CHAPTER 1 Introduction
1. G protein-coupled receptors
G protein-coupled receptors (GPCRs) constitute the largest superfamily of membrane receptors. An interesting feature of this receptor superfamily is the chemical diversity of their ligands, ranging from physical stimuli like light, odorants, acoustic and tactile stimuli, to ions (e.g., Ca2+) and chemical transmitters, such as ‘‘classical’’ biogenic amines (dopamine, noradrenaline, adrenaline and serotonin), acethylcholine, nucleosides and nucleotides (adenosine, ATP, UTP, sugar-nucleotides), peptide and protein hormones (chemokines, glucagon), lipids and eicosanoids (sphingolipids, leukotrienes) (Fig. 1). GPCRs are characterised by seven transmembrane (TM) a-helices and for this reason are also referred to as 7-TM receptors. The seven transmembrane helices (TM-1 through -7) connected by three intracellular loops (IL1, IL2 and IL3) and three extracellular loops (EL1, EL2 and EL3) (Baldwin, 1993). Two cysteine residues (one in EL1 and one in EL2) which are conserved in most GPCRs, form a disulfide link which is probably important for the packing and for the stabilization of a restricted number of conformations of these seven TMs. Aside from sequence variations, GPCRs differ in the length and function of their N-terminal extracellular domain, their C-terminal intracellular domain and their intracellular loops.
Ligand binding to GPCRs causes as yet poorly defined conformational changes in the receptor protein that promotes its interaction with distinct classes of G-protein heterotrimers (consisting of α-, β-, and γ-subunits) that are attached to the cytoplasmic surface of the plasma membrane (Neer 1995). This interaction triggers the exchange of GTP for GDP on the α-subunit, leading to the dissociation of the G-protein from the receptor and to a dissociation of the α-subunit from the βγ complex. The released G-protein subunits, α-GTP and free βγ are then able to interact with distinct effector enzymes and ion channels, eventually leading to the desired physiological response (Neer 1995). Structurally and phylogenetically, GPCRs have been classified into three different subfamilies, based on their sequence similarity to rhodopsin (type A receptors), calcitonin (type B) and glutamate metabotropic receptors (type C) (Howard et al., 2001; Kolakowski, 1994; Probst et al., 1992). Among different receptors, sequence similarity was found to be particularly high in the regions corresponding to the TM domains.
Fig. 1. Structure of GPCRs and diversity of messages which activate those receptors.
Modified from Bockaert and Pin, 1999.
GPCRs have been involved in a multitude of biological responses in all organs and systems including the central nervous system (CNS) and peripheral nervous system. In the latter, particularly important functions include neurotransmitter release, cell-to-cell communication, modulation of learning and memory, response to psycho-active substances, regulation of neuronal growth and differentiation and of glial responses. Pathological changes in GPCRs expression and function play established roles in neurological and neurodegenerative diseases, thus highlighting these receptors as interesting targets for drug development.
The progress of human genome sequencing has revealed the existence of several hundred orphan GPCRs (Fredriksson et al, 2003), that is, molecularly identified receptors that still lack a defined physiologically relevant ligand. The ‘deorphanization’ of these receptors and the identification of their roles is expected to clarify novel regulatory mechanisms of physiological phenomena and to unveil novel drug targets (Howard et al, 2001; Fredriksson et al, 2003; Mori et al, 2005).
1.1 Purinergic receptors
The first description of the extracellular signaling by purines was by Drury and Szent-Györgyi (1929), and purinergic receptors were defined in 1976 (Burnstock, 1976). After an early hint (Spedding and Weetman, 1976), receptors for purines were subdivided into P1 (adenosine) and P2 (ATP and ADP) receptors (Burnstock, 1978).
The P2 receptors for extracellular nucleotides are widely distributed in the body and participate inregulation of nearly every physiological process (Surprenant and North, 2009, Abbracchio et al., 2006). Of particular interest are nucleotide receptors in the immune, inflammatory, cardiovascular, muscular, and central and peripheral nervous systems. The ubiquitous signaling properties of extracellular nucleotides acting at two distinct families of P2 receptors – fast P2X ion channels and P2Y receptors (GPCRs) – are now well recognized. These extracellular nucleotides are produced in response to tissue stress and cell damage and in the processes of neurotransmitter release and channel formation. Their concentrations can vary dramatically depending on circumstances. Thus, the state of activation of these receptors can be highly dependent on the stress conditions or disease states affecting a given organ.
1.1.1 P2Y receptors
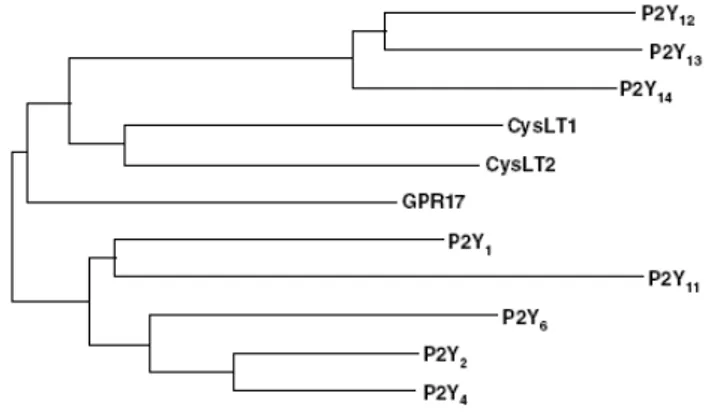
The numbering of unique human P2Y receptors (P2YRs) has some gaps – due to the fact that the assignment of numbers to certain putative P2YRs was later shown to be premature, with some of the previously designated sequences being P2Y species homologs and others being other types of receptors. As of today, there are eight accepted human P2YRs: P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14 (Abbracchio et al., 2006). The P2YRs belong to the rhodopsin family of GPCRs. Structurally related groups of GPCRs within the rhodopsin family (with some similarity in the amino acid composition) include cysteinyl leukotriene receptors, proteinase activated receptors, and glycoprotein hormone receptors. As shown in Fig. 2, the comparison of the amino acid composition reveals 2 subgroups within the family of functionally defined P2YRs: the first group consists of the P2Y1-, P2Y2-, P2Y4-, P2Y6-, and P2Y11-receptors and the second of the more recently cloned P2Y12-, P2Y13-, and P2Y14-receptors (Fig. 2). However, the similarity in average amino acid composition does not predict the pharmacological properties of a given subtype. Both groups
mentioned above contain adenine nucleotide- selective receptors as well as receptors that respond to uracil nucleotides or UDP-glucose.
Fig. 2. Phylogenetic tree of human P2Y-receptor subtypes as well as human
non-nucleotide and orphan receptors with sequence similarity. For comparison, the human P2Y1R
and P2Y11R share 33% identical amino acids.
