METODOLOGIE
3.1
La discarica controllata
Il termine “discarica controllata”, ampiamente utilizzato ogni qualvolta si parli di corretto stoccaggio definitivo dei rifiuti, non ha significato giuridico in quanto la legge nazionale italiana definisce solo la parola “discarica”. La necessità di specificare il termine in “discarica controllata”, indica che è esistito un periodo in cui lo smaltimento ultimo dei rifiuti avveniva in maniera non controllata, ovvero senza una normativa che contemplasse la protezione del suolo contiguo alle discariche, delle acque superficiali e sotterranee. Ciò rendeva le discariche forme diffuse di contaminazione a causa della dispersione del percolato o del biogas, prodotti dalla degradazione dei rifiuti. Lo sviluppo negli anni della normativa ha consentito il cambiamento della considerazione della discarica da semplice deposito di rifiuti a, appunto, discarica “controllata” in cui è posta l’attenzione sui tutti quei processi che avvengono all’interno della discarica e che possono essere possibili motivi di inquinamento.
La normativa di riferimento, in materia di discariche, è, in Italia, il D.L 36/2003 che definisce le caratteristiche tecniche che devono essere soddisfatte affinché una discarica possa essere considerata “controllata”. Dà disposizioni costruttive e gestionali per le tre tipologie di discariche (per rifiuti inerti, pericolosi e non pericolosi) affinché queste
abbiano un impatto nullo od il minore possibile sulle matrici ambientali. Circa gli impianti per rifiuti pericolosi o non pericolosi, sono indicate disposizioni in materia di:
•Ubicazione: la discarica non deve essere posta in zone soggette a vincoli paesaggistici, in aree naturali protette, in zone di rispetto, in aree di salvaguardia, in aree a rischio sismico o vulcanico, in aree esondabili o alluvionabili, in corrispondenza di forme di carsismo superficiale o dove i processi geologici superficiali potrebbero comprometterne la stabilità. Inoltre deve essere esaminata la distanza dai centri abitati, la collocazione in zone di produzione di prodotti alimentari particolari, o la presenza di rilevanti beni storici, artistici, archeologici;
•Protezione delle matrici ambientali: la discarica deve disporre di un sistema di regimazione e convogliamento delle acque superficiali, di impermeabilizzazione del fondo e delle sponde, di un impianto di raccolta e gestione del percolato, di un impianto di captazione e gestione del gas di discarica e di un sistema di copertura superficiale finale;
•Controllo delle acque e gestione del percolato: in relazione alle condizioni meteorologiche, devono essere adottate tecniche per limitare l’infiltrazione di acque meteoriche nella massa rifiuti. Il percolato e le acque devono essere trattati in impianti tecnicamente idonei al fine di garantire lo scarico nel rispetto dei limiti previsti dalla normativa vigente in materia;
•Protezione del terreno e delle acque: la protezione del suolo, delle acque sotterranee e delle superficiali, deve essere realizzata sia durante la fase operativa, mediante la combinazione della barriera geologica con un rivestimento impermeabile artificiale (sia per il fondo che per le sponde della discarica) e mediante un sistema di drenaggio del percolato, sia durante la fase post-operativa, attraverso anche la copertura della parte superiore;
•Controllo dei gas: le discariche che accettano rifiuti biodegradabili devono essere dotati di impianti per l’estrazione dei gas che garantiscano la massima efficienza di captazione e il conseguente utilizzo energetico. La gestione deve essere condotta in modo tale da ridurre al minimo il rischio per l’ambiente e per la salute umana; l’obiettivo è di non far percepire la presenza della discarica al di fuori di una ristretta fascia di rispetto;
•Disturbi e rischi: il gestore deve attuare misure idonee a ridurre al minimo i disturbi e i rischi provenienti dalla discarica e causati dall’emissione di odori, dalla produzione di polveri ed aerosol, dal rumore o dal traffico indotto o dalla presenza di uccelli, parassiti ed insetti;
•Stabilità: nella fase di caratterizzazione è necessario accertarsi che il substrato geologico non sia suscettibile a cedimenti tali da danneggiare i sistemi di protezione ambientale della discarica.
In figura 3.1 è mostrato come appare una discarica rispondente alla normativa.
Figura 3.1: Schema semplificato di una discarica controllata.
La discarica può essere considerata come un reattore biologico nel quale si hanno come flussi in ingresso, i rifiuti accumulati giornalmente, il materiale di copertura dei rifiuti e le acque, meteoriche, di infiltrazione sotterranea e di scorrimento, mentre come flussi in uscita, il percolato ed il biogas. Il primo è costituito da un complesso di prodotti liquidi derivati dalla lisciviazione dei rifiuti da parte delle acque in ingresso, mentre il secondo è un gas che si forma per decomposizione anaerobica dei rifiuti, ad opera di specifici microrganismi, e composto principalmente da CH4 e CO2 [Mora-Naranjo et al., 2004].
I flussi in ingresso ed i processi che avvengono all’interno del reattore influenzano le caratteristiche qualitative e quantitative di percolato e biogas che, a loro volta,
determinano l’entità degli impatti delle discariche sulle matrici ambientali. Pertanto, la minimizzazione degli impatti richiede il controllo dei due flussi e la conoscenza dei processi che avvengono all’interno del reattore. Per comprendere al meglio tali processi, è fondamentale riuscire ad interpretare e distinguere le diverse fasi in cui la decomposizione stessa si esplica, nonostante l’ecosistema che si crea all’interno del corpo rifiuti presenti caratteristiche molto eterogenee.
Sebbene la prima fase di degradazione del rifiuto avvenga in ambiente aerobico, è la digestione anaerobica che domina il processo sia nella durata, sia per il ruolo fondamentale che esercita nella stabilizzazione della matrice organica del rifiuto. Ancor prima che la cella della discarica sia completa, il rifiuto solido umido riceve, tramite l’apporto di idrometeore o la deposizione delle sostanze aerodisperse, svariati inoculi di batteri, attinomiceti e funghi. Questi organismi si sviluppano con una velocità di reazione che dipende dalle condizioni ambientali già presenti nella discarica, in particolare dal tasso di umidità, dalla temperatura, dallo stato nutrizionale, dal pH, e dalla massa volumetrica del materiale presente [Shekdar, 1997].
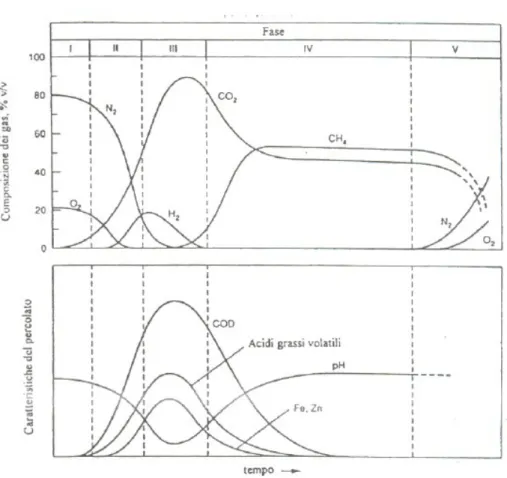
Numerosi studi sulle discariche hanno dimostrato che il processo di stabilizzazione dei rifiuti avviene attraverso fasi sequenziali e distinte che influenzano le caratteristiche quali-quantitative di biogas e percolato. Come mostrato in figura 3.2, il processo di degradazione dei rifiuti è sintetizzabile in cinque stadi [Farquhar e Rovers, 1973; Vallini et al., 1987; Christensen e Kjeldsen, 1989; Bogner et al., 1996; Rajedra D. Vajdya, 2002]:
Figura 3.2: Parametri che caratterizzano il biogas e il percolato nelle varie fasi del processo di degradazione. [Farquhar e Rovers, 1973; Christensen e Kjeldsen, 1989]
1. Stadio aerobico:
Si tratta di una fase di degrado iniziale che comincia nel momento in cui i rifiuti sono stoccati in discarica. La fase gassosa della cella rifiuti è costituita prevalentemente da azoto e ossigeno, quest’ultimo da inizio ai processi microbici aerobici degradando le proteine prima ad aminoacidi, quindi ad anidride carbonica ed acqua (i principali prodotti di questo stadio), nitrati e solfati. Segue un periodo di acclimatazione fino a quando non si sviluppa umidità sufficiente in grado di supportare le altre comunità batteriche. Durante questa prima fase, i quantitativi di percolato prodotto sono minimi ed il processo, poiché instabile, tende ad assorbire i liquidi presenti.
