2. Risultati e discussione
Le strutture tetrasaccariche 38 e 39 (Schema 12) sono state preparate seguendo una procedura simile a quella riportata da Yong-Xiang e coll.14 nella sintesi dell’unita pentasaccaridica della SP15C ed ha previsto tre passaggi chiave:
a) preparazione dei glicosidi 4OH-β-D-glucosamminici 12, ortogonalmente protetti al
C-3 e al C-6 e portanti in posizione anomerica un metile o un braccio spaziatore propilazidico, mediante apertura nucleofila dell’α-ossazolidina intermedia 42, facilmente ottenibile a partire dal cloridrato della D-glucosammina commerciale 40;
b) β-galattosidazione degli accettori β-D-glucosamminici 12 seguita da deprotezione
selettiva al C-6 a dare i derivati 6OH-β-D-lattosamminici 44.
c) β-lattosidazione al C-6 degli accettori disaccaridici 44 a dare i desiderati tetrasaccaridi 38 e 39. Schema 12 O OAc OAc OAc AcO O OR O NHAc O OAc OAc OAc AcO O O OAc AcO O OAc R2O AcO OR O NHAc AcO OAc AcO O O N AcO OAc O H OR O NHAc R2O OR1 O OAc OAc OAc AcO O OR O NHAc R2O OH AcO O AcO OAc NHAc OAc O H O O H OH OH 42 43 12 44 R= Me o CH2CH2CH2N3 38: R= CH2CH2CH2N3; R2=Bz; 39: R= Me; R2=Bz 41 40 NH3+Cl
-2.1. – Sintesi dei glicosil accettori 4-OH-ββ-ββ D-glucosamminici
Il primo obiettivo di questo lavoro di Tesi è stata la trasformazione del cloridrato della
D-glucosammina commerciale 40 nell’α-ossazolidina intermedia 42, che rappresenta un
ottimo precursore per la sintesi dei derivati 2-acetammido-2-desossi-β-D-glucopiranosidici
tipo 12 (Schema 12). L’α-ossazolidina 42 è stata efficacemente preparata (resa 95%) a partire dalla nota α-peracetil-D-glucosammina 4117 (Schema 12), facilmente ottenibile per
acetilazione convenzionale (Ac2O-Py) di 40, attraverso un metodo particolarmente efficiente18 basato sull’uso di trimetilsililtriflato (TMSOTf) come promotore acido e
conducendo la reazione in soluzione dicloroetanica anidra a 50°C. La reazione è riproducibile in scala di multigrammi e l’α-ossazolidina 42 è stabile se conservata a bassa temperatura ed in presenza di piccole quantità di Et3N.
Il gruppo acetammidico in posizione 2 di 41 è un ottimo gruppo partecipante e, in analogia a quanto descritto per gli O-acetati, la perdita dell’acetato in posizione anomerica come conseguenza dell’attivazione con TMSOTf è assistita anchimericamente dalla funzione ammidica in 2 con formazione del carbocatione ciclico 45 (Schema 13) che, a differenza dello ione 1,2-acilossonio, elimina un protone e si trasforma nella stabile α-ossazolidina 42. Schema 13 AcO O AcO OAc NHAc OAc N AcO O AcO O OAc N AcO O AcO O OAc H AcO O AcO OAc NH O AcO O AcO OAc NH O TMSOTf (1.14 eq) DCE anidro 41 42 (95%) + -H+ + 45 +
I dati analitici e spettroscopici di 42 sono in accordo con quanto riportato in letteratura.18b La formazione del ciclo α-ossazolidinico è evidenziata, nello spettro protonico, dalla presenza di un singoletto a δ 1.89 (3H) attribuibile al metile ossazolidinico e, nello spettro 13C NMR, da un segnale a δ 165.8, diagnostico per un doppio legame C=N, e dal segnale a δ 14.1, relativo al carbonio metilico.
I derivati α-ossazolidinici sono ampiamente usati nella chimica dei carboidrati, dato che l’apertura regioselettiva al C-1 per opera di numerosi nucleofili, condotta a temperature elevate (40-80°C) in solventi clorurati (CH2Cl2 o DCE) anidri ed in presenza di catalizzatori acidi quali TsOH, CSA19a e BF3⋅Et2O,19b fornisce i glicosidi 1,2-trans in maniera completamente stereoselettiva.
Visto che l’obiettivo finale di questa Tesi è rappresentato dalla sintesi dei tetrasaccaridi
38 e 39 (Schema 12), che differiscono esclusivamente per il sostituente in posizione
anomerica (Me o CH2CH2CH2N3), l’apertura nucleofila dell’α-ossazolidina 42 è stata condotta sia con metanolo sia con gli alcol 46a20a e 46b20b (Figura 10), entrambi precursori di β-glicosidi con un braccio spaziatore portante una funzione amminica necessaria sia per la coniugazione con una proteina immunogenica che per la preparazione di glycocluster. In particolare, mentre l’impiego dell’amminoalcol 46b presenta già la funzione amminica protetta, l’alcol 46a consente di introdurre uno spaziatore che per semplice sostituzione
(SN2) del gruppo tosilico con NaN3, seguita da riduzione, è facilmente trasformabile nel desiderato gruppo propilamminico.
Figura 10 TsO OH HO N H O O 46a 46b
Il 3-O-tosil-propandiolo (46a) è preparato per trattamento di un eccesso di 1,3-propandiolo commerciale con cloruro di tosile in CH2Cl2-Py, mentre il 3-N-t-butossicarbonil-amminopropanolo (46b) è ottenuto dal 3-amminopropanolo commerciale per trattamento con Boc2O in soluzione diclorometanica.
La preparazione del metil β-glicoside 47 (Schema 14) è stata realizzata applicando un protocollo riportato in letteratura,19b basato sull’apertura regioselettiva dell’α-ossazolidina
42 in MeOH anidro impiegando BF3⋅Et2O come promotore anomerico e conducendo la reazione a temperatura ambiente. Una volta evidenziata la scomparsa di 42 e la formazione di un prodotto prevalente (TLC), il trattamento della reazione fornisce un grezzo che, dopo purificazione cromatografica, permette di isolare 47 in resa non soddisfacente (50%). Allo scopo di ottimizzare la preparazione di 47 abbiamo ripetuto la glicosidazione utilizzando l’acido canfosolfonico (CSA) e conducendo la reazione in CH2Cl2 anidro alla temperatura di 80°C.19a In questo caso la reazione è estremamente veloce e dopo 45 minuti l’analisi TLC evidenzia la formazione di 47 come prodotto maggioritario. Dopo trattamento e purificazione flash cromatografica del grezzo di reazione si isola il metil β-glicoside 47 (resa 58%) avente dati analitici e spettroscopici in accordo con quanto riportato in letteratura.19b,21
L’apertura dell’α-ossazolidina 42 è stata, poi, condotta con gli alcoli 46a,b (Schema 14) in soluzione diclorometanica in presenza di CSA, secondo quanto decritto precedentemente nella preparazione di 47 ma utilizzando i setacci molecolari 4Å attivati in modo da garantire l’anidricità dell’ambiente di reazione. Mentre nel caso dell’amminoalcol
46b non si evidenzia la formazione del glicoside 48, nel caso dell’alcol 46a la reazione
risulta completa dopo 12 ore a 80°C (TLC). La flash purificazione del grezzo ottenuto dopo trattamento della miscela di reazione permette di isolare il β-glicoside 49 (resa del 69%) chimicamente puro all’analisi NMR. Nel caso della reazione con il nucleofilo azotato
miscela formata dall’accettore 46b e dal 2-acetammido-3,4,6-tri-O-acetil-2-desossi-β,α-D
-glucopiranosio, la cui formazione può essere strettamente correlata sia alla minore reattività dell’accettore 46b che alla presenza di acqua nell’ambiente di reazione, che funziona da nucleofilo competitivo rispetto a 46b nell’apertura nucleofila dell’α-ossazolidina 42. Schema 14 N AcO O AcO O OAc O AcO O AcO NHAc OAc R AcO O AcO NHAc OAc OMe O H NHBoc O H OTs 48: R=NHBoc (58%) 49: R= OTs (69%) 50: R=N3 (98%) 42 47 (58%) MeOH 46b 46a o NaN3, DMF
Per cercare di ovviare alla minore reattività dell’accettore 46b, l’apertura nucleofila di
42 è stata effettuata in soluzione diclorometanica ed in presenza di setacci molecolari,
impiegando il più efficiente TMSOTf quale promotore acido. La reazione è notevolmente lenta (5 giorni) e la purificazione flash cromatografica del grezzo, ottenuto dopo trattamento della reazione, fornisce il glicoside 48 (resa del 58%) chimicamente puro all’analisi NMR.
Dall’analisi delle rese ottenute nell’apertura regioselettiva dell’α-ossazolidina 42 sembra plausibile pensare che la minore polarità del nucleofilo 46a rispetto al derivato azotato 46b e al MeOH permetta di ottenere condizioni di reazione più anidre e non consenta, nella reazione con 46a, la formazione di quantità rilevanti di prodotti derivanti da apertura idrolitica di 42 ad opera di tracce di acqua, come viene evidenziato dall’analisi della TLC delle miscele di reazione con MeOH o 46b.