Adenine-nucleotide-selective human P2Y-receptors
The P2Y1R was first cloned from late-embryonic chick brain. Related sequences were then detected in mammalian species including man. The receptor mediates responses such as muscle relaxation and the release of endothelium-derived relaxing factors or prostaglandins. The P2Y1R also contributes to the ADP-induced platelet aggregation (Dorsam and Kunapuli, 2004). As shown in Table 1, the mammalian P2Y1R is selective for adenine nucleotides. When expressed in cellular systems, the human P2Y1R is activated by adenine diphosphate nucleotides (Table 1). 2-Methylthio-ADP (2- MeSADP) has a 10 times higher affinity at the human P2Y1R than ADP (Waldo and Harden, 2004). Its recently developed analogue, N-methanocarba-2-methylthio-ADP (MRS2365), even displays selectivity for the P2Y1R over the P2Y12R and P2Y13R (Chhatriwala et al., 2004). 2-Methylthio-ATP (2-MeSATP) and adenosine-(O-3-thiotriphosphate) (ATPγS) also act as agonists at the P2Y1R with potencies similar to that of ADP. However, ATP itself is a partial agonist with a reduced intrinsic activity when compared with that of ADP (Waldo and Harden, 2004). In agreement with this notion, ATP has been shown to antagonize responses to the activation of the P2Y1R by full agonists such as ADP or 2-methylthio-ADP (Leon et al., 1997; Hechler et al., 1998). The potent actions of ATP observed in studies on native P2Y1Rs in tissues (cf. Ralevic and Burnstock, 1998, 2003) are likely to be due to ADP
formed on the cell surface from ATP by the action of the ectoenzyme NTPDase2 (Alvarado-Castillo et al., 2002, 2005).
The P2Y11R is highly expressed in immunocytes and may play a role in the differentiation of these cells (Table 1; Communi et al., 1997). The human (but not the canine) P2Y11R is activated by ATP as naturally occurring agonist (Table 1). Only when measuring intracellular calcium levels, the recombinant receptor also responded to stimulation by UTP (White et al., 2003). The analogue 2-propylthio-h,g-dichloromethylene- D-ATP (ARC67085) is a very potent agonist at the P2Y11R.
The P2Y12R is expressed in platelets and neuronal tissues. It plays an important role in platelet aggregation (cf. Dorsam and Kunapuli, 2004) and also acts as an inhibitory neuronal receptor (Kubista et al., 2003). In human blood vessels, activation of P2Y12R contributes to vasoconstriction. The receptor is activated by adenine diphosphate derivatives with 2- methylthio-ADP being much more potent than ADP. Subtype-selective agonists are yet not known.
The P2Y13R is expressed in cells of hemopoietic origin as well as in neuronal cells. The P2Y13R responds to adenine diphosphate analogues, similarly as the P2Y12R. In some cellular systems, 2-methylthio-ADP was more potent than ADP, whereas under other experimental conditions both compounds were equipotent. This discrepancy may depend on multiple active conformations of the receptor with differences in affinity for 2-methylthio-ADP and ADP as well as differences in preference for G proteins (Marteau et al., 2003). ATP and 2-methylthio-ATP seem to be partial agonists with a weak potency at the P2Y13R (Marteau et al., 2003).
P2Y-receptors activated by uracil nucleotides or UDP-sugar derivatives
Early functional studies suggested the occurrence of receptors for uracil nucleotides distinct from receptors for adenine nucleotides (e.g., Häussinger et al., 1987; von Kügelgen et al., 1987). In addition, a receptor sensitive to both ATP and UTP was functionally identified and termed ‘‘P2U’’ (O’Connor et al., 1991). The functionally defined P2U receptor is likely to correspond to more than one of the cloned subtypes (see King et al., 1998).
P2Y2R mRNA has a wide tissue distribution and is found at highest levels in cells in the lung, heart, skeletal muscle, spleen, kidney, liver, and epithelia (Lustig et al., 1993). Studies on P2Y2R-deficient mice verified the important role of the receptor in regulating ion transport in epithelial cells (Leipziger, 2003). Some but not all of the
functionally defined P2U-receptors are likely to be in fact P2Y2Rs. Triphosphate nucleotides including UTP, ATP, uridine-(O-3-thiotriphosphate) (UTPγS), and ATPγS are full agonists at this receptor. When tested under conditions excluding enzymatic conversion of nucleotides, UDP and ADP did not activate the receptor (Nicholas et al., 1996), indicating that at least 3 phosphate residues are required for the activation of the receptor.
P2Y4R transcripts have a more restricted distribution; the receptor is expressed in the placenta and at lower levels in lung and vascular smooth muscle (Table 1; Erlinge et al., 1998). The human P2Y4R (but not the rodent orthologues) is highly selective for uracil triphosphate derivatives (Communi et al., 1995).
P2Y6Rs are widely expressed (Table 1). The P2Y6- receptor is a nucleoside diphosphate preferring receptor with UDP being 100-fold more potent than UTP (Nicholas et al., 1996; Table 1). Adenine nucleotides are almost inactive.
P2Y14R has a widespread distribution with highest expression in man in the placenta, adipose tissue, stomach, and intestine (Chambers et al., 2000). The rat orthologue has been shown to be expressed at highest levels in cells of hemopoietic origin. The receptor is activated by nanomolar and low micromolar concentrations of UDP-glucose, UDP-galactose, UDP-glucuronic acid, and UDP-Nacetylglucosamine (Chambers et al., 2000).
Studies with antagonists are more helpful for the pharmacological identification of a native receptor. Antagonist affinity constants of a receptor do not depend on the number of receptor proteins expressed on the cell surface or the signalling transduction pathways coupling to the receptor and hence can be used to identify the receptor. Up to now, however, only a restricted choice of subtype-selective P2YR-antagonists is available. Nevertheless, the combined use of subtype preferential agonists and antagonists will allow the pharmacological characterisation of a P2YR subtype. Reactive blue 2 (RB2l; Burnstock and Warland, 1987), suramin (Dunn and Blakeley, 1988; von Kügelgen et al., 1989), pyridoxal-5V-phosphate-6-azophenyl-2,4-disulfonate (PPADS; Lambrecht et al., 1992), as well as the suramin analogue 8-8V-[carbonylbis(imino-3,1-phenylene)]bis(1,3,5- naphthalene-trisulfonate (NF023; Lambrecht, 1996) have been used for several years to antagonize P2X and P2Y-receptors (for a review, see Jacobson and Boeynaem, 2010). Suramin, PPADS, and reactive blue 2 block more than one P2YR subtype. Suramin when given at high micromolar concentrations (e.g., 30–100 µM) affects all nucleotide-sensitive P2YRs with the exception of the P2Y4R (Wildman et al., 2003). PPADS and reactive blue also
block more than one subtype with most pronounced effects at the P2Y1Rs and the P2Y6Rs.