2. Stadio transitorio:
Questa fase è caratterizzata dal passaggio da condizioni aerobiche ad anaerobiche in quanto è consumato tutto l’ossigeno formandosi un ambiente via via più riducente. Si riscontrano, quindi, basse concentrazioni di nitrati e solfati che sono ridotti a solfuri metallici di ferro, manganese od altri metalli pesanti in quanto utilizzati come fonte di
ossigeno. I carboidrati si convertono a anidride carbonica ed acqua ed i grassi, attraverso la formazione intermedia di acidi volatili, s’idrolizzano ad acidi grassi e glicerolo. Pertanto, il risultato dell’idrolisi (reazione 1) è la solubilizzazione dei materiali in zuccheri, alcoli e lunghe catene di acidi grassi riducendo le dimensioni delle molecole organiche, e consentendo, di fatto, il trasporto attraverso le membrane cellulari dei microbi.
n(C6H10O5) +nH2O → nC6H12O6 (1)
La cellulosa, che costituisce la parte preponderante della frazione organica dei rifiuti, è degradata a glucosio, che è successivamente utilizzato dai batteri e convertito in CO2 e
H2O.
L’aumento progressivo della pressione parziale dell’anidride carbonica, che si dissolve in acqua formando un acido debole (reazione 2), oltre a diminuire il pH, può comportare la soluzione di altre sostanze minerali
CO2(g)+H2O → HCO3- + H+ (2)
Ne consegue che il percolato che si forma in questa fase di transizione, è caratterizzato da un abbassamento del pH e da un incremento del contenuto di COD, dovuto anche alla presenza delle sostanze organiche parzialmente degradate.
3. Stadio anaerobico non metanigeno o fase acida instabile:
Consumato tutto l’ossigeno, cominciano i processi biodegradativi anaerobici. Una gran varietà di prodotti può formarsi dal substrato organico di partenza che è in genere costituito da acidi grassi, zuccheri ed amminoacidi. Dal glucosio si possono formare gli acidi organici volatili come l’acido butirrico, propionico e acetico (reazioni 3, 4 e 5), per questo la fase viene anche detta di “acidogenesi”:
C6H12O6→ CH3(CH2)2COOH + 2H2 + 2CO2 (3)
C6H12O6 + 2H2→2CH3CH2COOH + 2H2O (4)
C6H12O6 + 2H2O → 2CH3COOH + 4H2 + 2CO2 (5)
Questi acidi e l’anidride carbonica disciolta, la cui formazione continua ad aumentare, accentuano le proprietà acide del percolato, il cui pH è generalmente compreso tra 5,5 e 6,5; è in questa fase, infatti, che il percolato presenta la concentrazione maggiore in metalli pesanti. Lo stesso avviene per il contenuto di COD e di acidi organici volatili.
In questo stadio ha inizio il processo di decomposizione anaerobica metanigena. Durante la fase iniziale (stadio metanigeno instabile) si assiste alla degradazione, da parte dei batteri acetogenici, delle lunghe catene di acidi grassi volatili ad acido acetico (per questo viene anche detta fase di “acetogenesi”):
CH3(CH2)2COOH + 2H2O → 2CH3COOH + 2H2 (6)
CH3CH2COOH + 2H2O → CH3COOH + 3H2O + CO2 (7)
Come conseguenza al consumo degli acidi organici, diminuisce la concentrazione di COD mentre il pH aumenta fino ad avvicinarsi alla neutralità. Questo comportamento comporta una riduzione dell’aggressività chimica del percolato ed una diminuzione delle concentrazioni dei composti inorganici: i metalli vengono, infatti, rimossi dal percolato attraverso la formazione di complessi, attraverso la precipitazione e, quindi, il trasporto in fase solida.
Il processo di degradazione anaerobica prosegue grazie batteri metanigeni acetofili che, degradando l’acetato, determinano una crescita progressiva della percentuale di metano; parallelamente si assiste ad una diminuzione della pressione parziale dell’anidride carbonica dovuta alla riduzione biogenica della CO2 da parte dai batteri metanigeni
idrogenofili. Questo avviene finché non si raggiunge l’equilibrio tra le frazioni volumetriche di CO2 e CH4 (fase metanigena stabile). Le reazioni 8 e 9 sintetizzano le
funzioni esplicate dai batteria metanigeni acetofili ed idrogenofili [Vallini et al., 1987]: CH3COOH → CH4 + CO2 (8)
CO2 + 4H2→ CH4 + H2O (9)
La percentuale di metano è piuttosto variabile, tuttavia compresa in un range del 45-65%.
5. Stadio di maturazione:
E’ lo stadio finale della stabilizzazione dei rifiuti in cui la quantità di nutrienti e il substrato disponibile alle famiglie batteriche iniziano a decrescere, con conseguente diminuzione dell’attività biologica. Anche la produzione di biogas decresce drammaticamente permettendo ai gas atmosferici di “entrare” nel corpo rifiuti; infatti, possono nuovamente presentarsi specie ossidate. La degradazione lenta della frazione organica più resistente può continuare con la produzione di sostanze humiche.
Dai processi di degradazione dei rifiuti appena sintetizzati, emerge che, a differenza del metabolismo aerobico, durante il quale la conversione della materia organica è quasi
sempre portata a termine da un’unica specie di batteri, il metabolismo anaerobico richiede diversi tipi di popolazioni batteriche, ciascuna delle quali ossida parzialmente una determinata classe di composti.
A seconda dello stadio di degradazione in cui si trova il rifiuto,
qualità e quantità di percolato e biogas subiscono variazioni rilevanti.
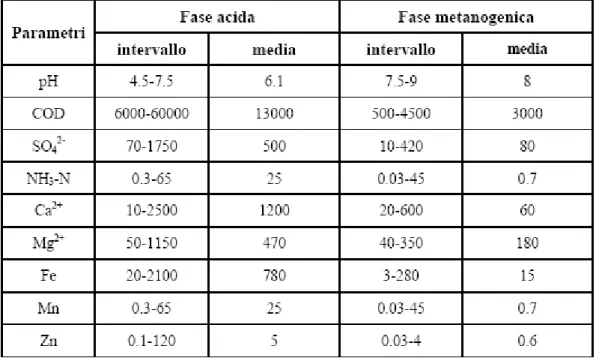
Oltre alla composizione merceologica, alle caratteristiche chimico-fisiche dei rifiuti ed alla loro tipologia di messa in dimora, i fattori che influenzano maggiormente la composizione del percolato sono il bilancio idrico che ne ha condotto alla formazione, il tipo di gestione dell’impianto e, appunto, l’età della discarica che determina il grado di stabilizzazione della sostanza organica [Ehrig, 1989b]. Per questo motivo, il range dei valori con cui si presentano i vari elementi chimici che lo costituiscono, è molo ampio e varia per le differenti discariche (tabella 3-1).
Tabella 3-1. Differenziazione dei campi di oscillazione dei parametri qualitativi del percolato tra fase di fermentazione acida e fase metanigena. Dati espressi in mg/L eccetto il pH [Ehrig, 1983, 1989; Andreottola et al., 1990].
In generale è possibile affermare che l’ossidazione della materia organica nella fase iniziale, che porta alla formazione di CO2, crea un ambiente acido nel mezzo acquoso,
un abbassamento del pH e la formazione di acidi organici [Christensen et al., 1994]. Tali reazioni provocano la solubilizzazione di sostanze inorganiche come i sali di Mg,
Fe, Mn, Zn e Cl [Christensen et al., 2000, 2001]. Dà ciò deriva un aumento delle concentrazioni dei cationi, della conducibilità, dell’alcalinità e della durezza totale. La concentrazione dell’azoto ammoniacale aumenta nei primi anni di attività e, successivamente, si mantiene costante su valori alti; il contrario accade per l’ossigeno disciolto [Ehrig, 1989].
Anche le variazioni nella qualità e nella composizione del biogas sono determinate da una molteplicità di fattori, biotici e abiotici [Andreottola e Cossu, 1988]. Determinanti risultano le caratteristiche ambientali quali precipitazioni, temperatura e umidità dell’aria, ventosità ed insolazione, e le caratteristiche dei rifiuti tra cui la composizione merceologica, la granulometria, la densità, gli eventuali pretrattamenti e l’umidità. Anche le modalità di gestione della discarica influenzano la qualità del biogas come le tecniche di estrazione, le modalità di deposito dei rifiuti, la geometria della discarica ed i materiali di copertura intermedia [Damiani e Gandolla, 1992]. Come si evince dal grafico in figura 3.2, il biogas consiste inizialmente in una miscela gassosa composta in prevalenza da CO2 e H2, prodotti nella fase di idrolisi ed acidogenesi (equazioni 2, 3, 4 e
5), una volta raggiunta la fase metanigena, i due prodotti predominanti diventano il CH4
e la CO2 (equazione 8 e 9).
La tabella 3-2 riporta la composizione del biogas durante la fase stabile della metanogenesi.
