Solutions
10X MMR NaCl 10M KCl 200mM MgSO4 100mM CaCl2 200 mM HEPES 500mM EDTA 10mM Dejelling solution Tris- HCl (pH 8.8) 0.2 mM DTT 3.2 mM Bleaching solution H2O2 1%-3% SSC 0.5x Formamide 5% MEM salts 10X MOPS (pH 7.4) 100 mM EGTA 2 mM MgSO4 1 mM MEMFA 1X MEM salts 1X formaldehyde 3.7%Hybridization buffer (Niehrs’ protocol)
Formamide 50% SSC 5X Torula RNA 1 mg/ml Heparin 0,1 mg/ml Tween-20 0,1% CHAPS 0,1% EDTA pH 8.0 10 mM Boeringer Block reagent 10 mg/ml
APB
Tris- HCl pH 9.5 100mM
NaCl 100 mM Tetramisole (Sigma) 5mM
Tween-20 0,1%
Hybridization buffer (in situ on sections)
Formamide 50% Dextran-sulphate 10% Torula RNA 1 mg/ml Denhart’s 1X Salts 1X 10X Salts NaCl 114 g Tris HCl 14.04 gr 1 Tris base. 34 gr NaH2PO4. 2H2O 7.8 gr NaH2PO4 7.1 gr 10X MAB Maleic acid 100mM NaCl 150mM pH 7.5 MABT MAB 1X Tween-20 0,1%
Protocols and techniques
All basic molecular cloning protocols were applied, with slight modifications, according to (Sambrook and Russell, 2001).
Purification of plasmid DNA
Plasmid DNA was extracted from bacterial cells (E. coli, DH5α: F-, φ80dlacZ∆M15 supE44, λ- ∆(lacZYA-argF) U169, deoR, endA1, gyrA96, hsdR17(rk-, mk+), phoA, thi-1, recA1, relA1) by alkaline lysis and purified by column chromatography by Nucleobond AX-100 columns (Macherey-Nagel) following the manufacturer’s protocol. The plasmids were linearized and used as templates for antisense probe or capped mRNA transcription.
Plasmids
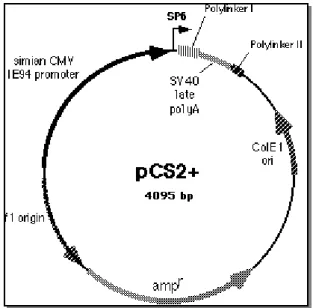
pCS2+ (Fig. 11) is a multi-purpose expression vector. It contains a strong viral
promoter (CMV IE94) for expression studies, followed by a polilinker and the poly A site from SV40 virus. SP6 and T7 promoters allow the transcription of sense or antisense RNA from cloned sequences; a second polylinker allows linearization for SP6 transcription. The plasmid is derived by the original pBluescript II KS+ (Rupp et al., 1994; Turner and Weintraub, 1994).
pCs2-Xrx1: the coding sequence of Xrx1 was cloned into pcs2+ in polylinker 1
between BamHI and XbaI sites, that were abolished. Plasimid is linearized with NotI for SP6 dependent capped mRNA synthesis and with HindIII for T7 polymerase dependent antisense probe transcription.
ephrinB-1: cloned in pBluescript KS, without the C-terminal aa. Plasimid is
linearized with XbaI for T3 dependent capped mRNA synthesis and with HindIII for T3 polymerase dependent antisense probe transcription.
pNPG152: contains an EcoRI fragment for an almost full-length nrp1 cDNA
(Richter et al., 1990). Plasimid is linearized with HindIII for T7 dependent capped mRNA synthesis and with NotI for T3 polymerase dependent antisense probe transcription.
EST clones for whole mount in situ hybridization analysis were obtained as cDNA clones from the NIBB consortium (http://xenopus.nibb.ac.jp/ ); the cDNAs were contained either in pBluescript SK-, between sites EcoRI and XhoI or in pCS105, between sites SalI and NotI (linearized with BamHI for antisense and transcribed with T3 polymerase).
Capped mRNA in vitro synthesis
Capped mRNAs were synthtized using the mMessage mMachineTM SP6 kit (AMBION), following the instructions provided by the manufacturer. After DNAse digestion to remove template DNA, the capped mRNAs were purified in phenol-chloroform and ethanol precipitated. The concentration of the RNA was estimated by agarose gel electrophoresis and spectrophotometry. Aliquots of 100ng were then stored at -80 °C.