I derivati 48 e 49, non riportati in letteratura, sono stati completamente caratterizzati dal punto di vista chimico-fisico e i dati NMR, ricavati utilizzando tecniche monodimenzionali (1H e 13C) e bidimensionali (DEPT-135, COSY, HETCOR), sono in accordo con le strutture proposte (vedi parte sperimentale). In entrambi i casi, nello spettro protonico, oltre alla presenza dei segnali caratteristici dei sostituenti anomerici, il doppietto a δ 6.43 o 6.52 (1H, J2,NH 9.5 o 9.1 Hz, NH) e il singoletto a δ 1.92 o 1.88 (3H, CH3) confermano la funzione acetammidica in posizione 2. Particolarmente indicativo, ai fini della determinazione della configurazione del legame glicosidico, è l’alto valore della costante di accoppiamento J1,2 (8.1 e 8.5 Hz), che indica un assetto assiale-assiale dei protoni H-1 e H-2.
La trasformazione dei derivati 47-49 nei glicosil accettori 4OH-β-D-glucosamminici 12, ortogonalmente protetti in posizione 3 e 6, prevede un’opportuna manipolazione dei
gruppi protettori realizzabile utilizzando condizioni sia basiche che acide, quest’ultime non compatibili con la funzione t-butossicarbonilica presente in 48. Questo ci ha, quindi, indotto a proseguire la nostra sequenza sintetica utilizzando i glicosidi 47 e 49 come materiale di partenza. Il glicoside 49, avente un buon gruppo uscente sull’aglicone, prima di essere sottoposto a qualsiasi manipolazione dei gruppi protettivi viene trasformato nel corrispondente azido derivato 50 (Schema 14) attraverso una classica reazione di sostituzione nucleofila tipo SN2 condotta in DMF anidra con NaN3 a 50°C ed in presenza di tetrabutilammonio ioduro (TBAI). La reazione risulta completa dopo 3 ore (TLC) e si forma unicamente il prodotto 50, che viene isolato in resa del 95% dopo purificazione cromatografica del grezzo di reazione.
L’azide 50, non riportata in letteratura, è stata completamente caratterizzata dal punto di vista chimico-fisico e i dati NMR sono in accordo con la struttura proposta (vedi parte sperimentale). L’avvenuta azidazione è confermata nello spettro 13C dal segnale a δ 48.8, schermato di circa 20 ppm rispetto allo stesso segnale di 49 (δ 68.7), attribuito al metilene legato al gruppo azidico.
Il prosieguo della sintesi ha previsto la desacetilazione di 47 e 50 (Schema 15) che è stata realizzata applicando le condizioni di Zemplen basate sull’uso di MeONa in soluzione di metanolica, ma operando alla temperatura di 0°C per evitare l’idrolisi del gruppo acetammidico. Schema 15 AcO O AcO NHAc OAc OR HO O O H NHAc OH OR O O O O H NHAc OR 47: R=Me 50: R=(CH2)3N3 MeONa-MeOH 51: R=Me (quantit.) 52: R=(CH2)3N3 (quantit.) 2-MP, CSA, CH2Cl2 53: R=Me (85% da 47) 54: R=(CH2)3N3 (88% da 50)
In queste condizioni la reazione risulta completa dopo 1-1.5 ore e si formano, come unici prodotti, i corrispondenti trioli 51 e 52 che vengono isolati per semplice neutralizzazione della miscela di reazione con resina acida seguita da filtrazione ed evaporazione del solvente.
La trasformazione dei trioli 51 e 52 (Schema 15) nei corrispondenti derivati 4OH-β-D
deprotetti al C-6, rende necessaria una protezione ortogonale nelle posizioni 3 e 6 con gruppi protettori stabili nelle condizioni di glicosidazione e rimovibili in condizioni che non intaccano né l’anello saccaridico né il legame glicosidico.
In generale, i gruppi protettori utilizzati nella sintesi oligosaccaridica possono essere suddivisi in permanenti quando rimangono installati fino al raggiungimento dell’oligosaccaride finale e temporanei quando vengono impiegati per ottenere un ossidrile libero nel corso della sequenza sintetica (nel nostro caso la posizione 6). I gruppi protettori permanenti devono poter essere introdotti e rimossi efficacemente e con un elevato regio-controllo e devono essere stabili nelle condizioni di rimozione ed installazione dei gruppi temporanei (ortogonalità dei gruppi protettivi). Il concetto di protezione ortogonale è definito come un set di classi di gruppi protettori completamente indipendenti tali che ogni classe può essere rimossa in presenza di tutte le altre. Un esempio di set di gruppi protettori ortogonali può essere rappresentato da: benzile (H2, Pd/C in MeOH), acetato o benzoato (MeONa-MeOH), TBDMS (TBAF-THF o AcOH aq.) e allile (PdCl2, MeOH). In generale, i gruppi protettori permanenti più utilizzati in sintesi oligosaccardica sono acetati, benzoati, benzili e acetali benzilidenici o isopropilidenici e la scelta deve considerare, oltre alla stabilità e un’agevole introduzione e deprotezione, anche la loro influenza sulla reattività delle molecole sulle quali sono installati, come già evidenziato nel Capitolo 1 a proposito della definizione di glicosidi armati o disarmati. In generale, per ottenere un’efficiente glicosidazione si deve riuscire a sfruttare al meglio le differenti proprietà dei gruppi protettori ed è quindi essenziale disporre di metodologie sintetiche che permettano di discriminare i vari ossidrili presenti sul substrato saccaridico al fine di installare regioselettivamente una data protezione su una determinata posizione. Nel caso dei trioli
51 e 52 vi sono due tipi di ossidrili: quello primario sul C-6 e i due secondari sul C-3 e C-4
che differiscono per la loro nucleofilicità. In particolare, il gruppo alcolico primario è più reattivo di quelli secondari e può essere funzionalizzato regioselettivamente con un gruppo protettore molto ingombrante come il t-butildimetilsilile (TBDMS), trifenilmetile (o tritile), pivaloile (Piv) etc.. Nel caso di gruppi alcolici secondari, generalmente la loro distinzione dipende dalla loro configurazione: quelli equatoriali tendono a reagire più velocemente di quelli assiali. A parità di disposizione non è possibile fare altre generalizzazioni, anche se è noto, ad esempio, che nel glucosio l’ossidrile equatoriale sul C-2 è più reattivo mentre quello sul C-4 è il meno reattivo.
Da queste considerazioni del tutto generali è possibile programmare la protezione ortogonale delle posizioni 3 e 6 dei trioli 51 e 52 attraverso una prima funzionalizzazione dell’ossidrile primario seguita da protezione regioselettiva al C-3, non facilmente realizzabile visto la disposizione equatoriale di entrambi i gruppi alcolici in posizione 3 e 4. Per ovviare a questo inconveniente siamo ricorsi ad un processo, molto comune nella sintesi dei carboidrati, che permette di proteggere più funzioni ossidriliche attraverso la formazione di gruppi protettori ciclici come gli acetali di tipo benzilidenico o isopropilidenico che, a causa della maggiore nucleofilia della posizione primaria, prediligono l’istallazione regioselettiva sulle posizioni C-4 e C-6 con formazione di un ciclo stabile a 6 termini. E’ da sottolineare che quando sono coinvolti due ossidrili secondari, la formazione di acetali isopropilidenici è favorita quando esiste una relazione 1,2-cis tra i due gruppi alcolici.
La trasformazione dei trioli 51 e 52 nei corrispondenti 4,6-O-isopropiliden derivati 53 e
54 (Schema 15) è stata possibile grazie all’uso del 2-metossipropene (2-MP) come agente
acetonante e dell’acido canfsolfonico (CSA) come catalizzatore acido,22 una metodologia ampiamente utilizzata per proteggere dioli vicinali e particolarmente efficiente quando, nella reazione di isopropilidenazione, è coinvolto un ossidrile primario.
Il meccanismo di reazione prevede un attacco iniziale del gruppo ossidrilico primario stericamente più accessibile sul viniletere protonato 55 (Schema 16) per dare un acetale intermedio aciclico protonato 55a (nello schema è riportata la forma protonata) che subisce un attacco nucleofilo intramolecolare da parte del gruppo ossidrilico più vicino a dare l’acetale ciclico 55b.22 Schema 16 OH R OH C+ OMe O R OH O+ Me H O R O + - MeOH - H+ 55 55a 55b
I trioli 51 e 52 sono stati quindi trattati con 2-MP in DMF anidra in presenza di CSA (10%). Dopo 3-5 ore a temperatura ambiente l’analisi TLC evidenzia, in entrambi i casi, la scomparsa del prodotto di partenza e la formazione di un prodotto nettamente prevalente. La neutralizzazione delle miscele con una soluzione acquosa satura di NaHCO3, seguita da estrazione con CH2Cl2 e purificazione flash cromatografica su gel di silice, fornisce i
corrispondenti alcol 53 e 54 in resa rispettivamente del 85 e 88% (calcolate a partire dai
tri-O-acetati 47 e 50), puri all’analisi NMR (1H, 13C).