Componente Formula A B
metano CH4 0-85% vol 45-65% vol
anidride carbonica CO2 0-85% vol 65-45% vol
monossido di carbonio CO 2.8% vol
idrogeno H2 0-30 ppm 0% vol (20-30%)
ossigeno O2 0-31.6 ppm 0% vol (max 20%)
azoto N2 0-82.5 ppm 10-0% vol (max 80%)
idrogeno solforato H2S 0-70 ppm tracce
acqua H2O 2-5% vol
ammoniaca NH3 0-0.35 ppm
Tabella 3-2: Componenti principali del biogas in discariche controllate durante la fase stabile di metanogenesi. Tra parentesi sono indicati i valori da considerarsi anomali [A: Perin (1984); B: Ham e Barlaz (1987)].
Oltre a questi componenti principali, esiste una vasta gamma di composti in tracce, anche nocivi e maleodoranti, tra cui idrocarburi alifatici e aromatici, composti clorurati del carbonio, aldeidi alifatiche e aromatiche, chetoni, acidi grassi, composti organici dello zolfo e dell’azoto e vapori metallici (mercurio) [Young e Parker,1983].
3.2
Il monitoraggio ambientale
Il monitoraggio, nel suo significato più ampio, può essere definito come un sistema di controllo edaccertamento continuo di una determinata situazione o dell’avanzamento di una pianificazione, mediante la raccolta puntuale e sistematica di dati. Pertanto consiste in un processo in continua evoluzione sia per quanto riguarda le conoscenze scientifiche richieste sia, nel monitoraggio ambientale in particolare, per quanto riguarda l’adeguamento alle politiche di gestione in un’ottica di sviluppo sostenibile.
Il monitoraggio della contaminazione di un qualsiasi sito inquinato o sorgente di inquinamento, deve rispondere ai seguenti obiettivi:
•caratterizzazione dei fenomeni di inquinamento;
•acquisizione delle informazioni necessarie alle fasi decisionali per gestire, tutelare e risanare le risorse coinvolte;
•verificare l’efficacia delle azioni già intraprese, confrontando la situazione attuale dei livelli di inquinamento con il trend degli stessi verificatosi nel corso degli anni. Nel caso di una discarica, gli impatti sull’ambiente che possono verificarsi sono molteplici e derivano soprattutto dalla migrazione del percolato e del biogas, che incidono sulla qualità delle acque e dell’aria circostanti con conseguenti pericoli per la salute [El-Fadel et al., 1997]. Se sulla matrice acqua può insistere solo il percolato, il cui sversamento può essere causato da una rottura delle opere di contenimento, l’aria può essere interessata da una pluralità di forme di emissione, sia di tipo convogliato che diffuso. Le emissioni convogliate provengono dalla combustione del biogas nei camini o nelle torce, si parla quindi di emissioni puntuali, mentre le emissioni diffuse sono dovute in gran parte al rilascio dalla superficie della discarica di biogas non captato e, in maniera minore, ai gas di scarico degli automezzi, alla dispersione dei materiali leggeri sollevati dal vento, alle emissioni di polveri e di sostanze odorigene.
Per tali motivi, la valutazione dell’impatto di una discarica consiste principalmente nel monitoraggio della qualità delle acque e dell’aria e si svolge in 4 fasi:
1.Pianificazione della campagna di monitoraggio , che è funzione degli obiettivi, della caratteristiche della zona da monitorare, del tempo di campionamento valutato, anche in base alla definizione numerica della rappresentatività chimica e statistica dei parametri da determinare;
2.Campionamento , che deve avvenire, per quanto possibile, secondo quanto stabilito in fase di pianificazione visto che è un atto unico e non ripetibile, durante il quale si deve evitare di alterare il campione sia nell’atto del campionamento, che nel trasporto e conservazione, e durante il quale, si prende nota di tutti i dati tecnici dei punti di campionamento e delle eventuali anomalie che si presentano;
3.Analisi dei campioni e determinazione degli analiti , eseguita sia in campo che in laboratorio attraverso la misura strumentale. A differenza del campionamento, la misura eseguita in laboratorio può, in molti casi, essere ripetuta;
4.Trattamento statistico e valutazione dei dati , che ha come obiettivo primario quello di trarre tutta l’informazione possibile dai dati ottenuti che per loro natura sono affetti da una certa incertezza (valutata ad es. tramite la deviazione standard) ed hanno un certo intervallo di validità ritenuta tale (intervallo di confidenza). Inoltre il trattamento statistico dei dati è utile per integrare parzialmente i vuoti spaziali e temporali del campionamento e contribuisce, insieme ad un’esperta valutazione, a distinguere eventuali errori umani e/o strumentali dalle reali anomalie.
La pianificazione della campagna di monitoraggio prevede la scelta dei parametri da analizzare per la caratterizzazione delle matrici coinvolte nell’eventuale inquinamento. La scelta di tali parametri deve avvenire in funzione del contesto in cui si trova la discarica, prediligendo i composti che in quella precisa condizione fungono da traccianti della contaminazione. In merito alla questione, come già affrontato nel paragrafo 1.4, la normativa non si dimostra un utile guida fornendo solo indicazioni sommarie, addirittura a volte non idonee, circa i parametri da monitorare e le procedure da applicare. Tuttavia, esaminando la letteratura scientifica sull’argomento e sulla base dell’esperienza maturata nel tempo dall’IGG-CNR nel monitoraggio delle discariche, è stato possibile mettere a punto una procedura in grado di far fronte a tali mancanze e lacune nella normativa.
Il primo passo consiste sempre nella valutazione complessiva dell’assetto geologico ed idrogeologico locale seguito dalla caratterizzazione chimica delle acque, del percolato, del biogas e dell’aria. La scelta dei punti di campionamento risulta un’operazione determinante al fine della buona riuscita del monitoraggio.
Per quanto concerne le acque, i punti di prelievo delle acque sotterranee devono essere definiti sulla base della caratterizzazione geologica ed idrogeologica dell’area, del modello concettuale del sito e delle caratteristiche degli acquiferi che si intende campionare (ad esempio superficie piezometrica, permeabilità, direzione prevalente del flusso, ecc..); in questo modo sarà possibile poter caratterizzare univocamente l’influenza del sito sulle caratteristiche complessive degli acquiferi in esame e la mobilità degli inquinati nelle acque sotterranee. I punti d’acqua devono interessare l’area interna ed esterna alla discarica così da verificare le caratteristiche delle acqua di falda “in ingresso” ed “in uscita”. Pertanto vanno individuati piezometri a monte ed a valle del sito (in senso idrogeologico) per ciascun acquifero considerato, comprendenti un “bianco” ovvero almeno un punto d’acqua distante abbastanza dal sito di smaltimento, che costituirà il valore di riferimento delle acque sotterranee. Anche per i corsi d’acqua superficiali è necessario caratterizzare la situazione chimica e ambientale a monte del sito, nel tratto mediano ed a valle, lungo il senso di scorrimento del corpo idrico, in modo da distinguere gli effetti derivanti dalla presenza di inquinamento preesistente da quello, eventuale, dell’impianto in esame.
In merito al monitoraggio dell’aria, la scelta dei punti di prelievo avviene in funzione della topografia, dell’estensione dell’area da monitorare, delle direzioni principali del vento od in funzione della presenza di recettori sensibili, come possono essere i centri abitati. Anche in questo caso è necessario individuare una stazione di campionamento che funga da bianco, usualmente posta molto lontano dalla discarica, così da caratterizzare il fondo ambientale.
La procedura messa appunto dall’IGG, frutto dell’esperienza maturata nel monitoraggio dei siti di smaltimento italiani, prevede che a seguito della caratterizzazione chimica del percolato, delle acque e del biogas, si provveda anche alla caratterizzazione isotopica. Come affronterò in dettaglio nel paragrafo 3.2.2.1 e successivi, le tecniche isotopiche rappresentano ad oggi uno strumento indispensabile nel monitoraggio ambientale in quanto permettono non solo di integrare le informazioni provenienti dalle analisi chimiche, ma forniscono anche informazioni altrimenti non ottenibili sull’origine ed i movimenti dei fluidi presenti e sono in grado di discriminare, spesso in maniera molto attendibile presenza ed origine di eventuali fenomeni di contaminazione.
3.2.1 Il monitoraggio chimico
3.2.1.1 Acque e percolato
Il monitoraggio chimico delle acque consiste nelle indagini idrogeochimiche delle acque sotterranee e superficiali, in corrispondenza delle sorgenti, dei corsi d’acqua e dei pozzi limitrofi all’area oggetto dello studio, presso i piezometri interni ed esterni alla discarica e, naturalmente, nella caratterizzazione chimica del percolato.
