56
3. RISULTATI E DISCUSSIONE
3.1 Obiettivi della tesi
I risultati comunque interessanti ottenuti nelle reazioni di addizione nucleofila con le aziridine raceme di tipo carba 2.23α e 2.23β, precedentemente illustrati (Capitolo 2), avevano indicato l’efficacia di questi sistemi per la costruzione di amminocarbazuccheri, sia mediante utilizzo di questi substrati quali pseudo glicosil donatori in reazioni di addizione 1,4 coniugate, sia mediante utilizzo di questi substrati quali piccoli eterocicli tensionati e quindi in grado di dare una classica reattività sul carbonio aziridinico C(3) secondo un processo SN2 per dare prodotti di addizione
tipicamente 1,2-anti. In entrambi i casi, la funzionalizzazione del doppio legame residuo, in posizione 1,2, oppure 2,3, avrebbe permesso infatti la trasformazione completa di questi sistemi in amminocarbazuccheri (Schema 3.1).
N BnO Ns SN2 Nu BnO N Nu Ns H 1,2-funzionalizzazione BnO N Nu Ns H X Y BnO SN2' Nu Nu N Ns H BnO Nu N Ns H 2,3-funzionalizzazione Y X 1 2 2 3 2.23 N BnO Ns SN2 Nu BnO N Nu Ns H 1,2-funzionalizzazione BnO N Nu Ns H X Y BnO SN2' Nu Nu N Ns H BnO Nu N Ns H 2,3-funzionalizzazione Y X 1 2 2 3 2.23
Amminoprotetto carbazucchero Amminoprotetto carbazucchero
57 Nella prima fase degli studi descritti sulle vinil aziridine di tipo carba, non era stato ritenuto necessario disporre delle aziridine 2.23α e 2.23β come enantiomeri puri e quindi le stesse erano state sintetizzate in forma racema. Verificata comunque la possibilità di poter sfruttare questi nuovi sistemi per la sintesi di amminocarbazuccheri, scopo della prima parte dellla mia tesi è stato quello di sintetizzare le N-acetil aziridine
3.1α e 3.1β enantiomericamente pure, poiché questo risultava importante per la sintesi
di molecole di interesse biologico (Fig. 3.1).
BnO BnO N N Ac Ac 3.1 3.1
Figura 3.1. Carba N-acetil aziridine 3.1α e 3.1β.
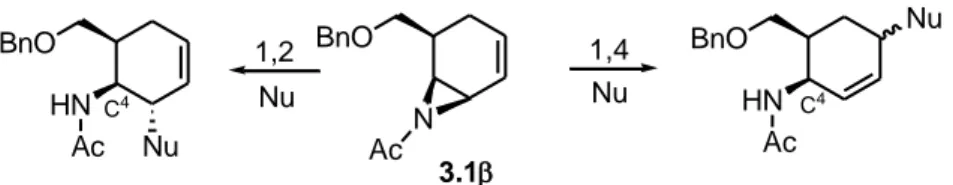
La scelta di introdurre come gruppo sostituente sull’azoto l’acetile nelle aziridine 3.1α e 3.1β, piuttosto che il nosile, come precedentemente fatto per i sistemi racemi 2.23α e 2.23β, è stata fatta sulla base della considerazione che la risultante funzionalità N-acetil ammino sul C(4), presente sui possibili sistemi derivanti dall’addizione nucleofila sia 1,2 che 1,4 (Fig. 3.2 dove per semplicità si mostra solo l’aziridina 3.1β) avrebbe rappresentato un gruppo nativo, naturalmente presente in molti amminocarbazuccheri naturali (vedi allosamidina, Fig. 1.3, della Introduzione).
BnO N Ac BnO Nu HN Ac 1,4 Nu BnO HN Ac Nu 1,2 Nu 3.1 C4 C4
Figura 3.2. Inserimento regio- e stereoselettivo della funzionalità N-acetilammino sul C(4) del
sistema carba 3.1β.
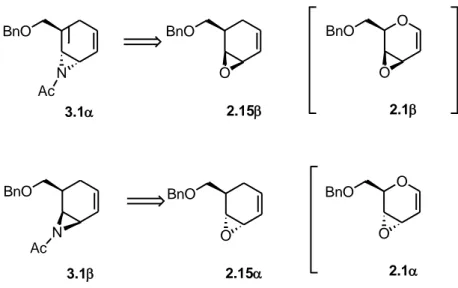
Sulla base dell’esperienza sintetica acquisita nella sintesi delle carba N-nosil aziridine raceme 2.23α e 2.23β, abbiamo individuato come precursori delle aziridine
3.1α e 3.1β, rispettivamente i carba vinil epossidi 2.15β e 2.15α, carba analoghi degli
58 BnO O BnO O 2.15 2.15 BnO BnO N N Ac Ac 3.1 3.1 O BnO O 2.1 O BnO O 2.1
Figura 3.3. Vinil carba epossidi 2.15β e 2.15α, precursori delle carba N-acetil aziridine 3.1α e 3.1β.
Primo obiettivo della mia tesi è stato quindi quello di sintetizzare i carba vinil epossidi 2.15α e 2.15β in modo enantioselettivo. Questo approccio, in realtà, era già stato indagato precedentemente,108 ma la sintesi era stata effettuata solo su piccola scala. Mi sono quindi occupata, inizialmente, di mettere a punto il processo sintetico su una scala decisamente maggiore che permettesse poi di elaborare i sistemi 2.15β e 2.15α nelle rispettive aziridine 3.1α e 3.1β. Sono stati così risolti alcuni problemi sintetici che abbassavano notevolmente la resa complessiva del processo di sintesi degli epossidi
2.15α e 2.15β, e al fine di realizzare una sintesi più sostenibile, sono stati ridotti i
processi di manipolazione e purificazione di molti intermedi sintetici e in un passaggio chiave (riarrangiamento di Claisen) abbiamo utilizzato la tecnica del microonde, che ha reso questo passaggio cruciale più agevole e facilmente realizzabile su una buona scala (500 mg di substrato di riarrangiamento in circa 20 minuti).