While the average amino acid composition does not predict the pharmacological properties of a subtype, both groups of P2YR subtypes mentioned above (and shown in Fig. 2) correspond to principal signalling transduction pathways of the receptors. The receptors of the first group (i.e., the P2Y1-, P2Y2-, P2Y4-, P2Y6-, and P2Y11-receptors) all couple via Gq proteins to stimulation of phospholipase C followed by increases in inositol phosphates and mobilization of Ca2+ from intracellular stores; the P2Y11R mediates in addition an increase in adenylate cyclase activity. The receptors of the second group, namely, the P2Y12-, P2Y13-, and P2Y14-receptor, all couple via Gi-proteins to inhibition of adenylate cyclase followed by a decrease in intracellular cAMP levels. In agreement with this general notion, the purified P2Y1R protein showed receptor activity when reconstituted with Gαq/β1γ2- or Gα11/β1γ2-proteins (Waldo and Harden, 2004) and the purified P2Y12R protein acted as receptor when reconstituted with Gαi2/β1γ2-proteins, but not when reconstituted with Gαq/ β1γ2-subunits (Bodor et al., 2003). The coupling of Gq coupled P2YRs to the Gq-protein may depend on a BBXXB motif (B for basic and X for non-basic residue) found at the C-terminus of these receptors (Ding et al., 2005). However, the coupling to signalling transduction pathways appears to be much more complex. For example, the Gi coupled P2Y12- and P2Y13-receptors mediate an inhibition of voltage-dependent N-type calcium channels in neuronal cells. An inhibition of N-type calcium channels, however, has also been demonstrated for the recombinant P2Y1R (Filippov et al., 2000). Moreover, P2Y12Rs clearly couple to inhibition of adenylate cyclase in platelets, but separate signalling transduction pathways including the phosphatidylinositol 3-kinase seem to be much more important for platelet aggregation. Hence, the demonstration of the coupling of a native P2YR to a distinct signalling transduction pathway does not allow its pharmacological characterization.
Table 1. Subtypes of P2 receptors and their ligands (potency at the human homologs
shown as pEC50, unless noted r = rat).Modified from Jacobson et al., 2010.
1.2 Cysteinyl-leukotriene receptors
Cysteinyl-leukotriene (cysLTs) are derived from the ubiquitous membrane constituent arachidonic acid and are members of a large family of molecules known as eicosanoids. These biologically active lipids are rapidly generatedat the sites of inflammation following a series of reactions initiated by cytosolic phospholipase A2 which release the arachidonic acid from the phospholipids present at the nuclear envelope. The arachidonic acid reacts with the enzyme 5-lipoxygenase (5-LO) after binding to the 5-LO-activating protein and results in the formation of 5,6-oxido-7,9-trans-11,14-cis-eicosatetraenoic acid or LTA4 (Brash et al., 1999; Boyce et al., 2008). LTA4 is converted to LTB4 by the enzyme LTA4 hydrolase, a cytosolic enzyme also exerting aminopeptidase activity and containing zinc at its catalytic center (
Rinaldo-Matthis and Haeggstrom, 2010). The loss of biological activity arises from catabolism
through 3 major mechanisms: the formation of N-acetyl derivatives of LTE4 , the reaction with hypochlorous acid to form sulfoxide and LTB4 and oxidation and elimination to form shortened metabolites. Alternatively, cysLTs can also be produced through transcellular metabolism from neutrophil-derived LTA4 by platelets and vascular endothelial cells. This transcellular biosynthesis is considered very important
because itcould generate remarkably high concentrations of cysLTs in a local environment, ultimately affecting organ function (Maclouf et al., 1989; 1996).
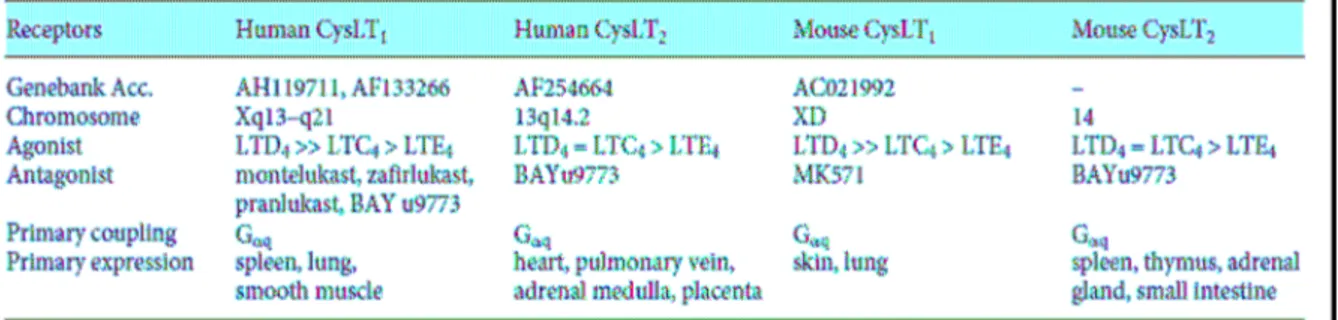
CysLTs exert their effect through cell surface receptors (Im 2009). According to the International Union of Pharmacology, the cysLT receptors (CysLTRs) have been mainly divided into two classes: CysLT1R that is sensitive to the classical antagonists and CysLT2R which mediates several effects that are not inhibited by the classical antagonists. The characteristics of human and mouse CysLT receptors have been summarized in Table 2. Hydrophobicity analysis of the deduced primary structure reveals that both CysLTRs have 7 hydrophobic transmembrane domains linked by the 6 hydrophilic loops, signifying a typical character of GPCRs (Hui et al., 2002; Capra et al., 2004).
The human CysLT1 receptor encodes a protein of 337 amino acids with a calculated molecular mass of 38 kDa, observed to migrate at a molecular weight of approximately 42 kDa as a monomeric form, but mostly present in dimeric and oligomeric forms, even in the presence of denaturing agents (Back et al., 2002; Capra et al., 2007). Human CysLT1R has the highest homology (32% amino acid identity) with the P2Y1R and the receptor for platelet-activating factor. Expression of the CysLT1R mRNA and protein was also demonstrated in normal peripheral blood eosinophils, subsets of monocytes, macrophages and pregranulocytic CD34+ cells (Figueroa et al., 2001). The human CysLT1R has been shown to be most highly expressed in spleen, peripheral blood leukocytes and less strongly in lung, small intestine, pancreas and placenta and little or no expression in the liver, colon, kidney, skeletal muscle, thymus, ovary, testis, heart and brain (Sarau et al., 1999; Back et al., 2002; Capra et al., 2007).
In cells transfected with the human CysLT1R, the rank order of potency for calcium mobilisation is LTD4 >> LTC4 > LTE4 (Lynch et al., 1999; Sarau et al., 1999; Nothacker et al., 2000; Takasaki et al., 2000), with LTE4 being a partial agonist (Nothacker et al., 2000; Takasaki et al., 2000). The rank order of agonist potency at the CysLT1R in isolated human bronchial preparations differs from that obtained in recombinant system (Muccitelli et al., 1987; Labat et al., 1992), but the reason for this difference has not been proposed.