Come già affrontato nel paragrafo 1.4, al fine della caratterizzazione chimica delle acque, la normativa individua alcuni parametri da analizzare con una frequenza minima semestrale (detti “fondamentali”); questi sono: cloruri, solfati, metalli (Fe e Mn) e azoto ammoniacale, nitroso e nitrico. Al fine di un “monitoraggio significativo”, il decreto dispone che siano analizzati almeno una volta l’anno anche altri parametri (detti “non fondamentali”) che sono: BOD, TOC, Ca, Na, K, fluoruri, IPA, metalli, composti organo alogenati, fenoli, pesticidi, solventi organici aromatici, azotati e clorurati.
Da esperienze dirette, in primis il caso di studio che presenterò nel paragrafo 4.3, emerge che la scelta di tali parametri risulta a volte inefficiente per individuare la contaminazione da percolato; questo perché a monte della scelta dei parametri indicatori della contaminazione, è necessario considerare tutti i vari fattori che partecipano alla definizione del chimismo specifico delle acque nella zona studiata.
In generale, la composizione chimica di un’acqua sotterranea è condizionata in minima parte dal chimismo delle precipitazioni atmosferiche, e in larga parte dall’interazione della roccia con il suolo e l’acquifero, che ne determinano l’appartenenza ad una particolare facies idrochimica. In zone antropizzate è però anche largamente condizionata dalla presenza di sorgenti di inquinamento derivanti da attività agricole, industriali, insediamenti urbani, presenza di strade ed altro. Per caratterizzare la composizione chimica di base di un’acqua, i parametri necessari (o “fondamentali” come scriverebbe la normativa) che devono essere considerati sono: cloruri, solfati, bicarbonati, sodio, calcio e magnesio ai quali generalmente si aggiungono nitrati e potassio. Tali analisi chimiche forniscono informazioni quantitative e qualitative circa la presenza di un composto disciolto nell’acqua e, poiché la composizione di un’acqua
cambia in funzione delle sue reazioni con l’ambiente, il suo chimismo consente anche di avere informazioni circa la sua storia ed origine.
Nel caso in cui si voglia, invece, valutare l’eventuale sversamento di percolato ed il miscelamento di questo con i sistemi idrici, oltre agli elementi sopra citati, è necessario esaminare gli elementi che più lo caratterizzano. Tra questi la scelta ricade su: nitriti, ammoniaca, COD e metalli pesanti (Fe, Mn, Cu, Zn, Ni, Cr, Pb, Cd, Al, B, As, Hg). Tali elementi fungono da traccianti della contaminazione in quanto, normalmente, sono presenti nel percolato in concentrazioni elevate o, anche se in tracce, con concentrazioni significativamente più alte del fondo naturale [Christensen et al., 1994, 1998, 2001; Siegel et al., 1990; Robinson, 1995, 2007]. Chiaramente, per poter considerare alcune sostanze chimiche come traccianti senza incappare in grossolani errori, occorre che il fondo naturale sia notevolmente diverso rispetto alle concentrazioni rinvenute nel percolato. Infatti una particolare composizione delle rocce dell’area o, ad esempio, l’utilizzo di fertilizzanti o la presenza di terreni adibiti al pascolo, possono trarre in inganno determinando, rispettivamente, alte concentrazioni di cloruri o di nitrati o di ammoniaca e COD nelle acque [Leone e Doveri, 2006; Cervelli 2006, 2007; Tazioli et al., 2002; Doveri et al., 2008b]. In questi casi, quindi, non è conveniente, anzi è fuorviante servirsi solo dei parametri elencati in precedenza, ne tantomeno dei parametri (fondamentali e non) indicati dalla legge, ma è necessario fare ricorso ad altri traccianti come gli isotopi di cui parlerò in dettaglio nel paragrafo 3.2.2.1 e successivi.
Oltre alle concentrazione dei vari ioni, si fa comunemente riferimento anche ai parametri chimico-fisici come il pH, la conducibilità, l’alcalinità e la temperatura, in genere misurati direttamente in campagna. Anche questi parametri caratterizzano significativamente il percolato in quanto variano in funzione dello stadio di degradazione del rifiuto (figura 3.2 e tabella 3-1, paragrafo 3.1), ma essendo i più suscettibili alle variazioni delle condizioni ambientali, non possono essere utilizzati come validi indicatori di contaminazione.
Una volta concluse le analisi chimiche e chimico-fisiche si provvede all’interpretazione ed alla presentazione dei dati attraverso grafici specifici che consentono di classificare le acque sulla base delle dominanze ioniche, e di evidenziare fenomeni di tipo idrodinamico od eventuali mescolanze di acque e percolato.
Queste informazioni consentiranno di contribuire alla conoscenza dell’assetto idrogeologico, di individuare l’eventuale interazione tra percolato e le acque naturalmente presenti nell’area, di valutare l’efficienza delle opere di contenimento, nonché di affinare il monitoraggio stesso.
I dati chimici vengono successivamente integrati con le analisi isotopiche che forniscono informazioni aggiuntive su origine storia ed eventuale contaminazione dei corpi idrici e sui processi che hanno contribuito alla definizione del chimismo del percolato.
3.2.1.2 Aria e biogas
Come descritto nel paragrafo 3.2, le emissioni gassose di una discarica sono di due tipi: convogliate e diffuse; per entrambe il D.L 36/2003 prevede un monitoraggio che sia in grado di quantificare l’emissione diffusa e di individuare eventuali fughe di gas esterne al corpo della discarica. La stessa normativa stabilisce che venga determinata la composizione chimica del biogas e la quantificazione del biogas prodotto. Per ottemperare a tali richieste, però, non sono forniti protocolli standardizzati impegnando implicitamente gli enti di ricerca, o referenti per l’ambiente, a sviluppare procedure comuni e condivise a livello nazionale per far fronte a tali mancanze.
Per la determinazione della composizione chimica del biogas, si esegue il campionamento usualmente presso il collettore di captazione; una conoscenza più approfondita circa la chimica e, soprattutto, circa lo stadio di degradazione dei rifiuti che determina la qualità del biogas, si raggiunge analizzando il biogas prelevato direttamente dal corpo rifiuti presso aree che risultano “particolari” da una precedente mappatura dei flussi diffusi dal suolo. I campioni aggiuntivi possono essere prelevati a profondità variabili in zone caratterizzate, ad esempio, da un basso flusso (rifiuto fresco), da flusso intermedio (copertura provvisoria) o da un flusso elevato che può verificarsi in corrispondenza di fratture della copertura. Pertanto, la conoscenza della composizione chimica del biogas convogliato (prelevato dal collettore) e diffuso (prelevato dal corpo discarica), fornirà informazioni indispensabili al fine di ottimizzare l’efficienza dell’impianto di captazione del biogas [Lippo et al., 2008].
Per il monitoraggio delle emissioni, la normativa prevede di analizzare mensilmente parametri come CH4, CO2, O2 e altri parametri quali H2, H2S, polveri totali, NH3,
mercaptani e composti volatili in relazione alla composizione dei rifiuti. La scelta ricade su questi composti perché o costituiscono i macrocomponenti del biogas o sono sostanze odorigene o sono microinquinanti generati principalmente dalla combustione del biogas o dalle attività di trasporto e lavorazione del rifiuto [Young e Parker, 1983]. Al fine di un attento monitoraggio ambientale, è necessario provvedere anche alla determinazione di parametri quali CO, NOx, SOx e diossine che rappresentano i prodotti della combustione del biogas sui quali esistono limiti di emissione.
Anche nel caso del biogas, le analisi isotopiche consentiranno di ottenere informazioni aggiuntive chiarendo non solo il particolare processo di produzione del metano e della CO2 che caratterizzano i diversi campioni di biogas prelevati, ma anche l’intensità e la
localizzazione dei fenomeni di ossidazione che avvengono a ridosso della copertura [Barker e Fritz, 1981; Raco et al., 2005].
In merito alla quantificazione della biogas prodotto, esistono diversi modelli numerici che si basano sulla qualità dei rifiuti e sulla cinetica di degradazione [Findikakis e Leckie, 1979; Findikakis et al., 1987; El-Fadel et al., 1988; Andreottola e Cossu, 1988; Manna et al., 1999; Nastev, et al., 2001; Hashemi, et al., 2002]; la normativa, pur stabilendo l’obbligo di quantificare la produzione di biogas, non specifica quale sia il modello di produzione da utilizzare impedendo, così, il confronto tra dati provenienti da diversi siti e la validazione degli stessi.
La stessa problematica si riscontra anche nella stima dell’emissione diffusa. L’IGG si è mosso in tal senso mettendo a punto una metodologia che impiega il metodo della camera di accumulo, di cui parlerò in dettaglio nel paragrafo 3.2.1.2.1, per eseguire misure puntuali del flusso diffuso, all’interfaccia aria-suolo, di CO2 e CH4.