3.2. Sintesi dei carba analoghi dei vinil epossidi derivati dal
D-allale e dal
D-galattale
Fra i vari approcci sintetici riportati per ottenere carbazuccheri otticamente attivi, abbiamo ritenuto particolarmente vantaggioso, per i nostri scopi, utilizzare, adattandolo alle nostre esigenze, il riarrangiamento di Claisen dei glicali descritto da Nagarajan e Sudha.109 Questo tipo di approccio ci sembrava particolarmente utile
59 poiché consentiva la trasformazione del sistema glicale 3.2 nel corrispondente sistema cicloesenico 3.3, in cui al posto dell’ossigeno endociclico dei glicali, si trova un CH2
metilenico (Schema 3.2). O HO OBn BnO OBn BnO HO 3.2 3.3
Schema 3.2. Trasformazione del sistema glicale in sistema carba glicale
La conservazione del doppio legame endociclico, nel nostro caso, era ovviamente fondamentale per costruire gli epossidi vinilici, analoghi degli epossidi derivati dal glicale. Non risultavano invece utili i due eteri benzilici in posizione C(3) e C(4), presenti in 3.3, perché la loro deprotezione, per idrogenazione catalitica, che avrebbe poi condotto tramite protezioni ortogonali, alla introduzione dell’anello ossiranico, avrebbe ridotto anche il doppio legame, non permettendo più la costruzione del sistema vinil ossiranico di nostro interesse.
La prima parte della via sintetica da noi elaborata, sulla base del conosciuto riarrangiamento di Claisen sopra citato, ha previsto quindi la trasformazione del triacetil D-glucale 3.4 nel suo carba analogo 3.5 (Schema 3.3), in cui la presenza dei p-metossi-benzil eteri in posizione C(3) e C(4), avrebbe garantito la loro deprotezione a gruppi ossidrilici, senza intaccare il doppio legame.
O AcO OAc AcO OPMB PMBO HO 3.4 3.5 OH HO RO 3.6
60 L’approccio da noi individuato e messo a punto per la sintesi del carba derivato
3.5, prevede inizialmente la sintesi del 3,4-di-O-(p-metossi benzil)-glucale (3.11)
secondo la procedura illustrata nello Schema 3.4.
CRL,(iPr)2O CH3COCH3 tampone fosfato DHP PPTS CH2Cl2 MeONa MeOH PMBCl NaH DMF AcOH/THF/H2O 45°C O OAc AcO AcO O OAc AcO HO O OAc AcO THPO O OH HO THPO O OPMB PMBO THPO O OPMB PMBO HO 3.4 3.7 3.8 (-)3.11 3.10 3.9 91% >99% >99% >99% 65%
Schema 3.4. Procedura sintetica per l’ottenimento dell’alcool 3.11
Il tri-O-acetil-D-glucale (3.4) viene monosaponificato regioselettivamente sull’ossidrile primario, utilizzando un’originale procedura messa a punto nel nostro laboratorio,110 che, sfruttando la lipasi CCL (lipasi da Candida Cylindracea) in tampone fosfato a pH 7.0, diisopropiletere e acetone, fornisce l’alcool 3.7 in alta resa (87%). Questa monosaponificazione regioselettiva enzimatica, inizialmente condotta secondo la classica procedura utilizzata da tempo nel nostro laboratorio, è stata modificata nell’ultimo periodo della mia tesi. La necessità di cambiamento è nata essenzialmente dal fatto che in commercio (ditta fornitrice Aldrich) non era più disponibile la Lipasi CCL (lipasi da Candida Cylindracea di tipo II) con attività ≈ 2 unità/mg, da noi originariamente utilizzata, ma era disponibile una lipasi equivalente alla precedente ma dotata di maggiore attività ossia la lipasi da Candida rugosa (tipo VII con un’attività ≥ 700 unità/mg). Anche la lipasi da Candida rugosa di tipo VII lavora in condizioni ottimali a pH 7.00 e a 25°C, come la Candida Cylindracea, ma essendo più attiva può essere utilizzata in quantità decisamente minori in presenza di un volume minore di
61 tampone fosfato a pH 7.00, con notevole miglioramento del work up di reazione. Infatti, mentre in precedenza utilizzando la Lipasi di tipo II, si formava facilmente nella fase di work up un’emulsione difficile da rompere, che abbassava notevolmente le rese del processo sintetico, con la lipasi da Candida rugosa di tipo VII, il work up (filtrazione su celite ed estrazione con AcOEt) è risultato estremamente agevole, non formandosi mai l’emulsione e con una resa del passaggio sintetico sempre molto alta (80-90%). Ottenuto l’alcool primario 3.7, questo è stato protetto selettivamente per trattamento con 3,4-2H-diidropirano (DHP) in presenza di piridinio p-toluensolfonato (PPTS) per dare il THP-derivato 3.8. Successivamente, la saponificazione di 3.8 con MeONa/MeOH portava al diolo trans 3.9, che doveva essere protetto come p-metossi-benzil etere sia sulla posizione C(3) che C(4). La reazione di protezione è stata effettuata mediante deprotonazione del diolo 3.9 con NaH in DMF a 0 °C; all’alcolato così ottenuto, è stato quindi addizionato p-metossi-benzil cloruro (PMBCl) e la miscela di reazione è stata lasciata a temperatura ambiente per 12 ore. Da sottolineare come l’uso di solventi diversi (THF o miscela 1:1 THF/DMF) non consenta di ottenere il prodotto 3.10 con rese significative e come il processo sintetico proceda fino all’ottenimento del glicale completamente protetto 3.10 senza alcuna purificazione. In questo modo, la resa complessiva del glicale 3.10 a partire dal tri-O-acetil-D-glucale 3.4 è pari al 91%. Una volta ottenuto il derivato 3.10 ho quindi proceduto alla deprotezione del gruppo acetalico con ottenimento del desiderato alcool primario 3.11.