The CysLT2 receptor has always been pharmacologically less defined than CysLT1R, mainly because of the lack of a selective antagonist. Human CysLTRs share only 38% amino acid identity with very low homology in the extreme carboxyl terminal. The human CysLT2R expression pattern is substantially different from that of human CysLT1R: it is highly expressed in the spleen and peripheral blood leukocytes,
but expression in the heart, adrenal gland and brain appears unique to this subtype. In the central nervous system, CysLT2R mRNA is highly expressed in several regions of the brain, with particular concentration in the hypothalamus, thalamus, putamen, pituitary and medulla (Heise et al., 2000). Its expression has also been reported in the granulocytes of the brain parenchyma and in neuron and glial cells appearing in the late stage of traumatic injury and in areas surrounding the tumors (Hu et al., 2005). CysLT2R expression is also widespread in most regions of the spinal cord (Heise et al., 2000). In the immune system, moderate expression of CysLT2R mRNA has been reported in the spleen, lymph nodes and peripheral blood leukocytes, with very strong expression in eosinophils (Heise et al., 2000).
At the CysLT2 receptor on human pulmonary venous smooth muscle preparations, the rank order of agonist potency is LTC4 = LTD4 > LTE4, with LTE4 being a partial agonist (Labat et al., 1993), which is similar to results obtained in cells transfected with the human CysLT2R (Muccitelli et al., 1987; Labat et al., 1992).
1.3. GPR17
1.3.1 Pharmacological profile
GPR17 is a previously orphan GPCR equally distant from the P2Y12,13,14 subgroup and the CysLT1 and CysLT2 group (Fig. 5).
Fig. 5 Phylogenetic tree showing the relationships between GPR17 and P2Yand CysLT receptors. Modified from Ciana et al., 2006.
The agonist-response profile of GPR17 is different from that of CysLTRs (Brink et al, 2003; Capra, 2004), and, for nucleotides, is intermediate between P2Y6 and P2Y14 receptors (Abbracchio et al, 2003; Burnstock and Knight, 2004). In 1321N1 cells, which do not respond to any P2YR or CysLTR ligands (Communi et al, 2001), human GPR17 expression induced the appearance of concentration-dependent responses to LTD4 and LTC4 (with LTC4 >> LTD4) and to UDP, glucose and galactose (with UDP-galactose=UDP>UDP-glucose). The mouse receptor more closely resembles the response profile of the human receptor (Lecca et al., 2008).At the rat receptor, UDP-glucose was more potent than UDP, and UDP-galactose had no effect; moreover, the relative potency of cysLTs was inverted.
Several established P2YR and CysLTR antagonists, namely, the P2Y1 R-selective antagonist 2'-deoxy-N6-methyladenosine 3',5'-biphosphate (MRS2179), the P2Y12/13R antagonist N(6)-(2-methyl-thioethyl)-2-(3,3,3-trifluoropropylthio)-, dichloromethylene-ATP (cangrelor), and the CysLT1R antagonists montelukast and pranlukast, were found to be able to counteract GPR17 activation in vitro (Ciana et al., 2006; Lecca et al., 2008). The non-hydrolysable analog of ATP, which does not act as a GPR17 agonist (Ciana et al., 2006) can instead act as an antagonist, as shown by complete and concentration-dependent blockade of UDP-glucose or LTD4 induced
GPR17 activation (Pugliese et al., 2009). GPR17 ligands, structure, pharmacological activity and affinity are summerized in Table 3.
Ciana and collaborators showed that GPR17 is coupled to Gi proteins and Gq proteins, in a similar way to many GPCRs (Milligan and Kostenis, 2006), leading to both adenylyl cyclase inhibition and [Ca2+]i increases in 1321N1 cells expressing human GPR17. By using an immunoprecipitation assay, allowing the identification of the specific G protein subtypes involved in GPCR effects, it has been confirmed that GPR17 indeed couples to Gi1,2 and, to a lesser extent, to Gq proteins (Pugliese et al., 2009). By utilizing patch-clamp studies, it has been also shown that in cells expressing GPR17, micromolar concentrations of UDP-glucose, UDP-galactose, and UDP lead to a significant increase of outward K currents (Pugliese et al., 2009).
GPR17 exists in two isoform, a “short” (GPR17-S) and a “long” (GPR17-L) receptor isoform of 339- and 367-amino acids, respectively, with the latter displaying a 28-amino acid longer NH2 terminus, as revealed by genomic analysis. The long isoform was found to be exclusively expressed in the brain, suggesting highly specific roles in the nervous system. The two isoforms of GPR17 may represent the response to a fine genic regulation. The characterization of the long receptor isoform in GTPγS binding assay led to an agonist-response profile that is highly similar to that obtained with the short receptor form. Although there are no significant changes in the EC50 values of cysLTs, UDP-glucose seemed slightly more potent at the long isoform with respect to the short one. Although no modeling studies are yet available for the long GPR17 isoform, authors speculate that the 28-amino acid-longer NH2 terminus in this splice variant may influence the binding affinity of ligand nucleotide agonists via a different conformation of this accessory binding site (in this respect see paragraph 1.3.2).
At variance from the [35S]GTPγS binding, no significant differences between the short and long GPR17 forms were found in the electrophysiological studies, likely due to different sensitivities of the two assays (Pugliese et al., 2009). The different functional properties of GPR17 isoforms have been partly confirmed recently (Benned-Jensen and Rosenkilde, 2010): the long isoform is only poorly (and in the case of UDP not at all) activated by uracil nucleotides, whereas the short isoform is activated with similar potencies, but slightly lower efficacies compared with the nucleotide receptors P2Y6R and P2Y14R (Benned-Jensen and Rosenkilde, 2010).
The “dual” pharmacological profile of GPR17 profile remains to be confirmed, as in literature conflicting data are reported. Recently, another group (Benned-Jensen and Rosenkilde, 2010) has confirmed that, at the level of G protein activation (using
[35S]GTPγS binding and further downstream by measuring CREB transcriptional factor activity) UDP-glucose, UDP-galactose and UDP activated human GPR17 short isoform dose-dependently with EC50 values in the micromolar range, which closely match those previously reported for this isoform (Ciana et al., 2006). In this work, the authors also have shown that this receptor, previously reported to be activated by cysLTs (Ciana et al., 2006; Lecca et al., 2008), is not activated, internalized or bound by these molecules. In constrast, Maekawa and collaborators could not obtain significant responses neither to cysLTs or UDP-glucose. Since heterodimerization of GPCRs modulates expression and/or function either negatively (McGraw et al., 2006; Jiang et al., 2007) or positively (White et al., 1998) in various transfectants, authors considered the possibility that GPR17 may associate with CysLT1R to control its function. They demonstrated that in stable cotransfections of GPR17 with CysLT1R, the robust CysLT1R -mediated calcium response and ERK phosphorylation to LTD4 is abolished. CysLT1 and GPR17 receptors, expressed in transfected cells, have been coimmunoprecipitated and identified by Western blots, and confocal immunofluorescence microscopy revealed that GPR17 and CysLT1R colocalize on the cell surface of human peripheral blood monocytes. Moreover, lentiviral knockdown of GPR17 in mouse bone marrow-derived macrophages (BMMs) increased both the membrane expression of CysLT1R protein and the LTD4-elicited calcium flux in a dose-dependent manner as compared with control BMMs, indicating a negative regulatory function of GPR17 for CysLT1R in a primary cells. Taken together, Maekawa and coworker findings suggest GPR17 as a ligand-independent, constitutive negative regulator for the CysLT1R, that suppresses CysLT1R -mediated function at the cell membrane.
