UNIVERSITÀ DEGLI STUDI DI ROMA
"TOR VERGATA"
FACOLTA' DI MEDICINA E CHIRURGIA
DOTTORATO DI RICERCA IN
IMMUNOLOGIA E BIOTECNOLOGIE APPLICATE
CICLO DEL CORSO DI DOTTORATO
XXIITitolo della tesi
Characterization of invariant NKT cell phenotype and
function in Wiskott-Aldrich Syndrome (WAS)
Nome e Cognome del dottorando
Michela Locci
A.A. 2009/2010
Tutor: Dr.ssa Anna Villa
ACKNOLEDGEMENT
I would like to acknowledge my mentor Anna Villa for supervising me and for making me grow up as scientist and as person.
A very special thank to the Elena Draghici, our super technician, and a very important friend to me. I would like to thank all the WAS group, in particular Marita Bosticardo and Marco Catucci, for the scientific interactions and for sharing with me all day at HSR-TIGET. I would also acknowledge Professor Alessandro Aiuti for the scientific suggestion during the lab meeting.
Finally, the most special thank to Rodrigo. Rorro, I would not have ever done this without you.
INDEX
1. ABSTRACT... 7
1. ITALIAN ABSTRACT ... 8
2. INTRODUCTION... 9
2.1 THE WISKOTT-ALDRICH SYNDROME ... 9
2.2. CLINICAL MANIFESTATIONS OF WAS ...10
2.2.1. Hemorrhages...10 2.2.2. Eczema ...11 2.2.3. Immunodeficiency ...11 2.2.4. Autoimmunity...13 2.2.5. Tumors ...14 2.2.6. Scoring system...15
2.3. MOLECULAR DEFECT CAUSING WAS ...16
2.3.1. Identification and characterization of the gene...16
2.3.2. Molecular functions of WASp...16
2.3.3. WAS gene mutations and genotype/phenotype correlation ...21
2.4. CELLULAR FUNCTIONS REGULATED BY WASp...23
2.4.1. Hematopoietic stem cells ...23
2.4.2. T lymphocytes ...23
2.4.3. B Lymphocytes...27
2.4.4. Natural Killer lymphocytes ...28
2.4.5. Myeloid cells ...28
2.4.6. Megakaryocytes and platelets...29
2.5 INVARIANT NATURAL KILLER T (iNKT) CELLS ...31
2.5.1 Identification of invariant Natural Killer T (iNKT) cells ...31
2.5.2 Distinctive features of iNKT cells ...31
2.5.3 Tissue Distribution and Localization of iNKT cells...33
2.5.4 Ligands of iNKT cells...34
2.5.5 Cell biology of lipid presentation by CD1d ...38
2.5.6 iNKT cell development...41
2.5.7 Intracellular signaling pathway regulating iNKT cell development...45
2.5.9 Signaling pathways involved in iNKT cell function ...52
2.5.10 iNKT cells in antimicrobial immunity...52
2.5.11 iNKT cells in antitumor immunity...53
2.5.12 iNKT cells in autoimmunity...54
2.5.13 iNKT cells in primary immunodeficiencies ...56
3. SPECIFIC AIMS...57
4. MATERIALS AND METHODS...58
4.1 PATIENTS...58
4.2 MICE ...58
4.3 CELL PREPARATION AND FLOW CYTOMETRY...58
4.3.1 Human cells...58
4.3.2 Murine cells...59
4.4 IN VITRO EXPANSION OF HUMAN Vα24 Vβ11 iNKT CELLS...60
4.5 GENERATION OF MIXED BONE MARROW CHIMERAS...61
4.6 ACTIVATED CASPASES ASSAY ...61
4.7 EVALUATION OF HOMEOSTATIC PROLIFERATION BY BrdU LABELLING ...61
4.8 IN VITRO CYTOKINE STIMULATION ...62
4.9 FUNCTIONAL CHARACTERIZATION OF was-/- iNKT CELLS...62
4.9.1 In vivo activation and cytokine production ...62
4.9.2 In vivo iNKT cell expansion following αGalCer stimulation ...62
4.9.3 Mice immunization and antigen-specific Ab titer measurement...63
4.9.4 In vitro activation of iNKT cells by αGalCer loaded DCs ...63
4.9.5 αGalCer presentation by Bone Marrow derived DCs (BM DCs) ...63
4.9.6 In vitro cytokine production by stimulated iNKT cells ...64