La formazione esclusiva di 53 e 54 conferma la regioselettività della reazione di acetonazione, in quanto la formazione del regioisomero acetonato in 3,4 prevedrebbe una fusione trans-diossolanica (5 termini) molto meno stabile di quella cis-diossanica (6 termini).
Mentre i dati analitici e spettroscopici di 53 sono in accordo con quanto riportato in letteratura;23 il derivato 54, non riportato in letteratura, è stato completamente caratterizzato dal punto di vista fisico-chimico e i dati NMR sono concordi con la struttura proposta. In particolare, la presenza dell’acetale 4,6-O-isopropilidenico è evidenziata, nello spettro protonico, da due singoletti a δ 1.46 (3H) e 1.34 (3H) attribuibili ai due metili e, nello spettro 13C, dalla presenza di tre segnali a δ 100.2 (carbonio quaternario), 29.4 e 19.4 (due CH3) diagnostici per un acetale ciclico di tipo diossanico.
I derivati 53 e 54 si sono rivelati degli ottimi precursori per la preparazione dei glicosil accettori 4OH-β-D-glucosamminici 12, variamente sostituiti nelle posizioni 3 e 6, necessari
per poter effettuare uno studio volto a trovare le condizioni ottimali per realizzare la successiva reazione di β-galattosidazione. In particolare, mentre sulla posizione 6 verranno introdotti gruppi temporanei eterei come il p-metossibenzile (PMB) o il t-butildimetilsilile (TBDMS), sulla posizione 3 sono installati gruppi permanenti sia di tipo estereo (acetato o benzoato) che etereo (benzile) con lo scopo di valutare la loro influenza sulla reattività della funzione alcolica al C-4 nella reazione di glicosidazione.
E’ da sottolineare che la protezione al C-3 di 53 e 54, sia come etere benzilico che come acetato o benzoato, permette di avere un gruppo ortogonale a quello isopropilidenico che consente l’idrolisi acida dell’acetale ciclico e formazione del derivato deprotetto in 4 e 6 sul quale realizzare la successiva protezione selettiva dell’ossidrile in posizione 6, più reattivo di quello in 4 perché primario.
La benzilazione del glicoside 54 (Schema 17) è stata effettuata secondo la metodica standard che impiega BnBr e KOH in THF umido in presenza di etere corona (18-crown-6). Il metodo consiste nella salificazione delle funzionalità ossidriliche mediante il sistema KOH/18-corona-6 in THF umido, seguita dal trattamento con un eccesso di bromuro di benzile (2 equivalenti per gruppo ossidrilico). Il KOH in presenza dell’etere corona forma una coppia ionica costituita dal sistema chelato etere-K+ e dall’OH- (Figura 11), nel quale l’anione ossidrile acquisisce una basicità maggiore: la sostituzione del K+, che con OH
-fornisce una coppia ionica molto stretta, con una struttura molto più grande quale il chelato etere-K+ esalta la reattività dell’anione OH- rendendolo capace di salificare quantitativamente le funzionalità ossidriliche, che sono quindi in grado di reagire con l’elettrofilo presente in soluzione.
Figura 11 O O O O O O K O O O O O O 18-corona-6 KOH OH +
-Dopo scomparsa in TLC (4 ore) del prodotto di partenza, work-up standard della miscela di reazione e purificazione flash cromatografica del grezzo, si isola il benzil derivato 56 (resa 75%) chimicamente puro all’analisi NMR (1H e 13C).
Il derivato 56, non riportato in letteratura, è stato completamente caratterizzato dal punto di vista fisico-chimico e i dati NMR sono concordi con la struttura proposta (vedi parte sperimentale). La presenza del gruppo benzilico è confermata, nello spettro protonico, da un multipletto a δ 7.37-7.24 (5 protoni aromatici) e da un sistema AB (δ 4.74 e 4.57, JA,B = 12.0 Hz) attribuibile ai protoni del gruppo metilenico. Anche lo spettro 13C conferma la struttura proposta e presenta, oltre ai segnali dei CH aromatici fra δ 129.1 e 128.3, due segnali a δ 74.2 e 140.0, attribuibili rispettivamente al CH2 benzilico ed al carbonio quaternario aromatico. Inoltre, il segnale relativo al C-3 (δ 79.9) risulta deschermato di circa 6.8 ppm rispetto al corrispondente segnale di 54 (δ 73.1) per la presenza del gruppo etereo in tale posizione.
L’acetilazione convenzionale (Ac2O-Py, t.a.) o la benzoilazione (BzCl, Py, 0°C) di 54 fornisce, dopo purificazione flash cromatografica dei grezzi di reazione, i derivati esterei
57 (97%) e 58 (96%) chimicamente puri all’analisi NMR (1H e 13C).
I derivati 57 e 58, non riportati in letteratura, sono stati completamente caratterizzati dal punto di vista fisico-chimico e i dati NMR sono concordi con le strutture proposte (vedi parte sperimentale). L’avvenuta acetilazione della posizione 3 di 54 è evidenziata, nello spettro protonico, da due singoletti, integranti ciascuno per 3 protoni, a δ 1.96 e 1.82 attribuibili rispettivamente ai CH3 del gruppo acetilico e acetammidico, la cui presenza è confermata anche nello spettro 13C da due segnali a δ 171.2 e 170.7 (2 x C=O) e da due segnali a δ 23.1 e 20.9 (2 x CH3). La presenza del gruppo benzoilico in 58 è confermata,
nello spettro protonico, da tre multipletti fra δ 7.98 e 7.04, integranti per 5 protoni aromatici e, nello spettro 13C, dai segnali dei CH aromatici fra δ 133.4 e 128.5 e da due segnali a δ 167.0 e 129.0, attribuibili rispettivamente al carbonio carbonilico ed al carbonio quaternario aromatico. Inoltre, per entrambi i derivati 57 e 58 la presenza del gruppo estereo in posizione 3 è confermata anche dalle risonanze del protone H-3 che risulta più deschermato (1.4-2 ppm), rispetto al corrispondente segnale di 54, a causa dell’effetto anisotropico del carbonile.
Schema 17 O O O O O H NHAc N3 O O O O RO NHAc N3 HO O N3 O RO NHAc OH 54 56: R=Bn (75%) 57: R=Ac (97%) 58: R=Bz (96%) 59: R=Bn (99%) 60: R=Ac (68%) 61: R=Bz (96%)
La rimozione dei gruppi O-isopropilidenici nei carboidrati è generalmente effettuata per idrolisi in ambiente reso acido da un’ampia gamma di reagenti acidi sia protici che di Lewis.Uno dei sistemi più utilizzati è senza dubbio quello che prevede il trattamento con soluzioni acquose di acido acetico a varia concentrazione e differente temperatura.
I composti 56, 57 e 58 (Schema 17) sono stati, quindi, sottoposti a idrolisi acida con AcOH acquoso al 70% alla temperatura di 40°C, condizioni in cui il legame interglicosidico è perfettamente stabile. Dopo eliminazione dei reagenti mediante co-evaporazione con toluene e purificazione flash cromatografica su gel di silice dei residui ottenuti, si isolano i corrispondenti dioli 59 (resa 99%), 60 (resa 68%) e 61 (resa 96%) chimicamente puri all’analisi NMR (1H e 13C). I derivati 59-61, non riportati in letteratura, sono stati completamente caratterizzati dal punto di vista fisico-chimico e i dati NMR sono concordi con le strutture proposte (vedi parte sperimentale).
Considerato che tutti i passaggi della sequenza sintetica illustrata negli Schemi 16 e 17 avvengono con rese molto elevate e portano alla formazione di un unico prodotto, è possibile effettuare le successive reazioni sui prodotti grezzi in modo da limitare le purificazioni cromatografiche. A tal scopo, la trasformazione del metil glicoside 51 nel derivato 63 (Schema 18) è stata condotta efficacemente (resa su tre passaggi del 58%), applicando la sequenza sintetica illustrata precedentemente per l’analogo derivato 61, senza effettuare purificazioni cromatografiche intermedie. I dati analitici e spettroscopici del metil derivato 63 sono in accordo con quanto riportato in letteratura.24
Schema 18 O O O O H NHAc OMe O O O NHAc OMe BzO HO O NHAc OH OMe BzO 53 62 63 (Resa 58% calcolata da 51) BzCl, Py AcOH aq 70%
A questo punto della sequenza sintetica si rendeva necessario funzionalizzare la posizione 6 con un gruppo ortogonale a quello presente in posizione 3. A tal scopo, come già precedentemente discusso, la nostra scelta si è orientata verso gruppi stabili nelle condizioni di glicosidazione come quello p-metossibenzilico o quello t-butildimetilsililico che, essendo molto ingombrante, può essere introdotto regioselettivamente sul C-6 sfruttando la maggiore nucleofilia dell’ossidrile primario.