Come indicato in letteratura,111 abbiamo utilizzato come protocollo di deprotezione quello che fa uso di AcOH aq. Così, il riscaldamento a 45°C per 12 ore di una soluzione del O-THP derivato 3.10 in AcOH/THF/H2O 1.5:2:1 conduce all’alcool
primario 3.11 quale unico prodotto che dopo purificazione, mediante flash cromatografia, viene ottenuto puro con una resa del 65%. La reazione condotta a temperatura ambiente, procede in modo analogo ma con tempi più lunghi (24 ore). E’ importante sottolineare che l’alcool primario 3.11 grezzo deve essere purificato per flash cromatografia o per cristallizzazione, immediatamente dopo la sua preparazione, in quanto eventuali tracce di acido presenti, possono portare ad una sua rapida decomposizione e una purificazione tardiva comporta spesso un drastico abbassamento delle rese con un recupero solo del 15% circa di alcool 3.11.
A questo punto il progetto sintetico prevedeva la trasformazione dell’alcool primario 3.11 nel diolo carbaciclico 3.12, che sarebbe poi stato elaborato nei desiderati
62 carba epossidi vinilici 2.15β e 2.15α (Schema 3.5). Il diolo trans 3.12 rappresenta un cruciale intermedio di tipo carba a partire dal quale, con un processo stereodivergente, si ottengono entrambi i vinil epossidi diasteroisomerici 2.15β e 2.15α.
O OPMB PMBO HO (-)3.11 (+) 3.12 (-) 2.15 OH HO BnO BnO O BnO O (-)2.15
Schema 3.5. Intermedi chiave 3.11 e 3.12 nella sintesi dell’epossido 2.15β e 2.15α.
La trasformazione dell’alcool primario 3.11 nel diolo carbaciclico 3.12, è stata realizzata sulla base dello schema sintetico qui riportato (Schema 3.6).
KHMDS THF IBX CH3CN 45°C Ph3PCH3I OPMB PMBO HO 1,3-diclorobenzene NaBH4 OPMB PMBO BnO NaH/BnBr DMF DDQ CH2Cl2 OH HO BnO (-) 3.11 3.13 (+) 3.14 3.15 (+) 3.16 (+) 3.12 OPMB PMBO H O (-)3.5 O OPMB PMBO HO OHC O OPMB PMBO O OPMB PMBO >99% >99% 88% 68% 65% microonde EtOH
63 L’ossidazione dell’alcool 3.11 è stata realizzata utilizzando come agente ossidante, l’acido 2-iodossi benzoico (IBX), sulla base delle caratteristiche riportate in letteratura.112
L’1-idrossi-1,2-benzoiodossol-3(1H)-one-1-ossido o acido iodossi benzoico (3.17) è da tempo conosciuto come agente ossidante, sebbene solo la sua trasformazione nel periodinano 3.18 (il ben noto periodinano di Dess-Martin DMP), ne ha decretato il largo impiego come agente ossidante, trasformandolo da reagente fortemente insolubile, in reagente molto solubile nei comuni solventi organici (Schema 3.7).
O I O HO O -: O I O AcO : OAc OAc Ac2O/AcOH 3.17 3.18 IBX DMP Schema 3.7. IBX e periodinano DMP
L’IBX è di per sé un agente ossidante di alcooli estremamente versatile e blando, in grado di ossidare dioli vicinali, senza rompere il legame C-C del glicol, 1,4-dioli a γ-lattoli e in grado di determinare la chiusura di ammino alcooli ad aminali ciclici. Uno dei problemi relativi all’uso dell’IBX, riguarda la sua preparazione, poiché le procedure in genere riportate comportano l’uso di agenti tossici o poco pratici, oppure forniscono un reagente di scarsa qualità che deve essere rigorosamente purificato. Recentemente, tuttavia,100 è stata riportata una procedura preparativa estremamente efficace, semplice e innocua, che prevede l’ossidazione dell’acido 2-iodobenzoico con Oxone® (2KHSO5
-KHSO4-K2SO4) a 70 °C per 3 ore. L’IBX così ottenuto è recuperato in alta resa (80%) e
con elevato grado di purezza (≥ 95%). Utilizzando questa procedura di Frigerio e coll.100 abbiamo quindi preparato di fresco l’IBX, e l’abbiamo utilizzato per ossidare l’alcool 3.11, conducendo la reazione in acetonitrile, a 45 °C. La reazione ha portato, in modo estremamente pulito, all’aldeide 3.13 desiderata (Schema 3.6), che è stata utilizzata senza ulteriori purificazioni per il passaggio successivo. La reazione di ossidazione con l’IBX procede in maniera efficace sia su piccola scala (200 mg), che su larga scala (5 g), risultando quindi particolarmente utile ai nostri scopi, che prevedevano
64 la preparazione di grandi quantità di aldeide. È da sottolineare che l’uso di temperature superiori a 45 °C è estremamente sconveniente, perché accanto alla formazione di 3.13, determina la formazione, in alte percentuali, (≈50 %), di p-metossi-benzaldeide (Schema 3.8). O OPMB PMBO H O OMe CHO IBX CH3CN T> 45°C 50% : 50% O OPMB PMBO HO H3CO PMB = 3.11 3.13 CH2
Schema 3.8. Miscela ottenuta per ossidazione di 3.11 a temperature superiori ai 40 °C Infatti, il residuo, p-metossi-benzilico, per effetto mesomerico di rilascio elettronico del sostituente p-OCH3 va facilmente incontro a processi di ossidazione a
carico del carbonio benzilico a dare l’aldeide corrispondente. Quando i gruppi alcolici in C(3) e C(4), sono protetti invece sotto forma di eteri benzilici, la corrispondente formazione di benzaldeide è ovviamente meno probabile, mancando l’effetto mesomerico del gruppo –OCH3, ed è quindi possibile condurre l’ossidazione anche a
temperature superiori ai 45 °C.