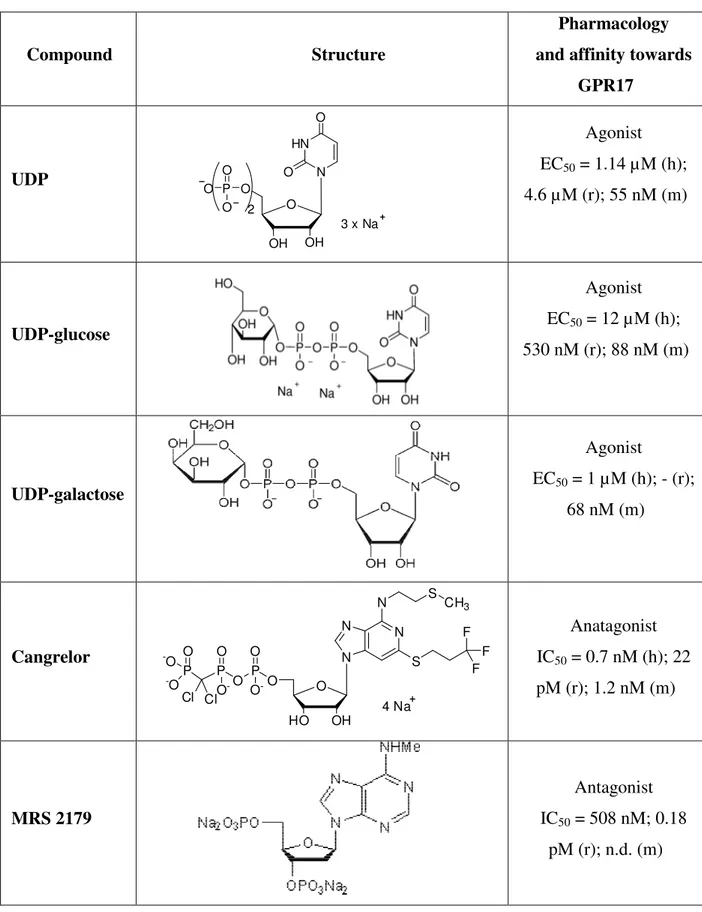
Table 3. Ligand specificity at human (h), rat (r) and mouse (m) GPR17 in GTPγS binding assay performed in 1321N1 transfected cells.
Compound Structure
Pharmacology and affinity towards
GPR17 UDP O OH HN N O O OH O P O O O 2 3 x Na Agonist EC50 = 1.14 µM (h); 4.6 µM (r); 55 nM (m) UDP-glucose Agonist EC50 = 12 µM (h); 530 nM (r); 88 nM (m) UDP-galactose Agonist EC50 = 1 µM (h); - (r); 68 nM (m) Cangrelor N N N N O OH HO 4 Na S F F F O P O O O -P O O -P Cl Cl O -O -O S CH3 Anatagonist IC50 = 0.7 nM (h); 22 pM (r); 1.2 nM (m) MRS 2179 Antagonist IC50 = 508 nM; 0.18 pM (r); n.d. (m)
ATP Antagonist IC50 = 3.1 µM vs UDP-glucose; 209.8 vs LTD4 (h); n.d. (r, m) LTC4 Agonist EC50 = 0.33 nM (h); 65 nM(r); 0.74 nMM (m) LTD4 Agonist EC50 = 7.2 nM (h); 5.9 nM (r); 0.63 nM (m) LTE4 Agonist EC50 = n.d. (h, r); 0.31 nM (m) Montelukast Antagonist IC50 = 60 nM; 196 nM (r), 61 nM (m) Pranlukast Antagonist IC50 = 10.5 nM (h); 31 nM (r); n.d. (m) n.d. = not done
1.3.2 GPR17 Molecular modeling and dynamics studies of the 3-D structure
A homology model of human GPR17 was built using as a template the X-ray crystal structure of bovine rhodopsine (bRh). The sequence of GPR17 consists of 339 aminoacids, corresponding to the human receptor sequence in its shorter isoform [GPCRDB: Q13304-2].
Multiple alignment of GPR17 with P2YRs, CysLTRs and bRh, showed the existence of two conserved cysteines among the various sequences (Cys104 and Cys181 in GPR17), which are conserved in the great majority of GPCRs. The receptor has an additional pair of cysteines which are conserved in all the P2Y and CysLT receptors; these two residues are positioned at the end of the Nt (Cys23) and at the middle of the EL3 domain (Cys269), respectively.
The dynamic simulations of a natural purinergic ligand and of two strong purinergic antagonists suggest the binding region corresponds to the well described nucleotide binding site of the other P2YRs, at least for some crucial interactions (e.g., with residue 6.55). Some important differences were, however, noticed, since only one of the three basic residues (Arg255) that are typically present in the binding pocket of P2YRs is conserved in GPR17. Thus, the so called "nucleotide binding site" is not defining a single binding mode and surely not a unique configuration of nucleotides bound to it. Initial docking and MD experiments suggest that, in a similar way to P2Y1R (Moro et al., 1999), there may be an additional "accessory" binding site also on GPR17, in a region located at the interface between the extracellular environment and the helical bundle. Here, some key aminoacids in EL3 and in Nt could drive the efficacy with which small ligands are guided into the helical bundle, and could consequently affect receptor response.
