 a
The concentration of test compounds that inhibited [3H]PK11195 binding to rat kidney mitochondrial membranes (IC50) by 50% was determined with six concentrations of the
displacers, each performed in triplicate. Ki values are the means SEM of three determinations.
b
C6 glioma cells were incubated for 2 h at 37 °C in the presence of each compound. Pregnenolone was quantified by enzymatic immunoassay, as described in the text. The values represent the means SEM of at least three determinations. For comparison, the effects of PK 11195 and Ro 5-4864 on pregnenolone production are also included. cData taken from ref. n. 51
33
Table 3. Receptor Binding Affinity of Compounds IIgg-IIzz for TSPO,
and their Stimulatory Effects on Pregnenolone Biosynthesis.
N H R5 R4 O N O R3 compd R3 R4 R5 Ki (nM) a increase in pregnenolone production vs control (%)b IIgg H H H 11 1.0 20 2 IIhh F H H 1.68 0.12 96 8 IIii Cl H H 1.30 0.05 136 11 IIll H Cl H 4.6 0.5 85 8 IImm Cl Cl H 3.33 0.3 171 14 IInn CH3 H H 2.64 0.1 86 7 IIoo NO2 H H 0.55 0.02 103 8 IIpp CF3 H H 1.0 0.1 n.d. IIqq H NO2 H 18.3 0.15 0 IIrr H OCH3 H 69.5 3.6 n.d. IIss H F H 1.33 0.2 n.d. IItt F F H 1.67 0.37 n.d. IIuu F Cl H 4.01 0.26 0 IIvv H H Cl 5.0 0.4 0 IIzz H H CH3 2.30 0.2 0 PK 11195 9.3 0.5 48 5 Ro 5-4864 23 3.1 41 4 Alpidem 0.5―7
34
Table 4. Receptor Binding Affinity of Compounds IIaaa-IIppp for PBR and
Their Stimulatory Effects on Pregnenolone Biosynthesis.
N R4 O R2 N R1 O R3 H compd R1 R2 R3 R4 Ki (nM) a Increase in pregnenolone production vs control (%)b IIaaa CH3 CH2CH3 H H 940 120 n.d. IIbbb CH3 CH2CH3 Cl Cl 9.54 1.29 0 IIccc CH3 (CH2)3CH3 H H 53.3 4.0 32 3 IIddd CH3 (CH2)4CH3 H H 12.1 1.0 36 4 IIeee CH2CH3 (CH2)3CH3 H H 12.6 1.0 43 5 IIfff CH3 (CH2)3CH3 Cl Cl 0.15 0.02 67 2 IIggg CH3 (CH2)4CH3 Cl Cl 0.18 0.02 31 2 IIhhh CH2CH3 (CH2)3CH3 Cl Cl 0.36 0.04 11 2 IIiii CH3 (CH2)3CH3 Cl H 11 1.0 n.d. IIlll CH3 (CH2)4CH3 Cl H 3.4 0.4 65 7 IImm m CH2CH3 (CH2)3CH3 Cl H 3.6 0.4 32 3 IInnn CH3 (CH2)3CH3 H Cl 3.9 0.5 70 7 IIooo CH3 (CH2)4CH3 H Cl 3.6 0.5 76 8 IIppp CH2CH3 (CH2)3CH3 H Cl 1.8 0.2 72 6 Idc (CH2)2C H3 (CH2)2CH3 H H 12.2 1.0 8 1 Iyc (CH2)2C H3 (CH2)2CH3 Cl Cl 0.62 0.06 49 6 Ipc (CH2)2C H3 (CH2)2CH3 Cl H 4.65 0.52 40 5 Ivc (CH2)2C H3 (CH2)2CH3 H Cl 2.80 0.3 10 2 PK 11195 9.3 0.5 48 5 Ro 5-4864 23 3.1 41 4 Alpidem 0.5 ― 7 a,b
35 The SAR data deriving from the biological results of the newly synthesized N,N-dialkyl-2-phenylindol-3-ylglyoxylamide TSPO ligands IIa-IIppp led to a refinement of our TSPO pharmacophore/topological model: (1) the R3 substituent has to be electron-withdrawing to reinforce a putative π-stacking interaction between the 2-phenyl and an electron-rich aromatic ring within the L1 pocket; (2) R4 has to be both electronwithdrawing and very small for optimal binding, a combination of properties featured only by fluorine; (3) substitutions in the 7-position of the indole nucleus (R5) do not produce any gain in affinity; (4) an aromatic moiety (R1/R2) is equivalent to an aliphatic moiety of similar size in interacting hydrophobically with the L3 or L4 lipophilic pocket; (5) the L3 and L4 pockets are probably different in their dimensions, as the best-performing substitution pattern on the amide nitrogen is obtained with R1 and R2 of different sizes.
1.1.11 Molecular Probes for the receptor characterization using Chemical and Physical Techniques: Radioligands and Fluorescent Ligands
Molecular Imaging is a new discipline that unites molecular biology and in
vivo imaging. It enables the visualization of the cellular function and the
follow-up of molecular processes in living organisms without perturbing them. The multiple and numerous potentialities of this field are applicable to the diagnosis of diseases such as cancer, and neurological and cardiovascular diseases. This technique also contributes to improve the treatment of these disorders by optimizing the pre-clinical and clinical tests of new medications. It is also expected to have a major economic impact due to earlier and more precise diagnosis. Molecular and Functional Imaging has taken on a new direction since the description of the human genome.