L’elaborazione dei dati prevede l’utilizzo di tecniche derivate dalla statistica classica e dalla più moderna geostatistica per la stima del flusso diffuso e per la costruzione delle mappe di isoflusso con relativa mappa dell’errore. Le due tecniche verranno descritte nel paragrafo 5.4 quando applicate per stimare il flusso diffuso della discarica di Komotini oggetto del presente studio.
Tra gli obiettivi del monitoraggio descritti nel paragrafo 3.2, è menzionato infatti anche quello di acquisire informazioni necessarie alle fasi decisionali per gestire, tutelare e
risanare le risorse coinvolte, oltre a quello di verificare l’efficacia delle azioni già intraprese. Pertanto, disporre delle mappe di isoflusso ottenute dall’elaborazione delle misure puntuali di CO2 e CH4, sommate ai campionamenti ed alle analisi chimiche ed
isotopiche del biogas, risulta estremamente utile, anzi indispensabile, per la pianificazione di ogni intervento riguardante la captazione del biogas e la copertura della discarica.
Queste operazioni, che non sono, purtroppo, richieste dalla normativa di riferimento, consentono di avere informazioni rilevanti ed aggiuntive sui processi che avvengono all’intero del reattore quale è la discarica, con conseguenti ricadute ambientali e gestionali [Lippo et al, 2008]. In particolare, contribuiscono ad ottenere il miglior recupero quantitativo possibile del combustibile, dove già esiste un sistema di captazione del biogas utilizzato, poi, a fini energetici, ed a ridurre l’impatto ambientale diminuendo l’emissione di gas-serra e dei gas nocivi da esso trasportati [Raco et al., 2008].
3.2.1.2.1 La tecnica della camera d’accumulo
Per quantificare l’emissione diffusa si può utilizzare il metodo della “soil gas survey”; tuttavia, dato che questa tecnica usa la legge di Fick, per correlare il gradiente di concentrazione al flusso è necessaria la conoscenza del coefficiente di diffusione in ogni punto di misura. Poiché la componente advettiva del flusso è rilevante, nelle discariche è preferibile utilizzare misure dirette di flusso [Cioni et al., 2002, 2003; Raco et al., 2006].
Il metodo della camera di accumulo, utilizzato nel presente studio per stimare emissione totale di biogas diffuso da una discarica partendo da misure puntuali di flusso, è stato impiegato nelle scienze agrarie fin dagli inizi degli anni 70 per misurare il flusso di CO2
dal suolo e quindi il tasso di respirazione [Witkamp, 1969; Kucera and Kirkham, 1971; Kanemasu et al., 1974; Parkinson, 1981]. Negli anni 90 la camera di accumulo è stata utilizzata per misurare flussi di N2O [Kising e Socolow, 1994] e per valutare l’output
totale di CO2 diffuso da aree geotermiche e vulcaniche [Tonani and Miele, 1991;
Il metodo statico della camera di accumulo è stato scelto rispetto ad altri [Trégourès et al., 1999] tenendo conto delle seguenti considerazioni:
· è in grado di fornire misure di flusso dai suoli a prescindere dalla conoscenza delle caratteristiche dei suoli stessi e dalla conoscenza del regime di flusso stesso. Pertanto, essa non richiede alcun coefficiente empirico (che tenga conto delle caratteristiche del suolo) per trasformare il gradiente di concentrazione misurato in flusso [Tonani e Miele, 1991];
· è molto più veloce di tutti gli altri metodi;
· la strumentazione risulta maneggevole e di facile utilizzo.
La strumentazione impiegata, messa a punto da IGG-CNR di Pisa, DST Università di Perugia e West Systems srl (Via Molise, 7 – z.ind. Gello – 56025, Pontedera (PI)), è stata ampiamente descritta e utilizzata in ambito vulcanologico [Chiodini et al., 1996, 1998].
L’attrezzatura specifica utilizzata in questo lavoro è costituita da 4 parti principali (Figura 3.3):
1. una camera alta 10 cm con una superficie di base di 560 cm2;
2. due strumenti IR non dispersivi; 3. un convertitore analogico-digitale; 4. un computer palmare.
Figura 3.3: La camera di accumulo [Raco et al., 2006; www.westsystems.com].
Lo strumento, di fatto, misura continuamente la concentrazione di CO2 e CH4 all’interno
di un recipiente (camera di accumulo) posto sul terreno, curando la perfetta adesione del bordo sul suolo. La camera è equipaggiata con una ventola per omogeneizzare i gas all’interno della stessa. Una piccola pompa a membrana provvede ad aspirare i gas dall’interno della camera: il gas viene fatto passare attraverso una piccola colonna contenente perclorato di magnesio (per eliminare l’umidità) e successivamente inviato alle celle degli spettrofotometri per poi essere di nuovo convogliato nella camera di accumulo. I valori di concentrazione misurati dagli spettrofotometri sono acquisiti dal convertitore analogico-digitale dotato di interfaccia seriale e quindi inviati al computer che crea una rappresentazione grafica della concentrazione di CO2 (e CH4) in funzione
del tempo. Il software permette di calcolare, direttamente sul terreno il valore della derivata della funzione nel suo tratto iniziale, cioè il flusso.
L’influenza delle variazioni dei parametri metereologici è ampiamente documentata sia per ciò che concerne le misure di concentrazione, sia per quelle di flusso [Hinkle and Ryder, 1987, 1988; Hinkle, 1994; King and Minissale, 1994; Pinoul e Baubron, 1996].
Le leggi fisiche che stanno alla base del trasporto di materia mostrano chiaramente che le variazioni di pressione atmosferica, temperatura del suolo e dell’aria, velocità del vento e umidità del suolo possono determinare importanti variazioni di flusso. Se il flusso è di tipo advettivo, ritenendo valida la legge di Darcy, una variazione di pressione atmosferica determina un cambiamento del gradiente di pressione; è quindi lecito attendersi un aumento di flusso quando la pressione atmosferica diminuisce. L’umidità del suolo modifica la permeabilità/porosità del terreno, per cui è ragionevole attendersi variazioni di flusso indotte, non solo dalle precipitazioni che producono comunque un certo carico idraulico, ma anche da variazioni di temperatura e/o umidità atmosferica che provocano processi di condensazione/evaporazione. Infine anche elevate velocità del vento possono influenzare le misure di flusso per effetto venturi.
La metodologia della camera di accumulo è risultata meno dipendente dalle condizioni atmosferiche rispetto ad altre quali ”airborne infrared thermometry”, “trace gas”, “micrometeorological”, “mass balance2”, “Eddy correlation”, come alcuni autori [Trégourès et al., 1999] hanno dimostrato confrontando sette diversi metodi per la misura del flusso.
Si cerca comunque di minimizzare l’influenza delle condizioni atmosferiche come la pioggia, l’umidità del suolo e dell’aria lavorando in condizioni di tempo stabile e secco. Ciò permette di poter effettuare la correzione per la temperatura dell’aria e, eseguendo le misure in condizioni di pressione atmosferica costante, non è necessario nemmeno apportarvi correzioni a quest’ultima.
3.2.2 Il monitoraggio isotopico
3.2.2.1 Breve introduzione sul ruolo degli isotopi nel monitoraggio ambientale
Fretwell's Law: “Warning! Stable isotope data may cause severe and contagious
stomach upset if taken alone. To prevent upsetting reviewers' stomachs and your own, take stable isotope data with a healthy dose of other hydrologic, geologic, and geochemical information. Then, you will find stable isotope data very beneficial."
Gli isotopi (dal greco isos = uguale, topos = posto) sono atomi dello stesso elemento chimico con diverso numero di neutroni, quindi masse diverse che comportano comportamenti chimico-fisici diversi e forze di legame diverse, proporzionalmente alla differenza di massa, nelle sostanze con abbondanze isotopiche differenti; si distinguono in isotopi radioattivi e stabili. Gli isotopi radioattivi (instabili), in natura ne esistono circa 1700, sono nuclidi che si disintegrano spontaneamente per formare altri isotopi emettendo particelle e/o energia; gli isotopi stabili, 270 sono quelli individuati, non decadono, perlomeno nella scala dei tempi dell’universo, ma possono anche essere prodotti ex novo durante il decadimento di quelli radioattivi.
La composizione isotopica degli elementi leggeri è normalmente riportata come valore δ (delta) ed espressa in parti per mille (‰), riferita ad uno standard di composizione nota o convenzionalmente convenuta:
δ (in ‰) = (Rx / Rs - 1) x 1000
dove R è il rapporto fra l'isotopo più pesante e quello più leggero (es. 18O/16O) e Rx e Rs
sono rispettivamente i rapporti del campione e dello standard.