L’aldeide 3.13 è piuttosto instabile ma può comunque essere conservata a bassa temperatura (-78 °C) per almeno 36 ore, ed è utilizzata come substrato per la reazione di Wittig, che fornisce l’alchene 3.14. In questo senso, l’ilide del fosforo ottenuta per reazione fra il metil-trifenil fosfonio ioduro (Ph3P+MeI-) e potassio esametildisilazide (KHMDS) in THF, viene fatta reagire con l’aldeide 3.13 per fornire il desiderato alchene esociclico 3.14. Inizialmente il work up della reazione veniva effettuato filtrando la miscela di reazione su buchner con silice e fluorisil®, estraendo poi con Et2O
e lavando con soluzioni sature di NH4Cl e NaCl. Questo trattamento consentiva di
ottenere un prodotto grezzo sufficientemente puro, ma ancora inquinato dalla presenza della Ph3PO, che si forma durante la reazione di Wittig, e che poteva interferire con le
65 grezza permetteva di ottenere un prodotto pulito, ma abbassava in modo consistente le rese (35%). Abbiamo allora messo a punto un trattamento che ci ha fornito un grezzo (1H NMR) estremamente pulito, costituito essenzialmente dall’olefina 3.14, ottenuto con rese ancora alte (88%) ed in cui il residuo di Ph3PO era inferiore al 5%. Il prodotto
così ottenuto è stato utilizzato, direttamente, senza ulteriori purificazioni, negli steps successivi migliorando complessivamente la resa del processo sintetico. L’olefina 3.14 rappresenta il substrato necessario per realizzare il passaggio cruciale dello schema sintetico, ovvero la trasformazione del sistema glicale nel corrispondente sistema carbaciclico. Il riarrangiamento termico di Claisen prevede, a livello concettuale, la trasformazione di un allil vinil etere in un composto carbonilico γ,δ-insaturo, come illustrato nello Schema 3.9.
O O H
Schema 3.9. Riarrangiamento termico di Claisen
Il glicale 3.14, con un doppio legame endociclico ed uno esociclico, rappresenta un particolare allil vinil etere che può comunque subire il riarrangiamento indicato. Il meccanismo ragionevolmente coinvolto nel riarrangiamento del vinil allil etere 3.14 nel corrispondente composto carbonilico γ,δ-insaturo, l’aldeide insatura 3.15, secondo le procedure di Claisen è mostrato nello Schema 3.10.
66 O O RO RO 1 2 3 4 5 6 7 O OR OR 1 2 3 4 5 6 7 O H OR H H OR 7 6 5 4 3 2 1 OR OR CHO RO RO CHO 1 2 3 4 5 6 7 1 2 3 4 5 6 7 RO RO 1 2 3 4 6 7 OHC 5 3.14 3.15 R=PMB RO OR 1 2 3 4 5 6 7 O OR RO
___
___
___
___
A B 1 2 3 4 5 6 7Schema 3.10. Riarrangiamento termico di Claisen applicato al sistema glicale
L’aldeide 3.15 così ottenuta, in cui il carbonio aldeidico C(1) deriva dal carbonio vinilico C(1) del sistema glicale 3.14, è instabile e deve essere immediatamente convertita, nel work up di reazione, nell’alcool primario 3.5, per suo trattamento con NaBH4 in THF/EtOH (Schema 3.6). Questa interessante trasformazione che rappresenta
un’applicazione del riarrangiamento di Claisen originariamente applicato da Sudha e Nagarajan97 all’analogo di 3.14 3,4 dibenzilato costituisce, come già detto, il passaggio cruciale dello schema sintetico che porta ai carba vinil epossidi 2.15α e 2.15β. Il riarrangiamento procede per via termica e in precedenza, nel nostro laboratorio, era stato realizzato riscaldando a 240°C l’olefina 3.13 in 1,3-diclorobenzene in fiala chiusa. Per raggiungere una temperatura così elevata e che si mantenesse stabile, abbiamo fatto uso, in alternativa al bagno di sabbia utilizzato da altri autori, di un olio di silicone, l’AP 100 (Aldrich), perfettamente idoneo allo scopo in quanto non infiammabile a tale temperatura. Tuttavia il riscaldamento a temperature così elevate rappresentava sempre un potenziale pericolo, tanto è vero che in un caso si è verificata una rottura della fiala con esplosione, che per quanto non abbia causato danni per la presenza costante di uno schermo protettivo, ci ha convinto della necessità di trovare procedure più sicure. Inoltre in queste condizioni, la reazione non poteva essere scalata, sempre per motivi di sicurezza (volume coinvolto), oltre un certo livello (250 mg).
67 Nell’ottica di realizzare un processo più sostenibile, abbiamo voluto verificare la fattibilità del riarrangiamento utilizzando la tecnica che fa uso della microonde. Così utilizzando uno strumento, presente nel nostro dipartimento, Microonde CEM Discover, abbiamo trovato le condizioni operative migliori per far avvenire il riarrangiamento in questione in maniera decisamente più sicuro, efficace e con un deciso risparmio energetico.