GPR17 has been also reported to bind to cysLTs (Ciana et al., 2006). The model suggests that neither the dimensions nor the dynamics of the receptor with or without ligands will accommodate the leukotrienic ligands in the nucleotide binding site. Therefore, this dual receptor most probably has a dual binding mechanism, according to the chemical entity that is going to bind. Based on modeling, the leukotrienic ligands may extend well beyond the nucleotide binding pocket, therefore also involving the extracellular loops. In this respect, in a second computational study (Parravicini et al., 2010) docking and MD have been applied to analyse the binding and the forced unbinding of the CysLTR antagonist pranlukast from GPR17, highlighting the importance of the conserved aromatic/hydrophobic cluster for the recognition of
pranlukast. The characterization of the binding site is even more uncertain, due to the flexible nature of the ligands for which the identification of the docked conformation to the CysLT1 and CysLT2 receptors has not been yet successful
In the same work, the authors developed also a mutant model, focusing their attention on Arg255, that has been proposed to play a crucial role in binding of other P2YRs to their nucleotide ligands (Erb et al., 1995; Van Rhee et al., 1995; Jiang et al., 1997, Moro et al., 1998; 1999; Hoffmann et al., 1999), and substituted this Arg with Ile (R255I). MD simulations, showing that the energy required to unbind UDP was significantly different between the wild-type (WT) receptor model and the mutated R255I, highlights the role of the basic residue Arg255 in the binding to nucleotides. No significant differences between the WT and the mutated receptor have been instead found for the unbinding of the leukotrienic ligand pranlukast from GPR17. MD simulations thus suggest that the mutation of Arg255, while influences the binding of nucleotides to GPR17, does not affect the binding of pranlukast, indicating that two different subsites are present on GPR17 and that the intermolecular interaction networks with the ligands are different between UDP and pranlukast. The existence of two different binding sites on this receptor, regardless of the agonist and antagonist nature of the ligands, is also consistent with the intrinsic difference in the chemical structure of the two classes of unrelated purinergic and leukotrienic ligands. Moreover, this hypothesis is also supported by the peculiar organization of the TM crevice, that, in GPR17, identifies two well defined areas with different hydrophilic/phobic surface profiles.
1.3.3 Physiological localization of GPR17
In the first published study, both human and rat GPR17 was found to be highly present in the brain and in other organs typically undergoing ischemic damage (kidney and heart) with very low expression in the liver and lung (Ciana et al., 2006).
A subsequent study (Lecca et al., 2008) focused on GPR17 expression in the mouse brain. Immunohistochemical studies with a specific, in-house made anti- GPR17 antibody showed that, in the intact rodent brain, GPR17 is expressed by neurons, as confirmed by co-staining with neuronal specific markers (i.e. NeuN and SMI-311). On the contrary, in a recent report, Chen and collaborators failed to find expression of GPR17 in neurons of adult mouse brain under basal physiological conditions (Chen et al., 2009). This discrepancy may reflect the specificity of the GPR17 antibodies used for
immunohistochemistry in the earlier studies. Conversely, no co-localization was found with cortical astrocytes, as shown by labelling with the specific marker GFAP (Lecca et al., 2008; Ceruti et al., 2010).
Based on previous data demonstrating that GPR17 is one of the 3 key genes expressed in human adult neuronal precursor cells (NPCs, Maisel et al., 2007), the presence of GPR17 was investigated in classical mouse brain neurogenic areas, i.e., the subventricular zone (SVZ) of lateral ventricles (LV) and the dentate gyrus (DG) of hippocampus. In the walls of LV, GPR17 is highly expressed in ependymal cells, which are in direct contact with ventricle cavities. None of these cells express either the astrocytic marker GFAP, the stem cell marker nestin or the neuronal precursor marker double-cortin (DCX), suggesting that they are not ‘‘classical’’ multipotent neurogenic cells or neuronal precursors; moreover, they did not incorporate bromodeoxiuridine, BrdU. These cells may thus represent an additional yet uncharacterized population of quiescent (or very slowly proliferating) precursors, or may be postmitotic SVZ-derived progenitors progressing along the oligodendroglial differentiation pathway. A similar population of ramified GPR17+ cells was also found in the hippocampus, another neurogenic area present in the adult brain (Lecca et al., 2008). In the same study the authors showed that in brain parenchyma GPR17 is segregated to a subset of oligodendrocytes precursor cells dispersed in both gray and white matter. Interestingly, in some of these cells, GPR17 immunoreactivity displayed an asymmetrical subcellular localization, suggesting GPR17 accumulation in the Golgi apparatus. This specific staining pattern may represent a well defined GPR17 maturation stage prior to distribution of the receptor to the plasma-membrane. The branched GPR17 + cells did not incorporate BrdU in their nuclei, suggesting that, in the intact brain, GPR17 decorates a subset of quiescent non-proliferating parenchymal oligodendrocyte precursors. Furthermore, no colocalization of GPR17 was found with more mature myelinating oligodendroglial markers. This suggests that GPR17 expression is specifically segregated to the early stages of the oligodendrocyte differentiating pathway, and that the receptor is turned down when these cells reach functional maturation. Interestingly, in corpus striatum, immature GPR17 precursor cells were often found physically associated to or inside myelin tracts, which have been previously shown to also contain immature cells positive for the transcription factor Olig2, a key regulator of oligodendrocyte development. These GPR17 + cells may represent a source of pre-oligodendrocytes able to turn into myelinating cells when necessary; alternatively, these cells could play a role in the local trophic control of myelination. In
primary cortical cultures, the exposure to GPR17 agonist (UDP-glucose or LTD4 for 72 h resulted in an approximately 3-fold increase of the number of mature oligodendrocytes cells. In treated cultures, the total number of cells was not increased, to suggest that activation of GPR17 is actually inducing the maturation of already existing precursors.
Very similar expression profiles of GPR17 have been found in the spinal cord (Ceruti et al., 2009; Chen et al., 2009), supporting a role for this receptor in both neurons and oligodendrocytes. Under physiological conditions, GPR17 is expressed by neurons and oligodendrocytes at different stages of maturation and by ependymal cells lining the central canal, but never by astrocytes.
As concern the different distribution between the two GPR17 splicing variant (the short, S, and the long, L one) Benned-Jensen and Rosenkilde have shown that for both isoforms the lowest expression is in thalamus, with gradually increasing expression levels through the hypothalamus, cerebellum, amygdala, cerebellar hemisphere, frontal cortex, hippocampus and putamen. For both isoforms, the putamen region exhibited relatively higher expression than in whole brain, while this was also observed for human GPR17-S in hippocampus (Benned-Jensen and Rosenkilde, 2010).
The long isoform hGPR17-L is expressed at higher levels in heart and kidneys compared with brain. Importantly, compared with the short isoform, hGPR17-L is expressed at much higher levels in the heart, and in the kidneys (with no detectable expression of hGPR17-S in the latter) indicating that GPR17-S plays a principal role in the brain, while GPR17-L probably adopts this role in the heart and kidneys. This differential expression profile suggests tissue-specific roles in humans and other primates.