Gli isotopi sono soggetti in natura a frazionamento isotopico: le proporzioni delle abbondanze relative dei vari isotopi stabili di uno stesso elemento non sono fisse, ma cambiano in seguito a processi fisici, chimici e biologici. Il frazionamento legato ai processi naturali è osservabile solo per gli elementi leggeri, fino alla massa atomica 40 circa, ed è fortemente dipendente dalla differenza relativa di massa, quindi massimo nel caso del rapporto 2H/1H.
Esistono due tipi principali di frazionamenti isotopici: frazionamenti all’equilibrio e frazionamenti cinetici [Kendall e McDonnell, 1998].
• Frazionamenti all’equilibrio: i processi di scambio all’equilibrio isotopico implicano una ridistribuzione degli isotopi di un elemento tra le varie specie o composti. Nelle condizioni di equilibrio isotopico, le velocità di reazione nei due sensi di ogni delle molecole caratterizzate da una specie isotopica sono uguali. Ciò non significa che la composizione isotopica dei due composti all’equilibrio sia identica, ma solo che i rapporti fra i differenti isotopi in ogni composto sono costanti a quella determinata temperatura.
Durante le reazioni di equilibrio, gli isotopi pesanti si accumulano preferenzialmente nelle specie o nei composti con il più alto stato di ossidazione.
Come “regola empirica”, fra fasi differenti dello stesso composto o specie differenti dello stesso elemento, più è denso il materiale, più tende ad essere arricchito in isotopi pesanti. Ad esempio, considerando gli stati fisici dell’acqua, si ha all’equilibrio che: δ18O
S > δ18OL > δ18OV
Durante i cambiamenti di fase, i rapporti tra isotopi pesanti e leggeri tra le molecole nelle due fasi cambia. Ad esempio, come condensa il vapor acqueo (un processo che può essere considerato all’equilibrio), gli isotopi dell’acqua più pesanti si arricchiscono nella fase liquida mentre gli isotopi leggeri rimangono nella fase vapore.
Il frazionamento all’equilibrio è fortemente dipendente dalla temperatura in maniera inversa: all’aumentare della temperatura le differenze isotopiche tra le due fasi tendono a diminuire. La separazione associata all’equilibrio durante una reazione di scambio isotopico tra due sostanze a e b, può essere espressa usando il fattore di frazionamento alfa (α):
α = Ra / Rb
dove Ra/b = il rapporto numerico di abbondanza fra le specie isotopiche considerate,
ad esempio 18O / 16O, rispettivamente per le fasi o nei composti a e b;
• Frazionamenti cinetici: i processi chimici, fisici e biologici possono essere considerati o come reazioni di equilibrio reversibili, o come reazioni cinetiche unidirezionali irreversibili. Nei sistemi al di fuori dell’equilibrio chimico ed isotopico, le velocità di reazioni nei due sensi non sono uguali, e le reazioni isotopiche possono, infatti, essere unidirezionali se i prodotti delle reazioni vengono fisicamente isolati dai reagenti. Tali velocità di reazioni sono anch’esse dipendenti dai rapporti delle masse degli isotopi e si parla di frazionamenti isotopici cinetici. I frazionamenti cinetici, a parità di condizioni di temperatura, sono generalmente più marcati di quelli all’equilibrio. In genere i legami tra gli isotopi leggeri si rompono più facilmente degli equivalenti legami degli isotopi pesanti, infatti gli isotopi leggeri reagiscono più velocemente e si accumulano nei prodotti causando un arricchimento in isotopi pesanti nei reagenti. E’ questo il caso del processo di metanogenesi, in cui il metano che si produrrà, sarà arricchito in isotopi leggeri (δ12C) rispetto al composto organico da cui si origina.
Negli studi ambientali, in particolare in quelli idrogeologici, gli isotopi principalmente utilizzati sono quelli della molecola d’acqua; essi si comportano come dei veri e propri traccianti in quanto i loro quantitativi non vengono alterati durante l’interazione acqua-roccia a bassa temperatura e, quindi, non dipendono dal tipo di acqua-roccia-acquifero. Poiché consistono in traccianti naturali, cioè intrinseci alla molecola d’acqua, sono utilizzati per ricostruire i percorsi seguiti dalle acque in acquifero, o per individuare l’interazione tra diversi circuiti o corpi idrici, per ottenere informazioni sulle condizioni idrodinamiche, ecc… [Doveri, 2006].
I frazionamenti del 2H e dell’18O che avvengono durante i processi di evaporazione e
condensazione, sono influenzati dalla temperatura; ciò determina la composizione isotopica delle precipitazioni [Gat e Gonfiantini, 1994; Gat et al., 1996]:
variazioni stagionali: le precipitazioni invernali hanno un contenuto medio più basso di isotopi pesanti rispetto alle precipitazioni estive;
effetto altitudine: le piogge originate ai piedi dei rilievi sono isotopicamente più pesanti delle piogge che cadono a quote maggiori;
effetto continentalità: le precipitazioni sulle zone interne di un continente sono isotopicamente più leggere rispetto a quelle che si verificano sulle zone litorali o sub-litorali;
effetto latitudine: la composizione isotopica delle precipitazioni ha contenuti minimi di isotopi pesanti su aree polari e contenuti massimi su aree tropicali.
Le concentrazioni di 2H e 18O nelle precipitazioni risultano, quindi, strettamente
correlate mediante la relazione 10 (figura 3.4) [Craig, 1961]:
Figura 3.4: La relazione tra δ18O e δ2H delle acque di precipitazione meteorica. I dati sono medie pesate
dei valori annuali delle precipitazioni monitorate nelle stazioni della rete globale dell’IAEA [Rozanski et
al., 1993].
Per definizione lo standard V-SMOW ha un δ18O ed un δ2H uguali a 0.
La linearità tra i contenuti di deuterio e di 18O consente di evidenziare alcuni processi di
“alterazione isotopica” delle acque meteoriche: la figura 3.5 mostra alcuni degli effetti che possono verificarsi in seguito a processi fisico-chimici che possono interessare la composizione isotopica delle acque.
Figura 3.5: Grafico della linea delle acque meteoriche che mostra agli effetti che possono verificarsi a seguito di alcuni processi chimico-fisici [Clark e Fritz, 1997 modificato da IAEA T.R.S, n° 228, 1983].
Per esempio l’evaporazione, che conduce ad un arricchimento degli isotopi pesanti nella fase liquida, determina uno spostamento diagonale al di sotto della linea delle acque meteoriche. Altri fenomeni, come l’interazione con le rocce di fluidi geotermici, interessano invece solo l’18O mentre, come affronterò nel dettaglio nel paragrafo 3.4.2,
reazioni come la metanogenesi coinvolgono solo il deuterio che subisce un forte arricchimento [Hackley et al., 1996].
A differenza del deuterio e dell’18O, il trizio è un isotopo radioattivo ed è prodotto
naturalmente dall’impatto di neutroni della radiazione cosmica con atomi di azoto presenti nell’alta atmosfera:
14
7N + 10n → 31H + 126C
L’isotopo è rapidamente ossidato a H3HO ed è incorporato nel ciclo idrologico; decade
radioattivamente con emissione di particelle β- (3H → 3He + e-) con un periodo di dimezzamento pari a 4500 giorni (circa 12.43 anni) [Unterweger et al., 1980; Doveri et al., 2008]. E’ espresso in concentrazioni assolute in Unità di Trizio UT (1 unità di trizio corrisponde a 1 atomo di 3H ogni 1018 atomi di H) [IAEA, 1983].
I quantitativi naturali di questo isotopo nelle acque (valori medi attuali annui compresi tra 3,5 e 5,5 UT, alle nostre latitudini [Doveri et al., 2005]) in passato sono stati fortemente alterati dall’uomo; in particolare i numerosi esperimenti termonucleari avvenuti in atmosfera tra gli anni 50 e 60, hanno comportato un forte incremento dei valori di trizio nelle acque meteoriche raggiungendo anche le migliaia di UT [Doveri et al., 2005]. Attualmente i contenuti di trizio in atmosfera, e quindi nelle acque meteoriche, sono tornati sui valori naturali.
Il fatto che il trizio sia radioattivo e che in passato i suoi contenuti nelle precipitazioni abbiano subito un forte incremento, consente di risalire all’età media delle acque sotterranee analizzate, ovvero ai tempi medi di permanenza delle stesse in acquifero; ciò può esser fatto con un semplice confronto tra i contenuti di trizio dell’acqua in studio e la curva dei contenuti di trizio registrati nelle precipitazioni degli anni passati, provvedendo ad attualizzare al momento dello studio quest’ultimi contenuti.