La p-metossibenzilazione regioselettiva in posizione 6 di 59 è realizzata attraverso una procedura ampiamente applicata nel nostro laboratorio, basata sulla formazione di intermedi ciclici stannilidenici 64 (Figura 12), seguita da apertura dell’acetale stannilidenico con un agente alchilante elettrofilo (BnBr, PMBCl, BzCl).25
Figura 12 OH (C) OH O (C) O SnBu2 R X O (C) O R SnBu2X OH (C) OR X n + Bu2SnO n - H2O n H2O n + SnBu2(OH)X 64
Gli acetali stannilidenici sono generalmente ottenuti per trattamento di un 1,2- o 1,3-diolo con circa un equivalente di Bu2SnO in toluene, MeOH o CHCl3 ed in condizioni di rimozione azeotropica dell’acqua che si forma durante la reazione. In genere questi derivati sono stabili, la loro manipolazione non richiede particolari precauzioni (atmosfera inerte, ambienti rigorosamente anidri), sono ottenuti in maniera quantitativa e non richiedono processi di purificazione. Essi reagiscono con vari reagenti elettrofili in solventi a bassa polarità (toluene, CHCl3) e la reazione risulta, nella maggior parte dei casi, completamente regioselettiva ed avviene con un meccanismo di tipo SN2 nel quale l’elettrofilo alchila sempre l’ossigeno meno ingombrato (nel nostro caso il primario) e quindi più nucleofilo.
Sulla base di queste considerazioni il diolo 59 (Schema 19) è stato sottoposto a reazione di stannilidenazione con quantità equimolari di Bu2SnO in toluene ed in condizioni di rimozione azeotropica dell’acqua mediante una colonna di Dean-Stark. Dopo 12 ore a riflusso del solvente, il derivato stannilidenico è stato trattato con Bu4NBr
(TBAB) e PMBCl (4 eq) e, dopo circa 4 ore, l’analisi TLC rivela la scomparsa del prodotto di partenza e la formazione di un prodotto prevalente. La successiva purificazione flash cromatografica su gel di silice del residuo, ottenuto per semplice evaporazione del solvente, consente di isolare il desiderato glicoside 65 (resa 65%) chimicamente puro all’analisi NMR. Schema 19 O N3 O H O RO NHAc OH O N3 O H O NHAc OPMB BnO O Sn O O O NHAc N3 Bu Bu BnO Cl OMe Br 59 65 59a Bu2SnO toluene PMBCl TBAB
Il derivato 65, non riportato in letteratura, è stato completamente caratterizzato dal punto di vista fisico-chimico e i dati NMR sono concordi con la struttura proposta (vedi parte sperimentale). In particolare, la presenza del gruppo p-metossibenzilico è confermata, nello spettro protonico, da due multipletti a δ 7.30 (7H) e 6.91 (2H) , da due singoletti a δ 4.47 (2H) e 3.77 (3H) attribuibili, rispettivamente, al metilene benzilico e al gruppo metossilico. Anche lo spettro 13C conferma la struttura proposta e presenta i segnali dei CH aromatici (δ 130.2 e 114.5), i segnali relativi ai due carboni quaternari (δ 160.1 e 131.6) e due segnali a δ 73.4 e 55.8 attribuibili rispettivamente al CH2 p-metossibenzilico ed al metossile. Inoltre, il segnale relativo al C-6 (δ 70.4) risulta deschermato di 7.7 ppm rispetto al corrispondente segnale di 59 (δ 62.7) per la presenza del gruppo etereo in tale posizione.
Un metodo molto efficiente di protezione regioselettiva al C-6 di unità saccaridiche è quello che prevede una reazione con un reagente ingombrante come il t-butildimetisilil cloruro (TBDMSCl). Generalmente questo tipo di reazioni sono condotte in DMF anidra in presenza di imidazolo o in piridina anidra.26 I dioli 60 e 61 (Schema 20) sono stati, quindi sottoposti, a sililazione regioselettiva utilizzando un equivalente di TBDMSCl, 2 equivalenti di imidazolo e conducendo la reazione in DMF anidra. Dopo trattamento della reazione e purificazione flash cromatografica dei grezzi di reazione si isolano, oltre ai dioli
60 (34%) e 61 (52%) non reagiti, i desiderati silil derivati 67 (resa 48%) e 68 (resa 37%)
chimicamente puri all’analisi NMR (1H e 13C). Risultati decisamente migliori sono stati ottenuti conducendo la reazione con TBDMSCl in piridina con la quale i dioli 59, 61 e 63
(Schema 20) sono stati trasformati efficacemente nei corrispondenti 6-O-silil derivati 66 (resa 89%), 68 (93%) e 69 (81%), isolati chimicamente puri all’analisi NMR.
Schema 20 O N3 O H O RO NHAc OH O N3 O H O RO NHAc OR' O H O NHAc OH OMe BzO HO O NHAc OTBDMS OMe BzO 59: R= Bn 60: R=Ac 61: R=Bz 66: R=Bn; R'=TBDMS 67: R=Ac; R'=TBDMS 68: R=Bz; R'=TBDMS 63 69 TBDMSCl Py TBDMSCl Py
I derivati 66-69, non riportati in letteratura, sono stati completamente caratterizzati dal punto di vista fisico-chimico e i dati NMR sono concordi con le strutture proposte (vedi parte sperimentale). In tutti i casi, la presenza del gruppo t-butildimetilsililico è confermata, nello spettro protonico, da un singoletto (9H) a circa δ 0.90, attribuibile ai metili del raggruppamento t-butilico, e da due singoletti fortemente schermati (δ 0.90, 0.08 e 0.07), integranti ciascuno per 3 protoni, assegnabili ai due metili legati all’atomo di silicio. Inoltre, la presenza del segnale relativo al gruppo ossidrilico in posizione 4 (δ 3.90-3.80) conferma la regioselettività al C-6 della reazione di sililazione.
2.2. – Sintesi di glicosil donatori galattosidici e lattosidici.
Ottenuta la preparazione dei glicosil accettori 4OH-β-D-glucosamminici 65-69, ci
siamo occupati della preparazione di donatori galattosidici e lattosidici opportunamente attivati in posizione anomerica, da impiegare nelle successive reazioni di β-galattosidazione e β-lattosidazione necessarie nel prosieguo della sintesi dei tetrasaccaridi
38 e 39.
Come ampiamente discusso nel capitolo 1, la formazione di un legame glicosidico richiede l’attivazione della posizione anomerica di un glicosil donatore. Il metodo dei tricloacetimmidati è ampiamente applicato perché questi composti presentano ottime proprietà glicosilanti e generalmente sono attivati con quantità catalitiche di promotore, al contrario di quanto accade per altri glicosil donatori che richiedono spesso quantità stechiometriche di attivatore. I tricloacetimmidati sono preparati per addizione dell’ossidrile emiacetalico al tricloroacetonitrile. La reazione con CCl3CN è reversibile, ma
è possibile avere un ottimo controllo della stereoselettività al carbonio anomerico: in presenza di una base debole (es. K2CO3) è possibile isolare il donatore β-70 (Schema 21) quale prodotto cinetico; mentre utilizzando base forti come NaH o DBU10 si ottiene prevalentemente il prodotto termodinamico corrispondente al più stabile α-70.
Schema 21 OH PO O PO OP OP O PO O PO OP OP O PO O PO OP OP PO O PO OP OP CHO O PO O PO OP OP NH CCl3 O PO O PO OP OP NH CCl3 M+ M+ α α α α-70 prodotto termodinamico β β β β-70 prodotto cinetico base forte base debole
E’ da notare che la possibilità di ottenere puro ciascuno dei due anomeri consente, quando la reazione di glicosidazione segue un meccanismo SN2, di formare stereoselettivamente il legame glicosidico. Il gruppo tricloroacetimmidato è molto labile e non permette nessuna manipolazione dei gruppi protettori presenti sulle altre funzionalità ossidriliche e per questo motivo l’introduzione di tale gruppo uscente rappresenta sempre l’ultimo stadio della sintesi di un glicosil donatore di questo tipo.
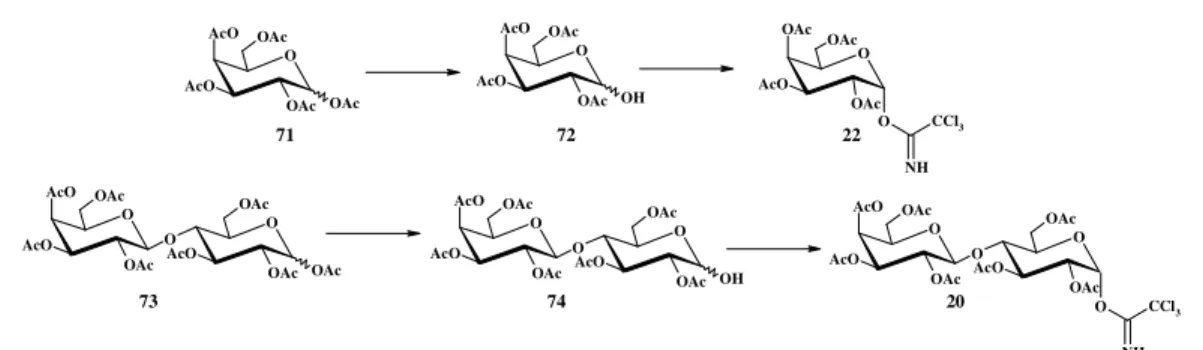
Sulla base di queste considerazioni sono stati preparati, come primo approccio, i tricloroacetimmidati mono- e disaccaridici 22 e 20 a partire, rispettivamente, dal peracetilgalattosio 71 e dal peracetillattosio 73 (Schema 22), facilmente ottenibili per acetilazione convenzionale (Ac2O/Py) del D-galattosio e del lattosio commerciali. Gli acetati anomerici sono più labili degli altri acetati e possono essere rimossi regioselettivamente con un blando trattamento basico. In particolare, il trattamento di 71 e
73 con idrazina acetato in DMF a riflusso fornisce i corrispondenti emiacetali 72 (resa
89%) e 74 (resa 64%) che, all’analisi NMR, si presentano come una miscela di anomeri α e β in rapporto 4:1, calcolato in base alle altezze relative ai carboni C-1 a δ 89.9/89.6 e 95.1/94.7 rispettivamente.