Abbiamo così osservato che irradiando 500 mg di olefina 3.14 in 4 mL di 1,3-diclorobenzene con una potenza di 300 W, alla pressione di 100 Psi con un “ramp time” di un minuto per 20 minuti si ottiene l’aldeide 3.15 che non viene isolata, ma immediatamente ridotta con NaBH4 per dare l’alcool carba 3.5 che viene così isolato
puro in buona resa (68%) dopo purificazione per flash cromatografia. Da sottolineare che la quantità massima di olefina in questo caso singolarmente riarrangiata era pari a 500 mg, ma ripetendo molto semplicemente il riarrangiamento per tre volte, in un ora era possibile riarrangiare 1.5 g di olefina. Secondo la procedura iniziale, per riarrangiare 1.5 g di olefina erano necessarie 6 ore con tutti gli inconvenienti sopra accennati.
L’alcool primario carbaciclico 3.5 (schema 3.6) è stato quindi benzilato sull’ossidrile primario, facendo uso del classico protocollo NaH/BnBr in DMF, che fornisce il carbaciclo insaturo completamente protetto 3.16 con resa superiore al 99% e sufficientemente puro da poter essere utilizzato nel passaggio successivo.
L’ultimo passaggio previsto per la sintesi del diolo 3.12, consisteva nella deprotezione nelle due funzionalità p-metossi-benzil eteree in posizione C(3) e C(4) (Schema 3.6). L’utilizzo di questo tipo di eteri, nel nostro caso, era particolarmente utile perché, come già detto precedentemente, la deprotezione poteva essere effettuata mediante un sistema a trasferimento elettronico (SET), che lasciava intatto il doppio legame endociclico come da noi richiesto. Come agente ossidante in grado di realizzare il sistema a trasferimento elettronico, abbiamo scelto il 2,3-dicloro-5,6-diciano benzochinone (DDQ).
68 MeO H O MeO MeO OR H MeO OR MeO OR H H MeO OR H Cl Cl O O CN CN Cl Cl O -CN CN O Cl Cl OH CN CN O O CN CN OH Cl Cl OH Cl Cl OH CN CN OR H OH . . . . .. ROH SET + H+ +H2O - H+ .. .
Schema 3.11. Meccanismo di deprotezione mediante SET (sistema a trasferimento elettronico)
con DDQ
Il meccanismo di deprotezione113 implica la formazione di uno ione ossonio che può essere catturato dall’acqua con formazione di un emiacetale da cui viene eliminato l’alcool desiderato con formazione delle p-metossi benzaldeidi (Schema 3.11).
Così il trattamento dell’olefina 3.16 in CH2Cl2/H2O 18:1 in presenza di DDQ
(1.5 equiv) fornisce dopo 2 ore a temperatura ambiente il diolo trans 3.12 con resa del 65% dopo rapida filtrazione su gel di silice.
Il diolo trans 3.12 rappresenta l’ intermedio sintetico dal quale origina la sintesi stereodivergente dei due epossidi 2.15α e 2.15β (Schema 3.12).
69 OH HO BnO BnO O BnO O (-)2.15 (+) 3.12 (-)2.15
Schema 3.12. Intermedio per la sintesi stereodivergente degli epossidi 2.15α e 2.15β Per la sintesi dell’epossido 2.15β, il diolo trans 3.12 viene trattato con pivaloil cloruro (PivCl), ossia con un cloruro acido stericamente ingombrato che reagisce più velocemente sul meno ingombrato e più reattivo gruppo ossidrilico allilico, a dare il mono pivaloil derivato allilico 3.19. La successiva mesilazione conduce al mesilato 3.20 che viene sottoposto a saponificazione con CH3ONa. La liberazione della specie
alcolato, innesca il processo di ciclizzazione sul carbonio adiacente portante il buon gruppo uscente a dare l’epossido 2.15β con resa dell’88% calcolata sul diolo 3.12 di partenza (schema 3.13). OH HO BnO OPiv HO BnO OPiv MsO BnO BnO O (+) 3.12 (-) 3.19 (-) 3.20 (-)2.15 PivCl DMAP, Py CH2Cl2 MsCl Py MeONa CH3CN >99% 88% >99%
Schema 3.13. Sintesi dell’epossido 2.15β a partire dal diolo 3.12.
Il diolo 3.12, come detto, è anche il sistema di partenza per l’epossido 2.15α. In questo caso per ottenere la desiderata stereochimica era necessario introdurre un buon gruppo uscente in posizione allilica, dovevamo procedere quindi alla sintesi, ad esempio, dell’opportuno mesilato allilico. Come noto in letteratura,114
i mesilati allilici alifatici secondari (SAAM) sono caratterizzati da un’alta reattività e instabilità, ciò nonostante è possibile sintetizzarli a bassa temperatura, utilizzando ad esempio anidride mesilica (Ms2O) e Et3N a 0°C. I SAAM sono troppo reattivi per essere isolati, ma
possono venire trasformati efficacemente, in situ, nei corrispondenti prodotti in seguito ad un processo di solvolisi (con H2O, MeOH o AcOH) o sostituzione con vari
nucleofili.
Facendo riferimento a questa procedura114 il diolo 3.12 in THF, Et3N e Py in
quantità catalitica (5%) è stato trattato a 0°C con Ms2O (1 equiv) per dare il mesilato
70 viene trattata con t-BuOK (2 equiv), che determina la formazione dell’alcolato sul C(4) di 3.21 che innesca il processo di ciclizzazione di tipo SN2 intramolecolare, con
inversione di configurazione sul C(3) allilico portante il buon gruppo uscente, a dare il desiderato epossido 2.15α (Schema 3.14) che viene isolato, praticamente puro in buona resa (60%). OH HO BnO OMs HO BnO BnO O Ms2O/ Et3N THF Py (5%) t-BuOK (+) 3.12 3.21 (-) 2.15 60%
Schema 3.14. Sintesi dell’epossido 2.15α a partire dal diolo 3.12.