Molecular imaging differs from traditional imaging in that probes known as biomarkers are used to help to image particular targets or pathways. Biomarkers interact chemically with their surroundings and in turn alter the image according to molecular changes occurring within the area of interest. This process is markedly different from previous methods of imaging which primarily imaged
36
differences in qualities such as density or water content. The ability to image fine molecular changes opens up an incredible number of exciting possibilities for medical application, including early detection and treatment of disease and basic pharmaceutical development. Furthermore, molecular imaging allows quantitative tests, imparting a greater degree of objectivity to the study of these areas.
Many areas of research are being conducted in the field of molecular imaging. Much research is currently centered on detecting what is known as a predisease state or molecular states that occur before typical symptoms of a disease. Other important veins of research are the imaging of gene expression and the development of novel biomarkers.
There are many different modalities that can be used for noninvasive molecular imaging: Magnetic Resonance Imaging (MRI), Optical Imaging (Fluorescent probes and labels are an important tool for optical imaging.), Single photon emission computed tomography (SPECT), Positron Emission Tomography (PET).
The founding principles of molecular imaging can be traced back to nuclear medicine procedures over the past few decades, with other technologies (e.g., optical, MRI) being adapted for molecular imaging by developing different types of molecular probes. At the widest level, there exist two classes of probes: nonspecific and specific. Nuclear medicine plays a key role in the latter class, as the signaling portion of specifically targeted probes. Probes that use antibodies, ligands, or substrates to specifically interact with protein targets in particular cells or sub cellular compartments include those used in most of the conventional radiotracer imaging methods, where the emphasis is on imaging final products of gene expression with radiolabelled substrates that interact with a protein originating from a specific gene. These interactions are based on either receptor-radioligand binding (e.g. binding of 11C-carfentanil to the mu opiate receptor) or enzyme mediated trapping of a radiolabelled substrate (e.g. 18 F-2-fluoro-2-deoxyglucose [18F-FDG] phosphorylation by hexokinase).
However, the main limitation of most of these specific approaches is that a new substrate must be discovered and radiolabelled to yield a different probe for each new protein target. With the significant difficulty, cost, and effort involved in
37 radiolabeling new substrates, along with the requirement for in vivo characterization of every substrate under investigation, more generalizable methods (i.e. those that can image gene product targets arising from the expression of any gene of interest) are preferred. In recent years, this issue has propelled the development and validation of molecular imaging reporter gene/reporter probe systems for use in living subjects together with other generalizable strategies.
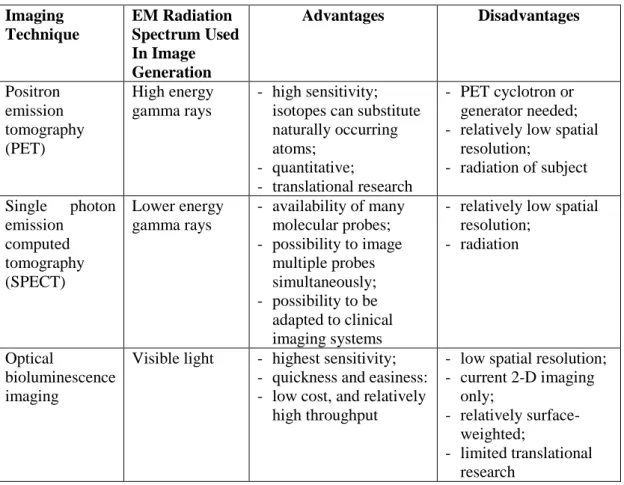
The various existing imaging technologies differ in five main aspects: (i) spatial resolution; (ii) depth penetration; (iii) energy expended for image generation (ionizing or no ionizing, depending on which component of the electromagnetic radiation spectrum is exploited for image generation); (iv) availability of injectable/biocompatible molecular probes; and (v) the respective detection threshold of probes for a given technology, Table 5.
Table 5. Key advantages and disadvantage of the main available imaging
modalities used in molecular approaches.
Imaging Technique EM Radiation Spectrum Used In Image Generation Advantages Disadvantages Positron emission tomography (PET) High energy gamma rays - high sensitivity; isotopes can substitute naturally occurring atoms; - quantitative; - translational research - PET cyclotron or generator needed; - relatively low spatial
resolution; - radiation of subject Single photon emission computed tomography (SPECT) Lower energy gamma rays - availability of many molecular probes; - possibility to image multiple probes simultaneously; - possibility to be adapted to clinical imaging systems
- relatively low spatial resolution;
- radiation
Optical
bioluminescence imaging
Visible light - highest sensitivity;
- quickness and easiness: - low cost, and relatively
high throughput
- low spatial resolution; - current 2-D imaging only; - relatively surface-weighted; - limited translational research


![Table 1. Binding dates of 2-phenylindol-3-ylglyoxylamides derivatives Ia-aa. [51] NR4 O NO R 1 R 2 H R 3 N R 1 R 2 R 3 R 4 TSPO a K i (nM) CBR b Ia (CH 2 ) 2 CH 3 H H H 81580 3293381 Ib (CH 2 ) 3 CH 3 H H H 116799 12% Ic CH 2 C](https://thumb-eu.123doks.com/thumbv2/123dokorg/7547784.108997/26.892.123.738.197.1001/table-binding-dates-phenylindol-ylglyoxylamides-derivatives-tspo-cbr.webp)