1.3.4 Pathological roles
a) Brain Ischemia
Based on GPR17 expression in organs typically undergoing ischemic dmage, the role of GPR17 has been explored in an established model of ischemic damage (the permanent MCAo, monolateral middle cerebral artery occlusion in the rat (Ciana et al., 2006) and in the mouse (Lecca et al., 2008). A differential role of GPR17 seems to depend on specific phases of response after the insult (sequentially, death of irreversibly damaged cells, clearance of dead cells, remodeling and repair). Upon ischemia induction, there is an early, time-dependent up-regulation of GPR17 in neuronal cells
both within and at the borders of the injured area. Numerous neurons overexpressing GPR17 are already evident inside the lesioned area 24 hours after MCAo. Several of these neurons colocalize with inducible HSP70, a marker of cellular stress, injury and death, suggesting a causal link between GPR17 up-regulation and neuronal death. To confirm this potential link, at 48 hours, GPR17+ neurons are no longer present inside the lesion suggesting that they have indeed undergone death. To further confirm a causative role of the receptor in induction of cell death inside the ischemic core, the in vivo knock down of GPR17 by either a pharmacological or a biotechnological approach markedly reduced the ischemic infarct volume. Starting from 48–72 hours after MCAo, at the borders of the lesioned area, many GPR17 + cells also strongly stained for IB4, a marker of activated microglia/ macrophages. One week after MCAo, these cells are detected in high amount inside the ischemic lesion. This pattern of IB4 positivity indeed reproduces the typical distribution of infiltrating cells in the ischemic brain (Stoll et al., 1998). Starting from 72 hours after MCAo, the number of ramified GPR17 + oligodendrocyte precursor cells is also significantly increased both around the injured area and in the corpus striatum. These data are in line with previous literature data demonstrating that, in a similar way to neurons, oligodendrocytes are very vulnerable to brain damage in vivo and proliferation of oligodendrocyte precursor cells is detected after ischemia around the lesioned area (Gregersen et al., 2001), likely as a response to demyelination. Thus, GPR17 as a ‘‘sensor’’ that is activated upon brain injury in several embryonically distinct cell types (neurons, microglia and adult neural precursor cells) that contribute to damage evolution and to the subsequent remodeling and repair. These findings are consistent with the emerging role played by endogenous cysLTs and nucleotides in the injured brain, when these molecules are released from damaged cells, rapidly diffuse in the extracellular environment and act as ‘‘danger signals’’ to alert responses to damage and start repair by ligating specific plasma membrane receptors on target cells.
b) Spinal cord injury
In a sequent study, the spatio-temporal changes of GPR17 expression following spinal cord injury (SCI). In a similar way to that reported for the MCAo model, SCI induces a dramatic andrapid death of GPR17+ neurons and oligodendrocytes inside the lesioned area. Starting from 24 h after SCI, activated microglia/ macrophages (expressing the specific marker of activation IB4 and whose totality co-expressed
GPR17) migrate towards and infiltrate into the lesioned area, with their number increasing over time after injury. One week p.i., most of these double-positive cells were found inside the lesioned area and receptor expression started decreasing. This suggests that GPR17 expression is increased along the first phases of microglia/macrophages activation and might favour their migration and infiltration to the lesioned area; when these processes are completed, GPR17 expression is down-regulated.
An additional interesting observation that further supports the apparently opposite effects (i.e. cell death and damage repair) mediated by GPR17 in the SCI model is represented by its expression on ependymal cells lining the central canal. These cells have been recently demonstrated to represent the true stem cells in the adult spinal cord, since they start proliferating after injury and produce oligodendrocytes, and more abundantly, astrocytes that migrate to the lesion site building a substantial part of the glial scar (Meletis et al., 2008). Also in the brain, where they are normally quiescent, ependymal cells can generate neuroblasts and astrocytes after stroke (Carlèn et al., 2009). GPR17 is constitutively expressed by spinal cord ependymal cells which start proliferating and expressing the marker of pluripotency GFAP upon induction of SCI. It is worth noting that GPR17 is one of the three genes that are exclusively expressed by neuroprecursor cells in the adult human hippocampus (Maisel et al., 2007), thus suggesting its possible role in driving precursor cell specification and differentiation after brain and SCI.
c) Demyelinating diseases
In another study, Chen et al. has substantially confirmed previous observations that GPR17 is mainly expressed by OPCs at different levels depending upon their differentiation state. It has been confirmed that in spinal cord and in the optic nerve GPR17 is downregulated during the peak period of myelination and in adulthood (Chen et al., 2009). Moreover, the authors demonstrated that in a mouse model of multiple sclerosis (demyelination injury induced by experimental autoimmune encephalomyelitis (EAE), GPR17 is upregulated in demyelinating lesions in the CNS. The fact that GPR17 expression is downregulated during active myelinogenesis and upregulated in demyelinating lesions raises the possibility that sustained expression of this receptor may negatively regulate oligodendrocyte myelination. To test this possibility, the
authors generated transgenic mice with GPR17 expression under the control of the promoter CNP1, leading to overexpression of the receptor in oligodendrocytes.
Starting at postnatal week 2, GPR17 transgenic mice began to experience generalized tremors, hindlimb paralysis and seizures that are reminiscent of those described for dysmyelinating mouse mutants (Nave, 1994). GPR17 overexpression not only blocked differentiation of neural progenitor cells into oligodendrocytes, but also inhibited terminal differentiation of primary OPCs. Conversely, GPR17 knockout mice showed early onset of oligodendrocyte myelination.
In the transgenic mouse model utilized by Chen et al., forcing GPR17 transcription in oligodendrocytes at a maturation stage when they physiologically downregulate its expression (i.e., CNPase+ cells) might have created conflicting intracellular signals leading to cell suicide. Thus the conclusions drawn based on a transgenic mouse model may lead to a misinterpretation of the native function of the receptor.
In a very recent study, Ceruti and collaborators evaluated the expression and function of GPR17 in primary mixed (astrocytes+OPCs) glial cultures grown in vitro under both standard culturing conditions or in conditions promoting cell reactivity, i.e., in the presence of growth factors (GFs) namely, EGF and bFGF (Williams et al, 2007), followed by a shift to a less permissive environment, able to promote the typical features of cell differentiation.
Growth factors (GFs) are among the first signals that have been associated to precursor cell survival and proliferation, and are therefore largely utilized both in vivo and in vitro to promote neurogenesis and gliogenesis (Hagg, 2005). Moreover, GFs are recognized as important players in the reaction of astrocytes to injury or noxious stimuli (Buffo et al, 2010). Thus, a crucial role for GFs in the genesis of new cells can be envisaged, since parenchymal precursor cells and reactive astrocytes can cooperate to the initial healing response of the damaged brain. Exposure to GFs can, in turn, regulate the expression of other key modulators of cell differentiation and survival, primarily due to GFs ability to modify the proliferative or differentiative status of precursor cells (Baldauf and Reymann, 2005). However, sometimes GFs can also act through a direct interaction with the signaling pathways activated by other receptors and factors. In this respect, extracellular purine and pyrimidine nucleotides have long been demonstrated to synergize with GFs in regulating the differentiation and proliferation of astrocytes
(Abbracchio et al.,1995; Neary and Zimmermann, 2009), and, more recently, of both embryonic and adult precursor cells (Grimm et al., 2009).