I derivati 72 e 74 sono stati trattati con CCl3CN in CH2Cl2 in presenza di quantità catalitiche di DBU. Dopo scomparsa dei prodotti di partenza (TLC, 1.5-2.5 ore), trattamento e purificazione flash cromatografica dei grezzi di reazione si ottengono i tricloroacetimmidati 2212 (resa 91%) e 2012 (resa 93%) aventi parametri spettrali in accordo con quanto riportato in letteratura. In particolare, nel caso del derivato 20, la presenza del gruppo tricloroacetimmidico in posizione anomerica è confermata, nello spettro 13C, dalla
presenza di due segnali a δ 160.7 e 90.6, attribuibili rispettivamente al C=N e al carbonio quaternario triclorurato (CCl3). Schema 22 OH AcO O AcO OAc OAc O OAc O AcO OAc OAc NH CCl3 OAc AcO O AcO OAc OAc OAc O O AcO OAc OAc AcO O AcO OAc OAc OH O O AcO OAc OAc AcO O AcO OAc OAc O O AcO OAc OAc AcO O AcO OAc OAc O NH CCl3 71 72 22 73 74 20
I tricloroacetimmidati sono relativamente stabili in condizioni basiche o neutre, ma reagiscono velocemente in condizioni acide e possono essere attivati con BF3⋅Et2O, TMSOTf e TfOH. Generalmente, le reazioni di glicosidazione promosse da questi attivanti procedono in buone rese e a basse temperature, anche se questi agenti sono altamente igroscopici e molto sensibili all’umidità. Queste caratteristiche, insieme al forte carattere acido, indicano una certa cautela sia nel loro uso che nell’introduzione di gruppo protettori acido labili sia sui glicosil accettori che sui glicosil donatori. La significativa labilità del gruppo tricloroacetimmidato costituisce probabilmente il principale motivo per cui i tioglicosidi sono i donatori più usati nella sintesi oligosaccaridica. Essi possono essere preparati, ad esempio, a partire dai corrispondenti tricloroacetimmidati, glicosil fluoruri ed esteri anomerici per trattamento con tioli (EtSH, PhSH) o loro precursori attivati (ad esempio PhSSiMe3) in presenza di acidi di Lewis come BF3.Et2O, ZnCl2, ZnI2.
Per verificare l’influenza del tipo di glicosil donatore usato nella reazione di β-galattosidazione dei derivati 4OH-β-D-glucosamminici ci siamo proposti di impiegare altri
donatori come il peracetilgalattosio 71, il 2,3,4,6-tetra-O-acetil-α-D-galattopiranosil
bromuro 13b commerciale e il tioderivato 75 (Figura 13), ottenuto per acetilazione convenzionale (Ac2O-Py) del tiofenil-β-D-galattopiranoside presente nel laboratorio dove è stata effettuata questa tesi.
Figura 13 SPh AcO O AcO OAc OAc Br AcO O AcO OAc OAc 13a 75
2.3. – ββββ-galattosidazione dei derivati 4OH-βββ-β D-glucosamminici 65-69
Generalmente, come già discusso nel capitolo 1, le reazioni di glicosilazione necessitano di condizioni particolarmente anidre dell’ambiente di reazione e sono influenzate da numerosi parametri come la temperatura, la quantità di donatore, la presenza o meno di setacci molecolari, il tempo di reazione, il tipo di promotore ed il tipo dei sostituenti presenti sia sul donatore che sull’accettore.
Auzanneau e coll.16 sottolineano che la glicosidazione di derivati 4OH-β-D
-glucosamminici, a parità di glicosil donatore (tricloroacetimmidato) e di promotore (BF3⋅Et2O), è fortemente influenzata: a) dal gruppo protettore all’azoto (esempio il gruppo ftalimmidico è migliore di quello acetammidico); b) dalla natura del sostituente al C-3 (gruppo etereo è preferito rispetto a quello estereo); c) dalla struttura dell’aglicone (si osserva un significativo calo di rendimento nella reazione di glicosidazione se è presente sull’accettore un gruppo azidoalchilico). Inoltre, visto che la reazione richiede alte temperature (40 °C) e quantità notevoli di promotore (2-5 equivalenti) non c’è compatibilità delle condizioni di reazione con la presenza in posizione 6 di gruppi acido labili come i p-metossibenzilici o t-butildimetilsililici. Nonostante questi dati bibliografici non incoraggianti ci è sembrato comunque opportuno effettuare uno studio metodologico volto alla ricerca delle condizioni ottimali per realizzare un’efficiente β-galattosidazione dei derivati 4OH-β-D-glucosamminici 65-69 contenenti sia il gruppo azidico sull’aglicone
sia la funzione acetammidica al C-2.
Le prove di β-galattosidazione degli accettori 65-69 (Schema 23, Tabella 1) aventi sostituenti diversi sia in posizione anomerica (Me, CH2CH2CH2N3) che sul C-3 (benzile, acetato, benzoato) e sul C-6 (p-metossibenzile, t-butildimetisilile), sono state effettuate utilizzando i più comuni metodi di attivazione dei tricloroacetimmidati (TMSOTf e BF3⋅Et2O), degli acetati anomerici (TMSOTf), dei bromuri anomerici (AgOTf) e dei tioglicosidi (NIS-AgOTf).
In tutte le reazioni di glicosidazione l’anidricità dell’ambiente è garantita sia dalla presenza di setacci molecolariattivati sia dalla formazione di un azeotropo con toluene ed evaporando la soluzione alla pompa meccanica munita di una trappola raffreddata a -78°C per eliminare le tracce di acqua presenti nell’accettore e nel donatore.
Le condizioni generali di reazione applicate alle glicosidazioni sono: a) il rapporto stechiometrico tra il donatore e l’accettore di 1.3-2.0; b) le quantità di promotore, aggiunto
in soluzione di CH2Cl2 anidro (1:10 v/v), i tempi di reazione e le temperature sono riportati nella Tabella 1; c) uso di CH2Cl2 e 1,2-dicloroetano (DCE) anidri come solventi.