Antisense labeled probes synthesis
Standard RNA syntheses from linearized plasmids using SP6, T7 or T3 RNA polymerases were carried out incorporating a digoxigenin conjugated ribonucleotide. In particular, transcription reactions were carried out in the presence of 1mM each of ATP, CTP and GTP, 350 µM UTP and 650 µM DIG-11 UTP for 2 hrs at 37 °C. After DNAase digestion to remove template DNA, the RNAs were purified in phenol-chloroform and ethanol precipitated. The
concentration of the RNA was estimated by agarose gel electrophoresis and spectrophotometry. Probes were then diluted in hybridisation mix at the final stock concentration of 10 µg/ml and stored at -20 °C.
Xenopus laevis embryos
In order to obtain embryos, Xenopus laevis females were pre-injected with 100 units of pregnant mare serum gonadotrophin (Folligon, Intervet) 4-11 days prior to egg collection, and with 800-1000 units of human chorionic gonadotrophin (Gonase HP 5000, Serono) the night before collection.
The next day, eggs were obtained by gently squeezing the frogs and then fertilized with testis homogenates. The embryos were cultured in 0.1X MMR. After half an hour from fertilization, jelly coats were removed by keeping the embryos for 5-10 minutes in dejelling solution. Embryos were staged according to Nieuwkoop and Faber (Nieuwkoop and Faber, 1967).
For whole-mount in situ hybridization procedure, staged embryos were fixed in MEMFA for 1 hour at room temperature, gradually dehydrated and stored in ethanol at -20 °C. For in situ hybridization on sections, embryos were fixed over-night in 4% paraformaldehyde in PBS at 4 C°, dehydrated in 20% Sucrose in PBS (Sigma) for 3-4 hours at room temperature, embedded in OCT Tissue-Tek (Tsuda et al.) and stored at -80C°.
Microinjections
For gain-of-function experiments, 50pg of wild-type Xrx1 mRNA were injected into both dorsal blastomeres of 8-cells stage embryos, using a Drummond ‘Nanoject’ apparatus. For loss-of-function experiments, 10pg of MoXrx1 morpholino antisense oligonucleotide were injeceted into both dorsal blastomeres at 4-cells stage embryos. Embryos were injected in 0.1X MMR and 4% Ficoll-400 (Sigma), and grown overnight at 14°C in the same solution. As experimental controls and for staging purposes, sibling embryos were grown in 0.1X MMR and 4% Ficoll-400 (Sigma) as well. Embryos for both experiments were collected at stage 13 but an aliquot was kept developing until stage 37/38, to assess phenotype.
Whole-mount in situ hybridization
By means of this technique, it is possible to detect gene expression domains into the whole embryo. Niehrs’ protocol, with slight modifications, was preferred because it is more sensitive than others and gives little or no background (Gawantka et al., 1995). Staged embryos were fixed in MEMFA for 1 hour at room temperature, gradually dehydrated and stored in ethanol at -20°C. On the first day of the whole mount protocol, the embryos were gradually rehydrated in PTw (PBS containing 0,1% Tween-20), treated with 10 µg/ml Proteinase K for 5’ and refixed in 4% paraformaldehyde for 20’. After washing in PTw, the embryos were prehybridized for two hours in hybridisation mix at 60-65°C. Then, embryos were incubated overnight in the probe mix (50 ng of antisense labeled RNA probe diluted in 600 µl of hybridization mix).
The next day, the embryos were washed at growing stringency to eliminate the unbound probe which could lead to background. After recovering the probe, the embryos were washed twice for 30’ in SSC 2X, CHAPS 0,1 % at 37°C and then twice for 30’ in SSC 0,2X, CHAPS 0,1 % at 60-65°C. After two washes, 10’ each, in TBSTX (TBS containing 0,1% Triton-X 100) the embryos were incubated for two hours at 4°C in blocking solution. In the meantime, the anti-DIG antibody was diluted to a ratio of 1:2500 in blocking solution and was incubated also at 4°C on a rocker. After this time, the embryos were incubated in the antibody solution for 4 hours at room temperature or overnight at 4°C. The unbound antibody was removed by 5 washes in TBSTX, one of them carried out O.N. at 4°C.
To reveal the probe, the embryos were washed twice for 10’ in an alkaline phosphatase buffer (APB), at pH 9.5, supplemented with 2mM tetramisole (SIGMA), an inhibitor of endogenous phosphatases. For the chromogenic reaction, embryos were incubated in BM-Purple AP substrate (Roche) until the staining reached the desired intensity (from some hours to several days, depending on the overall quality of the probe and the expression level of the gene under analysis).