E’ comunque da sottolineare che la formazione dell’epossido 2.15α non va a completezza: utilizzando 1 equiv di Ms2O, accanto al desiderato epossido 2.15α si
recupera circa il 20% di diolo 3.12 non reagito, ed utilizzando un leggero eccesso di Ms2O (1.2 equiv) si forma anche una piccola quantità (≈ 10%) del corrispondente
dimesilato sul C(3) e sul C(4). In entrambi i casi l’epossido 2.15α può essere efficacemente purificato per flash cromatografia con una resa che va dal 60% (nel caso dell’utilizzo di 1 equiv di Ms2O) al 75% (nel caso dell’utilizzo di 1.2 equiv di Ms2O).
Risulta comunque più conveniente utilizzare 1 equiv di Ms2O perché, anche se le rese in
epossido sono inferiori, il diolo 3.12 non reagito può essere efficacemente riciclato. La procedura da noi adottata per sintetizzare l’epossido 2.15α è comunque molto conveniente, anche se l’anidride mesilica è un reagente delicato che dà buoni risultati solo se aperta di fresco e se correttamente prelevata e conservata sotto atmosfera di gas inerte.
71
3.3 Sintesi dei carba analoghi delle N-acetil vinil aziridine derivate dal
D-allale e
D-galattale
I miglioramenti realizzati e precedentemente descritti nello schema sintetico che conduce agli epossidi 2.15α e 2.15β, avevano permesso di ottenere gli epossidi desiderati in modo più veloce e in quantità decisamente maggiore. Quest’ultimo aspetto era fondamentale, considerando che gli epossidi 2.15α e 2.15β sono i prodotti di partenza per la sintesi rispettivamente delle aziridine 3.1β e 3.1α e quindi avere una buona disponibilità degli epossidi avrebbe permesso di sintetizzare discrete quantità delle desiderate aziridine.
La sintesi delle aziridine 3.1α e 3.1β inizia con la trasformazione di entrambi gli epossidi 2.15β e 2.15α nei corrispondenti trans azido alcooli omoallilici.
Inizialmente l’epossido 2.15β era stato sottoposto a reazione di azidolisi secondo il classico protocollo NaN3/NH4Cl in MeOH/H2O che aveva condotto ad una miscela
costituita dall’azido alcool trans desiderato 3.22 (80% 1
H NMR) accompagnato dal prodotto regioisomero di addizione 1,4-anti, l’azido alcool trans 3.23 (20%
1
HNMR).115(schema 3.15) La formazione del prodotto 3.23 derivante da una addizione 1,4, in questo caso non di nostro interesse, abbatteva notevolmente la resa di una reazione semplice per la quale c’era la necessità di realizzare un’elevata conversione nell’azido alcool 3.22 desiderato. Il processo di azidolisi è stato così modificato ed è stato seguito un protocollo riportato in letteratura che fa uso di NaN3/THF/H20.116In
queste condizioni il trans azido alcool 3.22 è stato ottenuto come unico prodotto di reazione, con resa quantitativa (>99%) (schema 3.15).
BnO BnO O HO N3 BnO HO N3 + BnO HO N3 NaN3 THF/H2O NaN3 NH4Cl MeOH/H2O (-) 2.15 (+) 3.22 (+) 3.22 3.23 unico prodotto (20 : 80)
Schema 3.15. Sintesi dell’azido alcool 3.22.
L’azido alcool viene ridotto in fase eterogenea con PPh3 supportata su polimero
72 è stato quindi protetto a dare l’N-acetil derivato 3.25, per trattamento con 1 equiv di cloruro di acetile (CH3COCl) in CH2Cl2 a 0°C in presenza di Et3N (1.3 equiv). La
maggiore nucleofilicità del gruppo amminico in posizione allilica ha permesso la protezione regioselettiva del gruppo amminico rispetto al gruppo ossidrilico in C(4).
La successiva mesilazione del gruppo ossidrilico in C(4) conduce all’O-mesil-N-acetil derivato 3.26 che costituisce l’immediato precursore dell’aziridina 3.1α. Il trattamento di 3.26 con t-BuOK in benzene conduce alla desiderata N-acetil aziridina
3.1α con una resa del 93%. In queste condizioni di reazione, infatti, in considerazione
del pKa di un’ammide secondaria (pKa=17-18), il trattamento di 3.26 con t-BuOK determina la deprotonazione del legame N-H a dare il corrispondente anione 3.27 che, secondo un processo SN2 intramolecolare, entropicamente favorito, determina un
attacco nucleofilo sul C(4) adiacente portante il buon gruppo uscente a dare l’aziridina
3.1α (Schema 3.16). BnO HO N3 BnO HO NH2 BnO HO NHCOCH3 BnO MsO NHCOCH3 BnO N PPh3 supp THF CH3COCl (1 equiv) CH2Cl2/ Et3N MsCl, Py t-BuOK BnO MsO NCOCH3 OBn OMs NCOCH3 OBn N H3COC Ac (-) 3.1 benzene (+)3.22 (+)3.24 (+) 3.25 (+) 3.26 3.27 90% 96% 76% 93%
Schema 3.16. Sintesi dell’aziridina 3.1α.
In modo del tutto analogo ho proceduto alla sintesi dell’aziridina 3.1β. Anche in questo caso l’azidolisi dell’epossido 2.15α condotto con NaN3 in THF/H2O è
73 completamente regioselettivo e conduce al solo azido alcool trans 3.28, che viene successivamente ridotto ad ammino alcool 3.29 con PPh3 supportata. L’ammino alcool 3.29, anche in questo caso, viene protetto sulla funzionalità amminica con CH3COCl a
dare il trans N-acetil derivato 3.30 che per successiva mesilazione sul C(4) con MsCl/Py fornisce il trans N-acetil-O-mesilato 3.31, precursore ultimo della N-acetil aziridina 3.1β. La ciclizzazione base-catalizzata di 3.31 con t-BuOK in benzene conduce alla desiderata aziridina 3.1β con resa del 95% e con una resa complessiva del processo a partire dall’epossido 2.15α del 48% (schema 3.17).