Schema 23 X OAc O AcO OAc OAc Y HO O OR R2O NHAc OR1 OR O O R2O NHAc OR1 AcO O AcO OAc OAc OR O O R2O NHAc OR1 O AcO O AcO O OAc O AcO O AcO OAc OAc OAc AcO O AcO OAc OR AcO O R2O NHAc OR1 22: Y=H; X=CCl3OC=NH 13a: Y=H; X=Br 75: Y=SPh; X=H + + 82 + 76a: R= (CH2)3N3; R1=PMB; R2=Bn 76b: R= (CH2)3N3; R1=TBDMS; R2=Ac 76c: R= (CH2)3N3; R1=TBDMS; R2=Bz 76d: R= (CH2)3N3; R1=TBDMS; R2=Bn 77: R=Me; R1=TBDMS; R2=Bz 65-69 78a: R= (CH2)3N3; R1=PMB; R2=Bn 78b: R= (CH2)3N3; R1=TBDMS; R2=Ac 78c: R= (CH2)3N3; R1=TBDMS; R2=Bz 78d: R= (CH2)3N3; R1=TBDMS; R2=Bn 79: R=Me; R1=TBDMS; R2=Bz 80a: R= (CH2)3N3; R1=PMB; R2=Bn 80b: R= (CH2)3N3; R1=TBDMS; R2=Ac 80c: R= (CH2)3N3; R1=TBDMS; R2=Bz 80d: R= (CH2)3N3; R1=TBDMS; R2=Bn 81: R=Me; R1=TBDMS; R2=Bz Tabella 1
Prove R R1 R2 attivante Tipo donatore condizioni reazione glicoside (%) Ortoestere (%) 82 (%) 4-OAc (%) 1 (CH2)3N3 PMB Bn TMSOTf (0.01 eq) 22 (1.77eq), AW300, 0°C→ta (12h) 76a (11%) 78a (3%) -- 80a (72%) 2 (CH2)3N3 PMB Bn TMSOTf (3x0.01 eq) 22 (1.77 eq), 4Å MS, -30°C→ta (12h) 76a (tracce) 78a (47%) -- n.i. 3 (CH2)3N3 PMB Bn TMSOTf
(2.2 eq.) AW300, -20°C→ta 71 (1.1 eq), (tracce) 76a -- -- -- 4 (CH2)3N3 TBDMS Ac TMSOTf
(0.1 eq) 4Å MS, -40°C (4h) 22 (1.3eq), (tracce) 78b (59%) 78b -- n.i. 5 (CH2)3N3 TBDMS Ac TMSOTf (0.5 eq) 22 (1.3eq), 4Å MS, -40°C (4h) (tracce) 76b (37%) 80b -- n.i. 6 (CH2)3N3 TBDMS Ac BF3⋅Et2O (2 eq) 22 (5 eq), 40°C (1h) (tracce) 76b -- -- -- 7 (CH2)3N3 TBDMS Bz TMSOTf
(0.5 eq) AW300, -30°C→ta (1h) 22 (1.3eq), (27%) 76c (tracce) 78c 15% (20%) 80c 8 (CH2)3N3 TBDMS Bz TMSOTf
(0.5 eq) AW300, -30°C→ta (4h) 22 (1.5 + 0.3 eq), (26%) 76c (35%) 78c 9% (20%) 80c
9 (CH2)3N3 TBDMS Bz TMSOTf
(0.5eq) AW-300, -30→+10°C (4h) 22 (2 + 3 x 0.3 eq), (tracce) 76c (46%) 78c n.i. n.i. 10 (CH2)2N3 TBDMS Bz TMSOTf
(0.5eq) AW-300, -30°C→ta (12h) 22 (1.5 eq), (47%) 76c (tracce) 78c n.i. n.i. 11 (CH2)2N3 TBDMS Bz AgOTf
(2 eq) 13aAW-300, ta (1.6 eq), (tracce) 76c -- -- -- 12 (CH2)2N3 TBDMS Bz NIS-AgOTf (2-0.6 eq) 75 (2 eq), AW300, -30°C→ta (2h) 78c (29%) -- 10% -- 13 (CH2)3N3 TBDMS Bn TMSOTf (0.5eq) 22 (1.5eq), AW-300, -30°C→ta (20h) (53%) 76d -- <2% n.i. 14 (CH2)3N3 TBDMS Bn NIS-AgOTf (1.8-0.6 eq) 75 (1.8 eq), AW-300, -30°C→ta (2h) 76d (34%) -- <2 % -- 15 Me TBDMS Bz TMSOTf (0.5 eq) 22 (1.5eq), AW-300, -30°C→ta (20h) 77 (62%) 79 (tracce) <2 % n.i. 16 Me TBDMS Bz NIS-AgOTf
(1.8-0.6 eq) AW-300, -30°C→ta (2h) 75 (1.8 eq), (30%) 77 -- <2% -- n.i: prodotto non isolato ma evidenziato all’analisi TLC
Nelle prove di β-galattosidazione dell’accettore 65 (R1=PMB; R2=Bn), riportate in Tabella 1 (prove 1-3), effettuate in presenza di TMSOTf il desiderato disaccaride 78a è ottenuto in resa del tutto insoddisfacente (11%). L’uso dei setacci molecolari 4Å attivati (leggermente basici) al posto di quelli AW-300 (neutri), non conduce al disaccaride 76a (Schema 23) ma fornisce l’1,2-ortoestere 78a (resa 47%). Probabilmente i setacci molecolari 4Å, essendo leggermente basici, riducono l’acidità dell’attivante portando non al prodotto desiderato, ma all’intermedio ortoestereo la cui formazione, come già accennato nel capitolo 1, è reversibile e strettamente dipendente dall’acidità dell’ambiente di reazione. Per cercare di ovviare a questo inconveniente sono state aumentate le quantità di TMSOTf (da 0.01 a 0.1 eq) ma l’analisi TLC della miscela di reazione evidenzia la rimozione del gruppo p-metossibenzilico sul C-6 dell’accettore 65 con formazione del diolo 59 e altri numerosi prodotti che non sono stati analizzati. E’ da sottolineare che oltre al disaccaride 76a, si isola anche il 4-O-acetil derivato 80a (resa 72%), la cui formazione è spiegabile solo ammettendo un’apertura non convenzionale dell’intermedio ortoestereo
78a.
Generalmente, come già precedentemente detto nel capitolo 1, i derivati 1,2-ortoesterei costituiscono i prodotti secondari nelle reazioni di glicosidazione con tricloroacetimmidati promosse da TMSOTf e sono, come dimostrato in molte reazioni di glicosidazione, precursori del legame glicosidico 1,2-trans quando in posizione 2 dell’accettore, è presente un gruppo partecipante. Il meccanismo di riarrangiamento dell’ortoestere 78a a prodotto glicosidico 76a è illustrato nello Schema 24.27
Schema 24 O AcO OAc O O OR3 OAc O AcO OAc O O OAc O AcO OAc O O OR3 OAc O O AcO OAc OR3 O OAc O OH AcO OAc OX OAc O NHAc R2O OR1 AcO OR R3OH o H2O O R2O OR1 NHAc R O O O + + R3O-TMS+ b a TMSOTf R3= + 78a Tf strada a strada b TMSOTf TMS+ + Tf 80a 76a 83a 83b 84a: X=R3 84b: X=OH
In particolare se il riarrangiamento segue la strada a) il TMSOTf promuove la formazione di un carbocatione ciclico (83a), termodinamicamente stabile, che subisce
l’attacco del nucleofilo (RO-TMS+) sul C-1 dalla parte meno ingombrata con formazione dell’1,2-trans-glicoside 76a e conseguente rigenerazione del gruppo acilico.
Se invece il riarrangiamento dell’ortoestere 78a segue la strada b), si forma la specie carica aciclica 83b che può subire l’attacco sia dell’accettore sia dell’acqua, eventualmente presente nell’ambiente di reazione, con formazione dell’accettore acetilato 80a e del disaccaride deprotetto in posizione 2’ (84a) o del prodotto idrolizzato (84b).27
La reazione di β-galattosidazione condotta con i 6-O-t-butildimetilisilil derivati 66-69 ha fornito risultati migliori ed i dati più rilevanti sono riportati nella Tabella 1. In particolare è stata valutata, a parità di donatore 22, l’influenza sia dell’aglicone che del gruppo protettore presente sulla posizione 3 del glicosil accettore. Le glicosidazioni sono state condotte: a) utilizzando quantità diverse di TMSOTf (0.1-0.5 eq.); b) tempi e temperature di reazione variabili; c) promotori diversi come il BF3⋅Et2O.
In particolare, la glicosidazione del 3-O-acetil accettore 67 usando come promotore il TMSOTf in presenza di setacci molecolari 4Å attivati, in analogia a quanto osservato per
65, fornisce l’ortoestere 78b la cui resa (59 e 37%) è dipendente dalle quantità di TMSOTf
impiegate (prove 4 e 5, Tabella 1).
E’ da sottolineare che conducendo le β-galattosidazioni degli accettori 66, 68 e 69 a temperatura ambiente e a tempi di reazione lunghi (12-20 ore) i risultati sono stati decisamente migliori e i desiderati disaccaridi 76d, 76c e 77 sono stati isolati, rispettivamente, in resa del 53, 47 e 62%. In queste condizioni con un’analisi TLC accurata si osserva un veloce consumo sia del donatore che dell’accettore con formazione di un prodotto intermedio che si converte, a temperatura ambiente, nel β-disaccaride desiderato. Questa evidenza suggerisce e conferma che inizialmente si forma un intermedio ortoestereo che successivamente riarrangia all’1,2-trans glicoside isolato.
Da un’analisi dei risultati ottenuti (Tabella 1) il problema maggiore che abbiamo riscontrato in queste reazioni è costituito sia dalla formazione dei corrispondenti ortoesteri (Prove 2, 4, 5, 8 e 9) sia dalla presenza del disaccaride 82 a struttura per-O-acetil-β-D
-Galp-(1→2)-per-O-acetil-α-D-Galp (Schema 25). La formazione di 82, che complica la
purificazione del disaccaride desiderato a causa della similare mobilità su silice, è spiegabile ammettendo che l’emiacetale 72, che si forma per idrolisi del donatore 22, subisca uno shift di acile dalla posizione 2 a quella anomerica a dare l’accettore 72a, il quale subisce, nell’ambiente di reazione, β-galattosidazione in posizione 2.
Schema 25 O OAc O AcO OAc OAc NH CCl3 OAc O AcO OAc OAc OH OAc O AcO OH OAc OAc O AcO O AcO OAc OAc OAc AcO O AcO OAc 22 idrolisi 72 shift di acile 72a β β β β-galattosidazione al C-2 22 82
E’ da sottolineare che nelle glicosidazioni effettate a scopo preparativo, il β-disaccaride e l’eventuale derivato 82 sono stati isolati in miscela e sono separati solo successivamente, una volta realizzata la rimozione selettiva del gruppo t-butildimetilsililetereo in posizione 6 nelle condizioni descritte nel successivo paragrafo.