Oltre a costituire un parametro utile alla definizione delle caratteristiche idrodinamiche della circolazione, il trizio è uno dei migliori indicatori di contaminazione delle acque ad opera del percolato di discarica. L’argomento verrà affrontato in dettaglio nel paragrafo 3.2.2.3.
Oltre al deuterio, all’18O ed al trizio, nelle indagini ambientali vengono utilizzati anche
gli isotopi degli altri elementi maggiormente presenti nei sistemi idrologici, geologici e biologici, quali N, S e C: poiché sono elementi leggeri, la loro differenza di massa è relativamente elevata pertanto risultano misurabili i frazionamenti che avvengono durante i processi fisici, chimici e biologici.
Lo studio degli isotopi ambientali viene utilizzato come strumento di integrazione ai dati chimici e idrogeologici, ciò deriva dalla proprietà degli isotopi di essere traccianti naturali. Questa proprietà giustifica la differenza di filosofia sull’utilizzo dei dati isotopici rispetto a quelli chimici. Gli isotopi, infatti, si usano per determinare caratteristiche che la mera lettura delle concentrazioni chimiche non consente di apprezzare dando quest’ultime solamente un’indicazione dell’effettiva e quantitativa presenza di un determinato composto chimico disciolto nell’acqua [Tazioli, 2002]. Comunque, per evitare di violare la cosidetta legge di Fretwell, dobbiamo operare in modo che i dati isotopici delle acque siano usati contestualmente ai dati idrologici ed ai dati chimici degli elementi maggiori ed in traccia, così da procedere mediante approfondimenti successivi nel ricostruire i meccanismi geochimici ed idrologici che le caratterizzano. Infatti, una delle più potenti applicazioni delle misure isotopiche è il loro utilizzo nel discriminare e/o convalidare i modelli derivati dall’uso di altre tecniche [Kendall e McDonnell, 1998].
Gli isotopi di interesse nei problemi di inquinamento variano a seconda del tipo di sostanza che si considera inquinante per la zona in esame. In particolare:
• 34S viene utilizzato per lo studio degli effetti indotti dallo sversamento dei reflui
delle attività estrattive (metalli in primis) sulla qualità delle acque limitrofe, oppure nel tracciamento della deposizione del materiale atmosferico, nel tracciamento di fenomeni di contaminazione delle acque dovuto allo spargimento di fertilizzanti [Pilcher, 2005], nella descrizione di reazioni geochimiche o biologiche connesse alle pratiche di coltivazione, irrigazione o drenaggio dei terreni agricoli [Moncaster et al., 1999], nello studio della contaminazione da percolato [Van Breukelen et al., 2003]. In figura 3.6 sono rappresentati le concentrazioni in 34S nei vari composti;
Figura 3.6: Intervallo dei contenuti in 34S e dei composti di 34S in ambienti e materiali differenti. Lo
standard di riferimento è il CDT, meteorite di solfito di ferro dell’Arizona [Clark e Fritz, 1997 modificato da Krouse, 1980].
• 15N: viene utilizzato per indagare sull’origine dei composti azotati, come i nitrati
(NO3-) e l’ammonio (NH4+), distinguendo tra contaminazione da fertilizzanti
piuttosto che da liquame organico [Aravena et al., 1993; Wassenaar, 1995; McClelland et al., 1997; Andrews et al., 1998; Rogers, 1999]. Dai dati di letteratura si evince che per i nitrati disciolti nelle acque sotterranee valori di δ15N inferiori a
+8‰ rispetto allo standard N2 che corrisponde all’azoto atmosferico, sono tipici di
aree agricole, poiché il tenore di δ15N proveniente da fertilizzanti artificiali è
attorno a 0. Valori superiori sono, in genere, il risultato di nitrificazione dell’azoto contenuto in rifiuti animali o concimi o liquami [USGS, 2002]. Altri autori specificano che i rifiuti di origine animale hanno nitrati con valori di δ15N compresi
tra + 10‰ e +23‰, mentre i liquami organici tra +10‰ e +20‰ [Aggarwal et al., 1998]. L’δ15N nei nitrati viene studiato anche per indagare sugli sversamenti di
percolato di discarica nei sistemi idrici [North et al., 2006]. In figura 3.7 sono mostrate le concentrazioni in δ15N nei materiali naturali;
Figura 3.7: Range in 15N nei materiali naturali [Clark e Fritz, 1997].
• 13C: lo studio del δ13C nel carbonio inorganico disciolto (Dissolved Inorganic
Carbon, DIC) ovvero carbonati (CO3=), bicarbonati (HCO3-) ed anidride carbonica
(CO2) disciolti, è utilizzato per determinare l’origine fisica di tali composti. I valori
di δ13C nelle acque sotterranee si aggirano tra –15‰ e –10‰ rispetto allo standard
PDB (Pee Dee Belemnite, che corrisponde ad un carbonato biogenico marino dalla formazione Pee Dee nel South Carolina), mentre per le acque superficiali possono essere anche leggermente più elevati [Clark e Fritz, 1997]. Negli ambienti riducenti trova ampie applicazioni come, ad esempio, nel monitoraggio della contaminazione delle percolato di discarica in quanto l’attività batterica crea un intenso frazionamento isotopico, portando il δ13C a valori fortemente positivi (δ13C >
+15‰), per cui è possibile, una volta noto il fondo locale dell’ambiente, verificare l’esistenza o meno di inquinamento da percolato [Breukelen, 2003; Atekwana e Krishnamurthy, 2004; North et al., 2006; Van Doveri et al., 2008]. Un’ulteriore applicazione si ha nello studio dell’origine del metano e della CO2 del biogas di
discarica [Bogner, 1996; Hackley et al., 1996; Hornibrook et al, 1999; Fuganti, 2003; Raco et al., 2005] o per studiare i processi di ossidazione del metano sulla
copertura [De Visscher e De Pourcq, 2004; Mahieu, 2006]. In figura 3.8 è mostrata l’abbondanza del δ13C nei materiali naturali.
Figura 3.8: Intervallo dei valori in δ13C nei composti naturali [Clark e Fritz, 1997].
Le potenzialità delle tecniche isotopiche brevemente esposte, verranno affrontate con maggior dettaglio nei paragrafi successivi una volta applicate al monitoraggio di una discarica.
3.2.2.2. Gli isotopi ed il biogas
Nel reattore anaerobico quale è la discarica, avvengono una serie di reazioni irreversibili, mediate da batteri, che possono essere considerate reazioni cinetiche; tali reazioni consistono essenzialmente in reazioni redox e sono associate ad un ampio e riproducibile frazionamento isotopico [Clark e Fritz, 1997].
Durante la metanogenesi, il metano può essere prodotto o dalla fermentazione dell’acetato o dalla riduzione della CO2 [Barlaz et al., 1989]. Questi due processi,
entrambi supportati da batteri metanigeni, influenzano in maniera differente la composizione isotopica del metano e della CO2. L’ingresso iniziale di CO2
isotopicamente leggera (δ13C tra –10‰ e –20‰ [Clark e Fritz, 1997]) associata alla
ben presto surclassato dal costante input di CO2 isotopicamente pesante, associato alla
riduzione della CO2 e alla fermentazione dell’acetato (δ13C +1‰ ÷ +20‰ [Hackley et
al., 1997]). Infatti, durante la riduzione della CO2 i microbi utilizzano preferenzialmente
la CO2 che dispone del C più leggero per produrre CH4 arricchito in 12C, ciò fa si che la
CO2 residua diventi isotopicamente arricchita in 13C [Barker e Fritz, 1981].
Il miglior strumento per diagnosticare la provenienza del 13C appare essere il fattore di
frazionamento calcolato come:
Questo fattore di frazionamento è minore di 0,935 per il CH4 prodotto dalla riduzione
della CO2 mentre per la fermentazione dell’acetato è maggiore di 0,95 [Whiticar et al.,
1986; Lansdown et al., 1992]. Disponendo in un grafico la composizione in 13C del CH 4
del biogas contro quella della CO2, è possibile riconoscere anche le eventuali variazioni
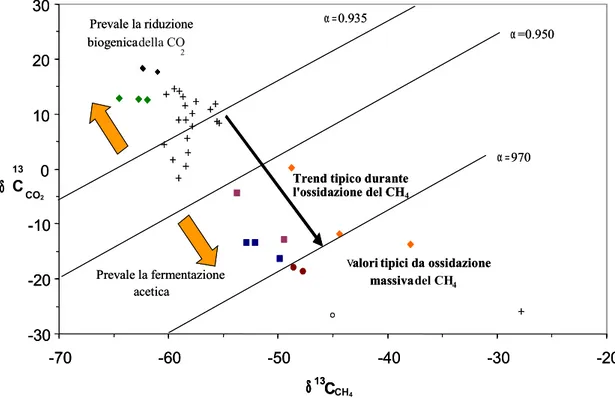
nella composizione isotopica del biogas, informandoci sulla fase in cui si trova la degradazione dei rifiuti (figura 3.9).