BnO N3 HO BnO O NH2 HO BnO NHCOCH3 HO BnO NHCOCH3 MsO BnO BnO N NaN3 THF/H2O PPh3 supp THF CH3COCl CH2Cl2 Et3N MsCl Py Ac t-BuOK benzene (-) 2.15 (-) 3.1 (-) 3.28 (+) 3.29 (+) 3.30 (+) 3.31 >99% 96% 83% 63% 95%
Schema 3.17. Sintesi dell’aziridina 3.1β.
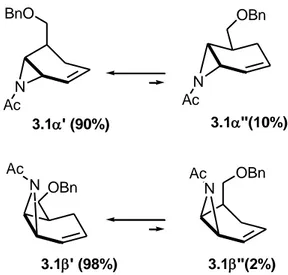
Calcoli teorici appropriatamente condotti su modelli semplificati hanno indicato che le aziridine 3.1α e 3.1β esistono fondamentalmente come un equilibrio 90:10 fra
3.1α' e 3.1α'',nel caso dell’aziridina 3.1α, e come equilibrio 98:2 fra 3.1β' e 3.1β'', nel
caso dell’aziridina 3.1β (Schema 3.18). Nel caso dell’aziridina 3.1β il conformero maggiormente presente all’equilibrio (3.1β') corrisponde a quello portante il gruppo sostituente –CH2OBn in posizione equatoriale, mentre nel caso del diasteroisomero
aziridina 3.1α, il conformero maggiormente rappresentato all’equilibrio (3.1α') corrisponde a quello portante la catena laterale in posizione assiale.
74 N N Ac Ac 3.1' (90%) 3.1''(10%) BnO OBn N Ac OBn 3.1' (98%) N Ac OBn 3.1''(2%)
Schema 3.18. Equilibri conformazionali delle N-acetil aziridine 3.1α e 3.1β.
Le aziridine otticamente attive così ottenute dovevano quindi essere sottoposte a reazioni di addizione nucleofila per valutarne la regio- e la stereoselettività. Era nostra intenzione utilizzare innanzitutto il MeOH come semplice O-nucleofilo modello e successivamente utilizzare un O-nucleofilo complesso ad alto valore biologico (del quale non si riporta la struttura per motivi brevettuali) che ci era stato fornito da un altro gruppo del nostro Dipartimento di Scienze Farmaceutiche.
Sulla base di quanto già verificato,117 sulle analoghe carba N-nosil aziridine viniliche derivate dal D,L-allale e D,L-galattale 2.23α e 2.23β rispettivamente (Fig. 3.4), i sistemi 3.1α e 3.1β da noi sintetizzati dovevano risultare stabili e conservabili, solo per precauzione, a bassa temperatura (-15°).C
N BnO Ns 2.23 N BnO Ns 2.23
Figura 3.4. N-nosil aziridine raceme 2.23α e 2.23β.
Col nostro grande disappunto abbiamo invece trovato che l’aziridina 3.1α, appena isolata risulta pura (1H NMR e 13C NMR), ma nel giro di pochi giorni, anche se conservata a bassa temperatura (-15°C), si decompone dando uno spettro protonico molto complesso. Analogamente l’aziridina 3.1β si è decomposta dopo solo poche ore (24 ore) dalla sintesi.
75 Non è stato quindi possibile completare i nostri studi che avevano come obiettivo ultimo l’ulteriore elaborazione del doppio legame in 1,2 (per i prodotti di addizione 1,2) e in 2,3 (per i prodotti di addizione 1,4) a dare amminocarbazuccheri non naturali funzionalizzati (schema 3.19 dove si mostra per semplicità solo l’aziridina 3.1β).
N BnO Ac SN2 Nu BnO N Nu Ac H 1,2-funzionalizzazione BnO N Nu Ac H X Y BnO SN2' Nu Nu N Ac H BnO Nu N Ac H 2,3-funzionalizzazione Y X 1 2 2 3 3.1
Schema 3.19. Elaborazione del doppio legame in 1,2 e in 2,3 dell’aziridina 3.1β.
Solo con l’aziridina 3.1α abbiamo potuto realizzare due reazioni di addizione nucleofila, prima che si decomponesse, per valutarne almeno in via preliminare la reattività.
Sulla base di quanto osservato precedentemente sulla corrispondente N-nosil aziridina racema 2.23α, che in presenza di quantità controllate di MeOH (6 equiv) e in soluzione a ridotta acidità (CH2Cl2/TsOH 0.01N) fornisce con completa regio- e
stereoselettività solo il corrispondente prodotto di addizione 1,2-anti (Schema 3.20), abbiamo ripetuto la stessa reazione sulla nostra aziridina 3.1α.
BnO N Ns 2.23 MeOH CH2Cl2/ TsOH 0.01 N BnO N H OMe Ns
Schema 3.20. Reattività del’aziridina racema 2.23α con MeOH.
Abbiamo però ottenuto un grezzo di reazione abbastanza complesso che anche dopo purificazione non ha evidenziato la presenza di prodotti che avessero incorporato
76 il gruppometossi (-OCH3) ad indicare come l’aziridina non avesse reagito col
nucleofilo.
Più interessanti, ma soltanto preliminari, sono stati i risultati ottenuti per reazione dell’aziridina 3.1α con il nucleofilo biologicamente attivo, che indicheremo genericamente con Ar-OH. In questo caso, a causa delle caratteristiche del nucleofilo e alla sua scarsa reattività in condizioni di catalisi acida abbiamo realizzato l’apertura dell’aziridina 3.1α in particolari condizioni basiche. Così all’aziridina 3.1α (2 equiv) in THF anidro è stata addizionata una soluzione del nucleofilo deprotonato con LHMDS(1 equiv) e in presenza di Crown 12 (2 equiv), per chelare il controione Li+ e ottenere quindi un alcolato più nucleofilo. Dopo 48 ore a temperatura ambiente si ottiene un grezzo che all’analisi 1
H NMR evidenzia la presenza di più prodotti di addizione.