L’isolamento degli ortoesteri 78a, 78b e 78c non doveva, in generale, costituire un problema visto che dall’analisi della letteratura27b si evince la possibilità di trasformare questi intermedi nei corrispondenti derivati disaccaridici per azione di un promotore acido come TMSOTf o AgOTf. Sfortunatamente, però, l’ortoestere 78b, sottoposto a trattamento con TSMOTf (0.3 equivalenti) in soluzione diclorometanica a temperatura ambiente, porta alla scissione della funzione ortoesterea con formazione dell’accettore 67 e del tetra-O-acetil derivato 72. Anche l’apertura dell’ortoestere 78c condotta come precedentemente descritto per 78b, ma in presenza dell’accettore 68 (0.3 equivalenti) ha fornito risultati deludenti e si recupera, oltre all’accettore 68 (in quantità doppia rispetto a quella usata) e allo stesso ortoestere 78c (12%), il 4-O-acetil derivato 80c (resa 44%) e il desiderato disaccaride 76c (resa 47%). Questo risultato conferma i due possibili riarrangiamenti degli ortoesteri descritti nello Schema 24.
Analizzando tutti i risultati ottenuti nelle β-galattosidazioni degli accettori 65-69, utilizzando come promotore il TMSOTf e come donatore il tricloacetimmidato 22, è stato evidenziato che le condizioni ottimali per ottenere i derivati disaccaridici sono: a) 1.5 equivalenti di donatore 22; b) la presenza di setacci molecolari AW-300 attivati; c) 0.5 equivalenti di TMSOTf aggiunto a -30°C; d) evoluzione della reazione a temperatura ambiente; e) tempi lunghi di reazione, che riducono notevolmente la formazione dell’indesiderato disaccaride 82.
Il confronto dei risultati ottenuti con gli accettori 66 e 68, portanti rispettivamente un gruppo etereo (benzilico) e uno estereo (benzoilico), evidenzia che le rese nei corrispondenti disaccaridi 76c (47%) e 76d (53%) sono del tutto paragonabili, ed il cambiamento della protezione sul C-3 da estere ad etere non fornisce gli incrementi di resa attesi. La piccola differenza osservata è probabilmente attribuibile ad un differente
ingombro sterico e non alle differenze elettroniche tra le due tipologie di gruppi protettori in posizione 3. Invece, come già osservato da Auzanneau e coll,16 il tipo di aglicone presente sull’accettore influenza notevolmente l’andamento della glicosidazione: passando, infatti, dal gruppo 3-azidopropilico a quello metilico si osserva un incremento di resa dal 47 al 62%.
Per cercare di migliorare le rese delle glicosidazioni sono stati utilizzati anche i donatori 71, 13a e 75 che richiedono sistemi di attivazione diversi, ma i risultati ottenuti sono stati del tutto insoddisfacenti. In particolare, nella glicosidazione tra gli accettori 65 e
68 e i donatori 71 e 13a utilizzando, rispettivamente, come promotore TMSOTf e AgOTf
si osserva, in entrambi casi, la rimozione della protezione al C-6 (PMB o TBDMS) a causa delle elevate quantità (2 equivalenti) di promotore necessarie ad attivare i donatori 71 e
13a. Infine, anche l’uso del tioglicoside 75 nella β-galattosidazione di 66, 68 e 69
promossa dal sistema NIS-AgOTf, che aveva fornito soddisfacenti risultati nella sintesi dell’unità trisaccaridica costituente il polisaccaride capsulare SP19F,28 non ha prodotto i risultati sperati e i corrispondenti β-disaccaridi 76c, 76d e 77 sono stati isolati in rese modeste (29-34%).
I disaccaridi 76a, 76c, 76d e 77, non riportati in letteratura, sono stati completamente caratterizzati dal punto di vista chimico-fisico e i dati NMR, ricavati utilizzando tecniche monodimensionali (1H e 13C) e bidimensionali (DEPT-135, COSY, HETCOR), sono in accordo con le strutture proposte (vedi parte sperimentale). In tutti i casi, nello spettro 13C, particolarmente significativo è il segnale relativo al C-4 (δ 75-76) che risulta deschermato di circa 7 ppm rispetto al corrispondente segnale dell’accettore (δ 68-69) per la presenza dell’unità β-galattosidica in tale posizione. Particolarmente indicativo, ai fini della determinazione della stereoselettività della reazione di glicosidazione, è l’alto valore della costante di accoppiamento J1’,2’ (7.3-7.5 Hz), che indica un assetto assiale-assiale dei protoni H-1' e H-2'.
Gli ortoesteri 78a, 78b e 78c, non riportati in letteratura, sono stati completamente caratterizzati dal punto di vista chimico-fisico e i dati NMR sono in accordo con le strutture proposte (vedi parte sperimentale). In particolare, la funzione ortoesterea è evidenziata, nello spettro protonico, da un singoletto (δ 1.51-1.68), integrante per tre protoni, assegnabile al metile e da un basso valore della costante di accoppiamento J1’,2’ (4.4-4.8 Hz), che indica un assetto equatoriale-assiale dei protoni H-1' e H-2'. Confermano
la struttura la presenza, nello spettro 13C, dei segnali attribuibili ad un carbonio quaternario (δ 122.6-123.7) e ad uno metilico (δ 26.7-127.0) tipici per un gruppo ortoestereo.
Il disaccaride 82 presenta dati analitici e spettroscopici in accordo con quanto riportato in letteratura.29 In particolare, nello spettro protonico, particolarmente significativo risulta il doppietto a δ 6.25 attribuibile al protone H-1, che risulta fortemente deschermato a causa dell’effetto anisotropico del carbonile estereo presente nella posizione 1. Inoltre, il basso valore della costante di accoppiamento J1,2 (3.8 Hz) indica un assetto equatoriale-assiale dei protoni H-1 e H-2. Anche in questo caso la β-galattosidazione è confermata dall’alto valore della costante di accoppiamento J1',2' (7.7 Hz) che indica la configurazione 1',2'-trans.
2.4. – Preparazione degli accettori 6OH-lattosamminici
A questo punto della sequenza sintetica si è resa necessaria la rimozione selettiva della protezione sul C-6 dei derivati lattosamminici 76a, 76c e 77, in modo da liberare la funzione alcolica primaria su cui operare la β-lattosidazione che avrebbe condotto ai desiderati tetrasaccaridi 38 e 39. A tal scopo, come precursori di accettori 6OH-lattosamminici sono stati scelti i disaccaridi 76c e 77, che avrebbero permesso, nel caso volessimo ottenere i tetrasaccaridi completamente deprotetti, un unico trattamento con quantità catalitiche di MeONa in soluzione metanolica nello step finale della sequenza sintetica.
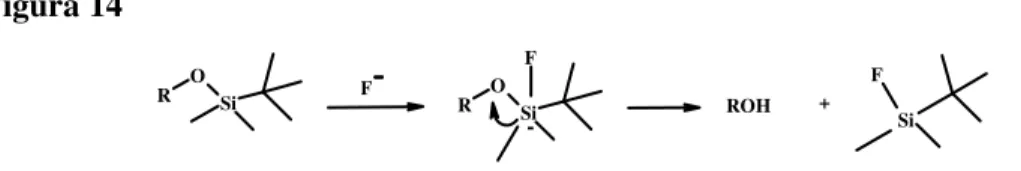
Dati di letteratura hanno evidenziano che i silileteri possono essere rimossi selettivamente mediante trattamento con ioni fluoruri, ad esempio con tetrabutilammonio fluoruro (TBAF) in soluzione di THF,26 senza influenzare gli altri gruppi protettori presenti sul substrato. In generale nel caso dei tri-alchilsilileteri, come il t-butildimetilsilile, la stabilità nei confronti degli acidi e delle basi è fortemente dipendente dal tipo di sostituenti presenti sul silicio ed è riportato che il grado di stabilità di questi composti è il seguente: TMS<TES<TBDMS<TIPS.
Il meccanismo di rimozione di un etere sililico per opera di ioni fluoruri è illustrato nella Figura 14. L’attacco nucleofilo dell’anione fluoruro sull’atomo di silicio, reso elettrofilo dalla presenza di orbitali ibridi d vacanti, porta ad una specie pentavalente che evolve verso l’alcol e il t-butildimetilsililfluoruro per rottura del legame Si-O, che avviene molto velocemente a causa della formazione del forte legame Si-F.
Figura 14 R O Si R O Si F ROH Si F F -+
Sulla base di queste premesse il disaccaride 76c (Schema 26) è stato sottoposto a reazione con TBAF in THF anidro alla temperatura di 0°C. Dopo 24 ore, l’analisi TLC evidenzia la scomparsa del prodotto di partenza e la formazione di due prodotti (Rf 0.39 e 0.34) visibili alla lampada UV. Dopo trattamento della reazione e purificazione flash cromatografica del grezzo di reazione si ottiene una frazione che all’analisi NMR risulta costituita dal desiderato disaccaride 85 e dal diolo 86 in rapporto 35:65 calcolato sulla base degli integrali dei segnali relativi ai protoni H-4’ a δ 5.11 e 5.04 rispettivamente.