In this study (Ceruti et al., 2010) mixed astrocytic/precursor cells cultures were used; these cultures can be maintained upon culturing conditions resembling the environment of reactive astrocytes, and thus more closely mimic the in vivo situation where cells communicate with each other and influence their own reaction to external stimuli. For example, in cultures grown in the presence of GFs (medium +GFs) a significantly higher total number of cells was detected with respect to cells grown in medium -GFs, indicating active astrocytic proliferation as detected after brain injury in vivo (Buffo et al, 2010).The authors demonstrated that GPR17 is never expressed by cortical astrocytes irrespectively of the culturing condition in vitro, thus confirming previous in vivo and in vitro observations (Ceruti et al, 2009; Ciana et al, 2006; Lecca et al, 2008). GPR17 expression is instead specifically confined to a population of O4-espressing OPCs, and results more abundantly expressed in the absence of GFs and under differentiating conditions. This suggests that receptor expression is specifically modulated by the proliferation/differentiation state of the cells, although we cannot definitely exclude a direct role of GFs on GPR17 transcription or an indirect role played by the surrounding reactive astrocytes. Indeed, the presence of both bFGF and EGF is required to down-regulate GPR17 expression, suggesting a synergy between the two GFs in keeping cells to a less differentiated state. In the used mixed astrocytes/precursor cultures express three out of seven P2X receptor channels (namely, the P2X4, P2X6, and P2X7 subtypes) and the whole family of G protein-coupled P2YRs at the mRNA level. Concerning P2X receptors, it is worth mentioning that, although clear evidence for their role in modulating astrocytic functions has been provided in vitro, no direct demonstration of their functionality is currently available in vivo where they are upregulated upon induction of reactive astrogliosis (for review, see Verkhratsky et al, 2009). Thus, results obtained in vitro could more closely resemble an “activated” state, more than a physiological situation. Interestingly, while no significant differences between cultures grown in medium −GFs and +GFs were detected in the expression of the seven P2X or of the P2Y receptor subtypes belonging to the subgroup 1 (namely, P2Y1,2,4Rs), a reduced amount of the P2Y6 and, more significantly, of the P2Y12,13,14Rs was found in the presence of GFs, i.e., in the same conditions where GPR17 is expressed at very low levels. Notably, the latter P2YR subtypes are included in the subgroup 2 of the P2YR family, and are very similar to GPR17 (Ciana et al.,2006). Since the work was focused on GPR17 and due to the lack of really reliable antibodies,
we cannot exclude that these receptors were expressed by the astrocytic population, representing the vast majority of cells in our cultures. Nevertheless, it may well be that some biochemical, transcriptional or structural features shared between P2Y12,13,14R and GPR17 are at the basis of the modulation of the expression of all theses structurally related receptors exerted by GFs.
Upon shifting to differentiation medium (DM), a different behaviour was observed for cells initially grown in the presence or in the absence of GFs. In cultures grown in medium +GFs, GPR17+ cells started to appear after 24 hours in DM and underwent a linear increase in number over time. All these cells were also positive for the pre-oligodendrocyte marker O4; very few mature CNPase+ or MBP+ were seen, which never coexpressed GPR17. On the contrary, cultures grown in medium −GFs showed a much higher degree of differentiation, with a lower increase in the number of O4/GPR17 double-positive cells over time. Moreover, in these cultures, a subpopulation of O4+/GPR17- pre-oligodendrocytes, which has not been found in cultures grown in medium +GFs, was also detected. Again, more mature oligodendrocytes never stained positive for GPR17. The differential receptor expression on pre-oligodendrocytes also reflected in cell sensitivity to high ATP concentrations, with induction of cell death only in cells expressing GPR17. Activation of the receptor with its known agonist UDP-glucose instead promoted a trend to increase in the number of MBP+ cells, thus confirming previous observations obtained in a different experimental model in vitro (Lecca et al, 2008). These results are particularly interesting at the light of in vivo data showing that, under pathological conditions, high extracellular concentrations of adenine nucleotides, especially ATP, are reached due to increased excitotoxic neurotransmission and to the leakage of the cytoplasmic content from dying cells (Melani et al, 2005). It has been demonstrated that ATP can antagonize the activation of GPR17 induced by uracil derivatives (Pugliese et al, 2009), thus counteracting the beneficial and pro-survival role possibly played by this receptor under physiological conditions. This could contribute to the death of GPR17-expressing cells at the site of injury; this hypothesis is also consistent with the present data showing that O4+ cells undergo massive cell death only when coexpressing GPR17, and are instead partially preserved by the toxic action of ATP when they do not express this receptor.
Since GPR17 undergoes a progressive down regulation along with cell maturation, the authors hypothed that a basal, tonic activation of GPR17 is needed to maintain precursor cell survival and function, and also to drive their differentiation towards a more mature phenotype. This basal activation could be guaranteed by the
continuous release of uracil nucleotides as additional cargo molecules during the delivery of glycosylated proteins from the endoplasmic reticulum to the plasma membrane (Lecca and Ceruti, 2008; Sesma et al, 2009). In line with this hypothesis, UDP and UDP-glucose induced the generation of outward K+ currents in cells heterogously expressing GPR17 (Pugliese et al, 2009). K+ currents represent an important protective mechanism that can preserve cell viability in the brain (Gribkoff et al, 2001). Notably, due to the high concentrations of glycosylated proteins in myelin sheets, a higher physiological release of GPR17 agonists can be envisaged at sites with ongoing active myelination (Jennemann et al, 2005). This is also in agreement with the general protective role played by uracil nucleotides and sugar-derivatives in the brain (Lecca and Ceruti, 2008), and could open new perspective in the evaluation of the role of uracil nucleotides in promoting physiological myelination with possible pharmacological outcomes for demyelinating diseases.
d) Allergic pulmonary inflammation
As mentioned before, cotransfection of GPR17 with CysLT1 receptor in several cell lines suppressed CysLT1R-mediated calcium flux in response to LTD4, and knockdown of GPR17 in mouse bone marrow derived macrophages markedly increased the calcium flux in response to LTD4 in a dose-dependent manner (Maekawa et al., 2009). The authors of this study thus recently explored the role of GPR17 in allergic pulmonary inflammation, a condition in which the involvement of cysLT/CysLT1R have already been shown (Figueroa et al., 2001; 2003; Parameswaran et al., 2004).
In this study, it has been shown that GPR17 negatively regulates the CysLT1R -mediated inflammatory cell accumulation in the bronchoalveolar lavage fluid and lung, the levels of IgE and specific IgG1 in serum, and Th2/Th17 cytokine expression in the lung after intranasal sensitization and challenge with the house dust mite (extract of Dermatophagoides farinae [Df]) in mice. Sensitization of naive wild-type recipients with Df-pulsed bone marrow-derived dendritic cells of each genotype or sensitization of each genotype with Df-pulsed wild-type bone marrow-derived dendritic cells and Df challenge revealed markedly increased pulmonary inflammatory and serum IgE responses for GPR17-deficient mice as compared with wild-type mice and reduced responses in the genotypes lacking CysLT1R. These findings reveal a constitutive negative regulation of CysLT1R functions by GPR17 in both the Ag presentation and downstream phases of allergic pulmonary inflammation (Maekawa et al., 2010).