Figura 3.9: Composizione isotopica del C della CO2 e del CH4 coesistenti nelle emissioni dal suolo di
alcune discariche italiane. [Raco et al., 2005].
α = 13C/12C (CH4) 13 C/12C (CO2) α = 13C/12C (CH4) 13 C/12C (CO2) -30 -20 -10 0 10 20 30 -70 -60 -50 -40 -30 -20 δ13 CCH4 α =0.935 α=0.950
Trend tipico durante l'ossidazione del CH Prevale la fermentazione acetica Prevale la riduzione biogenicadella CO 2 α =970
Valoritipici da ossidazione massivadel CH4 -30 -20 -10 0 10 20 30 -70 -60 -50 -40 -30 -20 δ13 C δ 13 C CO2 α =0.935 α=0.950
Trend tipico durante l'ossidazione del CH4 Prevale la fermentazione acetica Prevale la riduzione biogenica α =970
Valoritipici da ossidazione massiva
Uno spostamento verso concentrazioni minori in 13C della CO
2 accompagnato da un
arricchimento in 13C del CH
4, può presentarsi in seguito a processi di ossidazione
batterica del metano [Barker e Fritz, 1981; Raco et al, 2005], come mostrato in figura 3.11.
Valori isotopici del carbonio sia nel CH4 che nella CO2 più elevati che in analoghi
processi fermentativi naturali sono comunque tipici dei gas da discariche e vengono attribuiti alla condizione termica del processo di fermentazione, più elevata di quella comune nei sistemi naturali [Clark e Fritz, 1997].
Anche il frazionamento del 2H può aiutare a caratterizzare la sorgente di metano infatti i
contenuti in 2H sono stabiliti durante la metanogenesi dai contenuti in 2H del substrato
organico e delle acque che partecipano alla reazione [Schoell, 1980]. 3.2.2.3 Gli isotopi ed il percolato
La composizione isotopica dei percolati si distingue nettamente da quella delle acque superficiali e sotterranee. Nell’ambiente per lo più anaerobico quale quello di una discarica, avvengono processi di scambio isotopico con i gas prodotti durante la fermentazione tra cui idrogeno e metano [Siegel et al., 1990; Hackley, 1996; Tazioli et al., 2002; Doveri, 2008].
Durante l’ossidazione del metano che genera acqua, avvengono frazionamenti isotopici; tali frazionamenti determinano un arricchimento in deuterio nell’acqua in neoformazione portando il percolato ad avere valori che si discostano verticalmente dalla linea delle acque meteoriche [Hackley et al., 1996; Fuganti et al., 2003]. Gli eventuali processi evaporativi che hanno caratterizzato le acque in ingresso alla discarica, determinano invece l’arricchimento in 18O. La risultante di questi due processi
fa sì che si osservi per il percolato uno spostamento diagonale al di sopra della retta delle acque meteoriche mondiali e del mediterraneo centrale (figura 3.10) [Hackley et al., 1996, Leone e Doveri, 2006; Doveri et al., 2008].
Figura 3.10: Disposizione del percolato sul grafico 18O-2H [Leone e Doveri, 2006].
La composizione isotopica del percolato, se confrontata con la composizione isotopica delle acque di falda circostanti la discarica, consente di stimare la provenienza dell’acqua che penetra nella discarica e, in presenza di un forte variazione del contenuto in deuterio, la quantità di percolato che penetra nella falda [Fuganti et al., 2003].
Come già accennato nel paragrafo 3.2.2.1, anche il trizio viene utilizzato per studiare la contaminazione delle acque sotterranee ad opera del percolato. Questo isotopo, infatti, una volta presente nelle vernici luminescenti è oggi fortemente utilizzato nei display od in altri oggetti a cristalli liquidi, che comunemente sono presenti nei rifiuti smaltiti in discarica, insieme ad altre fonti non bene identificate [Kisalu et al., 1991; Hicks et al., 2000]. L’alterazione e degradazione di questi materiali porta ad arricchire enormemente in trizio il percolato che, di conseguenza, è ben rintracciabile nei corpi idrici circostanti la discarica. Ad oggi, nella maggior parte dei casi, a seguito del naturale decadimento si riscontra un contento di trizio nelle acque sotterranee naturali inferiore al 10÷20 UT mentre nei percolati di discarica, i contenuti in trizio sono mediamente compresi tra 20 e 1500 UT [Rank et al., 1992; Calestani et al., 1999; Fuganti et al., 2003; Tazioli et al., 2004; Doveri et al, 2008], raggiungendo in alcuni casi valori anche di 10000 UT
[Doveri et al, 2008] (figura 3.11). Questa netta differenziazione consente di rilevare anche basse percentuali di mescolamento del percolato con le acque.
Figura 3.11: Abbondanza del trizio nelle piogge di diverse età, negli acquiferi profondi e superficiali e nei percolati. [Doveri et al., 2006, 2008].
Inoltre, il fatto che non subisca alterazioni durante i processi chimico-fisici e batterici, rendono questo isotopo il tracciante principe della contaminazione da percolato. Queste proprietà ci vengono in aiuto nei casi in cui i parametri chimici previsti dalla normativa non ci consentono di discriminare se la contaminazione sia attribuibile al percolato od ad un’altra causa naturale od antropica.
Nonostante non sia efficace come il trizio, anche il contenuto in 13C nel DIC delle acque
è utile per monitorare un’eventuale contaminazione da percolato [Hackley et al., 1996]. Nel percolato si riscontra generalmente un forte arricchimento del 13C nel DIC infatti
durante la degradazione anaerobica si produce CO2 arricchita in 13C contestualmente a
CH4 impoverito. L’aumento di alcalinità dettato dall’attiva metanogenesi, conduce il
percolato ad avere valori positivi in 13C del DIC che variano da +10‰ a +20‰
[Hackley et al., 1996; Fuganti et al., 2003]. Tali valori sono in netto contrasto con quelli riscontrati delle acque non contaminate che si aggirano tra -10‰ e - 20‰ [Clark e Fritz, 1997; Doveri et al, 2008].
Il 13C può dare indicazioni utili anche sullo stato di maturazione del rifiuto [Hackley et
al., 1996]. La CO2 che si forma nel corpo rifiuti, e conseguentemente i DIC da essa
0.1 1 10 100 1000 10000 100000 piogge anni “60 piogge attuali percolati falde prof./confin falde superf. U T
Contenuto tipico in trizio delle acque e del percolato
acque infiltrate negli anni “60, 70
0.1 1 10 100 1000 10000 100000 piogge anni “60 piogge attuali percolati falde prof./confin falde superf. U T
Contenuto tipico in trizio delle acque e del percolato
originati, saranno più o meno arricchiti in 13C in funzione della composizione isotopica
del 13C della materia organica di partenza [Hackley et al., 1996]. In particolare valori
che approssimano quelli del materiale organico di partenza, circa -15‰ ÷ -30‰ [Clark e Fritz, 1997], tendono a caratterizzare le fasi aerobiche iniziali e soprattutto terminali
δ13C, mentre valori più elevati corrispondono alla fase prettamente anaerobica del
degrado [Barker e Fritz, 1981].
In conclusione, grazie alle metodologie isotopiche, utilizzate da supporto ed integrazione alle indagini chimiche su acque, composti disciolti e gas nell’ambito del monitoraggio di una discarica di RSU, limitandosi ad i soli isotopi dell’idrogeno, carbonio ed ossigeno, si possono trarre informazioni circa: l’origine ed il comportamento delle acque nell’area (18O, 2H e 3H), movimenti delle acque interne
all’impianto (18O, 2H e 3H), perdite e contaminazioni verso l’esterno (2H, 3H, 13C),
determinazione dello stato di maturazione (2H e 13C) e caratteristiche delle emissioni dei


![Figura 3.3: La camera di accumulo [Raco et al., 2006; www.westsystems.com].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7307760.87963/19.892.198.718.154.607/figura-la-camera-accumulo-raco-www-westsystems-com.webp)


![Figura 3.7: Range in 15 N nei materiali naturali [Clark e Fritz, 1997].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7307760.87963/28.892.244.690.151.526/figura-range-n-materiali-naturali-clark-fritz.webp)
![Figura 3.8: Intervallo dei valori in δ 13 C nei composti naturali [ Clark e Fritz, 1997].](https://thumb-eu.123doks.com/thumbv2/123dokorg/7307760.87963/29.892.213.731.244.563/figura-intervallo-valori-δ-composti-naturali-clark-fritz.webp)