La rimozione del gruppo acetilico in posizione 2’ è spiegabile ammettendo che la basicità dello ione fluoruro, accentuata dall’anidricità dell’ambiente di reazione, è sufficiente ad idrolizzare, oltre al TBDMS, anche gruppi acetati particolarmente reattivi come quello in posizione 2’.
Schema 26 O N3 O O NHAc OTBMS AcO O AcO OAc OAc BzO O O N3 O NHAc OH AcO O AcO OAc OAc BzO O O N3 O NHAc OH AcO O AcO OH OAc BzO 76c 85 86 + TBAF THF 85:86=35:65 AcOH aq 70%, 70°C resa 90%
Questo inconveniente è stato superato effettuando la desililazione selettiva di 76c e di
77 in AcOH acquoso26 al 70% alla temperatura di 70°C. Queste condizioni consentono,
dopo trattamento della reazione e purificazione flash cromatografia del grezzo, di isolare i 6-OH glicosidi 85 e 87 (Schema 27) in rese del 90% e 83% rispettivamente.
Schema 27 O O NHAc OTBDMS AcO O AcO OAc OAc BzO OMe O O NHAc OH AcO O AcO OAc OAc BzO OMe 77 87 AcOH aq 70%, 70°C resa 90%
Gli accettori disaccaridici 85 e 87, non riportati in letteratura, sono stati completamente caratterizzati dal punto di vista chimico-fisico e gli spettri NMR, in accordo con le strutture
proposte (vedi parte sperimentale), evidenziano la scomparsa dei segnali relativi al gruppo
t-butildimetilsilico.
Nel caso del diolo disaccaridico 86, isolato insieme a 85, l’analisi NMR della miscela consente di identificare solo alcuni segnali significativi di 86. In particolare, nello spettro protonico, particolarmente significativo è il multipletto a δ 3.70 attribuibile al protone H-2’ dell’unità galattosidica, che risulta schermato di circa 1.40 ppm rispetto al segnale del corrispondente protone di 85 (δ 5.10) per l’assenza della funzione esterea in tale posizione. Inoltre, ricordando che la presenza di gruppi esterei provoca un piccolo effetto di deschermo sui carboni in alfa ed un più rilevante effetto di schermo su quelli in beta, nello spettro 13C di 86 si osserva che i segnali assegnati ai carboni C-1’ (δ 104.2) e C-3’ (δ 73.6) dell’unità galatto risultano più deschermati di 2 ppm rispetto ai segnali 1’ (δ 101.3) e C-3’ (δ 71.5) di 85 a causa dell’assenza dell’acetato in posizione 2’.
2.5. – ββββ-lattosidazione dei derivati 6OH-lattosamminici 85 e 87
L’ultimo passaggio della nostra sequenza sintetica delle unità tetrasaccaridiche 38 e
39 (Schema 28) prevede una β-glicosidazione fra gli accettori 6OH-lattosamminici 85 e 87
e il tricloroacetimmidato lattosidico 20 che, prevedibilmente, dovrebbe presentare minori difficoltà rispetto a quanto descritto precedentemente nella β-galattosidazione degli accettori 4OH-glucosamminici. Ciò è spiegabile dal coinvolgimento di un gruppo alcolico primario più reattivo e più accessibile di uno secondario e dall’assenza di gruppi acido-labili, come PMB o TBDMS, che consente l’uso di quantità superiori di promotore. In particolare, visti i risultati ottenuti nelle precedenti β-galattosidazioni e quelli riportati in letteratura,12,15 per evitare la formazione di specie intermedie come quelle ortoesteree che avrebbero diminuito le rese in tetrasaccaride abbiamo deciso di utilizzare il BF3⋅Et2O come attivante del centro anomerico del donatore 22.
Sulla base di queste premesse un eccesso (1.4 equivalenti) di glicosil donatore 20 (Schema 28) è stato sottoposto a glicosidazione con gli accettori 85 e 87 (1 equivalente) impiegando BF3⋅Et2O (1.2 equivalenti) come promotore e CH2Cl2 anidro come solvente. La reazione, mantenuta anidra per la presenza di setacci molecolari AW-300 attivati, è condotta a partire dalla temperatura di -15°C per poi evolvere a temperatura ambiente. Dopo 4 ore l’analisi TLC ha evidenziato, in entrambi i casi, la scomparsa sia del donatore
trattamento delle reazioni di glicosidazione e le purificazione flash cromatografiche dei grezzi permettono di isolare i tetrasaccaridi 38 e 39 in rese soddisfacenti (74 e 70% rispettivamente) e chimicamente puri all’analisi NMR (1H e 13C).
Schema 28 O OAc OAc OAc AcO O OR O NHAc O OAc OAc OAc AcO O O OAc AcO O OAc BzO O OAc OAc OAc AcO O OR O NHAc OH BzO O OAc OAc OAc AcO O O OAc AcO O OAc CCl3 NH 20 + 85: R=CH2CH2CH2N3 87: R=Me 38: R=CH2CH2CH2N3 (resa 74%) 39: R=Me (resa 70%) BF3.Et2O, CH2Cl2 AW-300
I tetrasaccaridi 38 e 39, non riportati in letteratura, sono stati completamente caratterizzati dal punto di vista chimico-fisico e i dati NMR sono in accordo con le strutture proposte (vedi parte sperimentale e Figure 15-22 per il derivato 39). In particolare, gli spettri 1H e 13C NMR, pur permettendo un’immediata identificazione dei sostituenti (PhCOO, CH3CON, CH3COO, OCH2CH2CH2N3, NHAc,e OMe), presentano delle difficoltà quando si tenta un’interpretazione completa dei segnali protonici glicosidici che risuonano tutti in una zona molto ristretta dello spettro (5.3-3.4 ppm). Una buona risoluzione degli spettri, ottenuta usando solventi a bassa viscosità come il CD3CN, abbinata all’uso di tecniche bidimensionali (DEPT-135, COSY, HETCOR), hanno permesso di assegnare inequivocabilmente sia la struttura che la configurazione anomerica dei derivati 38 e 39. Generalmente, per strutture tetrasaccaridiche i segnali dei protoni anomerici (H-1, H-1', H-1'', H-1'''), per la loro semplice struttura (doppietti) e la loro posizione nello spettro (δ 5.0-4.4), più deschermati degli altri protoni piranosici, costituiscono il punto di partenza per l’interpretazione completa di un oligosaccaride.
Nel caso di 38 e 39 la presenza di ben 11 gruppi acetato sulle strutture rendono più difficile l’identificazione dei 4 protoni anomerici, in quanto questa zona risulta molto affollata per la presenza dei protoni piranosici deschermati dall’effetto anisotropico del carbonile acetilico. In entrambi i casi, nello spettro 13C, particolarmente significativo è il segnale relativo al C-6 dell’unità glucosamminica (δ 68.8) che risulta deschermato di circa 8 ppm rispetto al corrispondente segnale dell’accettore (δ 61.0) per la presenza dell’unità β-lattosidica in tale posizione.
Nonostante le difficoltà di interpretazione degli spettri protonici, oltre all’identificazione di tutti i protoni piranosici legati ai carboni funzionalizzati come acetati, sono stati individuati 4 doppietti relativi ai 4 protoni anomerici a δ 4.70 (H-1', J1′,2′ 7.9 Hz,), 4.56 (H-1''', J1″′,2″′ 7.8 Hz), 4.55 (H-1, J1,2 8.4 Hz) e 4.53 (H-1'', J1″,2″ 7.7 Hz,). Ai fini della determinazione della configurazione relativa al carbonio anomerico lattosidico C-1'' di 38 e
39, è stato considerato diagnostico il valore della costante di accoppiamento J1'',2'' di 7.7-7.9 Hz tipica di una disposizione trans dei protoni H-1'' e H-2'' (β-anomero).
2.6. – Sviluppi futuri
Con la preparazione dei tetrasaccaridi 38 e 39 è stato raggiunto lo scopo prefissato in questo lavoro di Tesi. Il tetrasaccaride 38 verrà utilizzato in successive ricerche volte alla preparazione di glycocluster coniugati con un opportuno peptide immunogenico che saranno testati, dalla Novartis Vaccines & Diagnostics di Siena, con lo scopo di valutare la loro attività immunogenica contro le infezioni da Streptococcus Pneumoniae 14 (SP14). In particolare, l’approccio sintetico a queste macromolecole è basato su una sequenza sintetica (Schema 29) che prevede: a) scambio degli acetati e del benzoato con gruppi benzilici; b) riduzione del gruppo azidico ad ammina; c) inserzione di uno spaziatore dicarbossilico per reazione con il cloruro dell’adipato di metile e successiva trasformazione dell’estere metilico ad acido carbossilico; d) coupling con strutture dendrimeriche (PAMAM) portanti sulla superficie un numero variabile (2,4, 8, etc.) di gruppi amminici; e) deprotezione totale e coniugazione con una proteina immunogenica (Schema 29).
Schema 29
Inoltre, i risultati ottenuti in questo lavoro di tesi saranno una buona base di partenza per realizzare la zwitterionizzazione del frammento tetrasaccaridico del polisaccaride SP14 in modo da ottenere derivati del tipo 88 e 89 (Schema 30), per i quali sarà valutata la loro