La purificazione su TLC preparativa ha permesso di isolare tre prodotti che sulla base del loro spettro protonico (1H NMR) verosimilmente risultano essere i due prodotti di addizione 1,4 sin e 1,4 anti (3.32 e 3.33) e il possibile prodotto di addizione 1,2-anti
3.34 (Schema 3.21). Tuttavia le piccole quantità di prodotti isolati (la reazione era stata
condotta su una scala molto piccola: ≈ 20 mg di aziridina e i prodotti di addizione hanno PM elevato) non hanno permesso una caratterizzazione completa dei prodotti di reazione. BnO N Ac (-) 3.1 Ar-OH LHMDS THF Crown 12 BnO N H O Ac Ar BnO N H O Ac Ar + + BnO N H Ac O Ar 3.32 3.33 3.34
Schema 3.21. Reazione dell’aziridina 3.1α con nucleofilo complesso.
Questo risultato preliminare, comunque promettente, dovrà necessariamente essere ripetuto una volta che l’aziridina 3.1α, sia stata nuovamente preparata, tenendo necessariamente ed opportunamente conto della sua instabilità anche a -15°C.
77
3.4 Sintesi ed elaborazione degli epossidi diasteroisomerici 3.35α e 3.35β
Nel laboratorio dove ho svolto la mia tesi, recentemente era stato sintetizzato un carbaglicoside del tipo 3.36 (Fig. 3.5), in cui la porzione arilossi legata al carbazucchero ancora una volta derivava da un nucleofilo complesso ad elevato valore biologico, che
per motivi ancora legati a copertura brevettuale, indicheremo genericamente con Ar-OH. OBn OH BnO O BnO Ar 3.36
Figura 3.5. Struttura generica del carbaglicoside 3.36.
Di questo carbaglicoside doveva essere valutata in vitro e in vivo l’attività antiproliferativa, ma per poter far questo era necessario procedere alla debenzilazione degli eteri benzilici in C(3), C(4) e C(6). La debenzilazione catalitica (H2/Pd) anche in
condizioni blande, tuttavia si era dimostrata fallimentare per la natura stessa strutturale sulla porzione proveniente dal nucleofilo. Del resto altri metodi di debenzilazione (BCl3, TMSI) si erano dimostrati incompatibili con la struttura del gruppo arilossi
introdotto sul C(1).
Abbiamo quindi ritenuto utile procedere alla sintesi del carbaglicoside 3.37 (Fig. 3.6), analogo di 3.36 in cui al posto del benzile fossero presenti sul C(3), C(4) e C(6) gruppi p-metossibenzilici (-OPMB) facilmente rimovibili in condizioni ossidanti (DDQ), piuttosto che riducenti.
OPMB OH PMBO O PMBO Ar 3.37
Figura 3.6. Carbaglicoside analogo di 3.36.
A livello retrosintetico (Schema 3.22), 3.37 poteva ragionevolemente provenire dall’apertura trans-diassiale dell’epossido 3.35 con il nucleofilo Ar-OH. A sua volta l’epossido 3.35 poteva esser pensato provenire dall’olefina 3.38 facilmente ottenibile
78 dall’alcool primario 3.5 che avevamo sintetizzato in buone quantità per ottenere le vinil N-acetil aziridine 3.1α e 3.1β viste in precedenza (vedi anche Schema 3.6).
OPMB OH PMBO O PMBO Ar 3.37 OPMB PMBO PMBO 3.35 O OPMB PMBO PMBO (+) 3.38 OPMB PMBO HO (-) 3.5
Schema 3.22. Retrosintesi del carbaglicoside 3.37.
A partire dall’alcool 3.5 abbiamo quindi proceduto alla sintesi dell’epossido 3.35 (Schema 3.23). OPMB PMBO PMBO (+) 3.35 O OPMB PMBO PMBO (+) 3.38 OPMB PMBO HO (-) 3.5 PMBCl DMF MCPBA CH2Cl2 OPMB PMBO PMBO (+) 3.35 O + 40 : 60
Schema 3.23. Sintesi degli epossidi 3.35α e 3.35β a partire dall’alcool primario 3.5.
Così, l’alchenolo 3.5 è protetto sul C(6) con p-metossibenzil cloruro (PMBCl) in DMF a temperatura ambiente a dare l’olefina completamente protetta 3.38, che per epossidazione con mCPBA al 70% in CH2Cl2 forniscei due epossidi 3.35α e 3.35β in
rapporto 40:60 (1H NMR). A questo riguardo è interessante notare come l’analoga reazione di epossidazione condotta sulle corrispondenti olefine tri-O-benzil protette fornisce solo il 10% di epossido β. I due epossidi 3.35β e 3.35α sono stati poi separati per flash cromatografia.
L’epossido 3.35β (2 equiv), di maggiore interesse, è stato quindi messo a reagire in THF anidro con il nucleofilo Ar-OH (1 equiv) deprotonato con LHMDS (1 equiv) per otto giorni a 80°C per fornire il desiderato prodotto di addizione 3.37, ma con una resa molto bassa (10%). Purtroppo la piccola scala sulla quale è stata realizzata la reazione e la resa estremamente bassa delle stesse non hanno permesso di proseguire nel processo sintetico che a questo punto avrebbe previsto la deprotezione di tutte le funzionalià ossidriliche O-PMB protette.
79 Quindi questa fase finale della mia tesi sperimentale deve intendersi come uno studio preliminare per ottenere questi prodotti coniugati estremamente interessanti. A questo scopo, opportuni studi sono attualmente in corso nel nostro laboratorio al fine di migliorare le rese del processo sintetico che conduce all’epossido 3.35 e da questo al prodotto di addizione 3.37 e alla sua successiva deprotezione.