In situ hybridization on sections
Stage 42 embryos were embedded in Tissue-tek (Tsuda et al.) resin and cryostat-sectioned to be further processed for in situ hybridisation. Cryostat sections were thawn and dried at room temperature. In situ hybridisation was performed as published, with slight modifications (Kanekar et al., 1997). On the first day, sections were incubated overnight in the probe mix (1 µg/ml hybridisation mix) at 65 °C in a humified chamber (50% formamide, 1X salts). The next day, sections were washed 30’ for 3 times at increasing stringency in washing solution to eliminate the unbound probe which could lead to background. After two washes, 30’ each, in MABT, the sections were incubated for two hours at RT in blocking solution. After this time, they were incubated in the antibody solution overnight at RT.
The unbound antibody was removed by 5 washes, 30’ each, in MABT. To reveal the hybridized probe, sections were washed twice for 10’ in alkaline phosphatase buffer (APB), pH 9.5 supplemented with 2mM tetramisole (SIGMA), an inhibitor of endogenous phosphatases. For the chromogenic reaction, sections were incubated in NBT-BCIP AP substrate (Roche) until the staining reached the desired intensity, then mounted with Aqua poly-mount (xxx) and stored at 4°C for microscope analysis.
Bleaching of embryos
The embryo pigmentation was eliminated by exposing dehydrated embryos in bleaching solution to a strong light source until adequately depigmented. To stop the reaction, the embryos were transfered back in 100% Ethanol and stored at -20C°.
RNA extraction and RT-PCR analysis
To guarantee the maximum yield and purity for subsequent PCR screening and microarray experiments, TRIzol reagent (Invitrogen) was used to extract RNA from stage 13 embryos, following the manufacturer protocol with slight modifications. Extracted total RNA quality was assessed by gel electrophoresis and UV spectrophotometry.
Total RNA was retro-transcribed using random hexamers as primers and the SuperScript III reverse transcriptase, following the protocol provided by the manufacturer (Invitrogen). Starting total RNA for each reaction was 1µg.
Primers used for PCR screening were the following:
Gene Forward oligo Reverse oligo
Xrx1 CAAATGGAGGAGGCAAGAGA CCCACACTGGTAAGGAGGAG
ODC ACCCGAATGCAAAGCTTGTT TCCAGGGCTGGGTTTATCAC XBF-1 TCAACAGCCTAATGCCTGAAGC GCCGTCCACTTTCTTATCGTCG Xhoxb-9 TACTTACGGGCTTGGCTGGA AGCGTGTAACCAGTTGGCTG Xhairy2 GGCGTCAACACCGAAGTGAG ATCTTGTGGCCGAAGGT
XZic2 CACCCCATTCAGCCTACTCT CGTCCTACCGAACACCTCTC
Tab. 1: forward and reverse oligonucleotides used for thePCR screening.
PCR reactions were assembled as follows: • GoTaq© polimerase (Promega) 1.25 U
• 5x GoTaq©buffer 5 µl • dNTP mix (10 mM each) 0.5 µl • cDNA template 2 µl • Forward primer 1.0 µM • Reverse primer 1.0 µM • H2O to 25 µl.
Morpholino antisense oligos
To attain the functional knock-down of the Xrx1 gene, a Moxrx1 morpholino antisense oligonucleotide was injected into Xenopus blastulae.
MoXrx1 :
5’-TCAGGGAAGGGCTGTGCAGGTGCAT-3’ (Stock Solution 1.0 mM) MoXrx1 binds in position +1 of the Xrx1 coding sequence.
Microarray experiments
The Affymetrix microarray platform was used for microarray experiments. Extracted total RNA from embryos was amplified and biotin-labeled strictly following the protocols offered by Affymetrix. The resulting cRNA was fragmented to 200bp fragments and hybridized onto Xenopus laevis genome arrays, following Affymetrix protocols and using an Affymetrix Hybridization oven 640 and Fluidic station FS-400. The hybridized Genechips were scanned with a GeneChip® Scanner 3000.
Microarray data analysis
Raw intensity data and single chip QC reports were obtained with the GCOS (GeneChip Operating Software) package by Affymetrix. Global QC analysis was performed using the Bioconductor software package (Gentleman et al., 2004), in the R programming environment. Data resuming and normalization were performed using the Li-Wong model-based algorithm implemented into the dChip software (Li and Wong, 2001; Schadt et al., 2001), and the GC-RMA algorithm (Wu et al., 2004), as implemented in the Bioconductor software package.
Differential gene expression between clusters of samples was calculated after importing the normalized data into the MeV software (Saeed et al., 2006).
Data annotation was performed via the NetAffx Analysis Center online database by Affymetrix (http://www.affymetrix.com/analysis/index.affx) .
Probe to transcript mapping was performed using the BaseRank and AutoProbe custom developed softwares (Biasci et al., unpublished).