4.9.7 Measurement of TCR Avidity...64
4.10 STATISTICAL ANALYSIS...65
5. RESULTS ...66
5.1 CHARACTERIZATION OF Vα24 Vβ11 iNKT CELLS FROM WISKOTT-ALDRICH SYNDROME PATIENTS...66
5.1.1 Lack of Vα24 Vβ11 iNKT cells in WAS, but not in XLT patients ...66
5.1.4 WASp is expressed by all human Vα24 Vβ11 iNKT cells ...75
5.2 PHENOTYPICAL CHARACTERIZATION OF iNKT CELLS FROM was -/-MICE ...76
5.2.1 WASp is expressed by murine iNKT cells...76
5.2.2 Reduced iNKT cell frequency and number in was-/- mice ...77
5.2.3 Regular phenotype and subset distribution of was-/- iNKT cells ...80
5.2.4 Developmental block of was-/- iNKT cells...83
5.2.5 Evaluation of apoptotic cell death and homeostatic proliferation in murine iNKT cells...88
5.2.6 Proliferation of was-/- iNKT cells in response to IL-15...91
5.3 FUNCTIONAL CHARACTERIZATION OF MURINE was-/- iNKT CELLS.93 5.3.1 Reduced in vivo cytokine production upon αGalCer injection in was-/- mice ...93
5.3.2 Functional impairment of in vivo activated was-/- iNKT cells...95
5.3.3 Reduced help to antigen specific humoral responses by in vivo activated was-/- iNKT cells...98
5.3.4 Contribution of was-/- DCs to was-/- iNKT cell functional defect...99
5.3.5 Normal ability of was-/- DCs to present αGalCer...101
5.3.6 In vitro cytokine production ability of was-/- iNKT cells...102
6. DISCUSSION ...106
6.1 WAS PATIENTS ARE DEVOID OF iNKT CELLS ...106
6.2 IMPAIRED DEVELOPMENT OF MURINE was-/- iNKT CELLS...108
6.3 REDUCED FUNCTION OF MURINE was-/- iNKT CELLS ...112
6.4 CONCLUDING REMARKS ...114
7. BIBLIOGRAPHY ...116
1. ABSTRACT
WAS protein (WASp) is a key regulator of actin cytoskeleton in hematopoietic cells. Mutations of WAS gene cause the Wiskott-Aldrich Syndrome (WAS). Although WASp is involved in various immune cell functions, its role in invariant NKT cells (iNKT) has never been investigated. Defects of iNKT cells could contribute to the pathogenesis of several WAS features, such as recurrent infections and high tumor incidence. Indeed, we found a profound reduction of circulating iNKT cells in WAS patients, directly correlating with the severity of clinical phenotype. To better characterize iNKT cell defect in the absence of WASp, we analyzed was-/- mice. iNKT cell number is significantly reduced in thymus and periphery of was-/- mice as compared to wt controls. Moreover analysis of was-/- iNKT cell maturation reveals a complete arrest at the CD44+NK1.1- intermediate stage. Notably, generation of BM chimeras demonstrated a was-/- iNKT cell autonomous developmental defect. The lack of WASp does not affect IL-15 signaling, which is important in iNKT cell development. Conversely WASp is required for the control of homeostatic proliferation. was-/- iNKT cells are also functionally impaired, as suggested by the lower expansion and reduced secretion of IL-4 and IFN-γ upon in vivo activation. Furthermore, in vitro assays suggest that the functional defect of WASp null iNKT cells is TCR-mediated and indicated that the defective IL-4 production is due to a
was-/- iNKT cell autonomous defect, whereas the lower IFN-γ production is caused by
an inefficient crosstalk between was-/- iNKT cells and was-/- DCs. Altogether, these
results demonstrate the relevance of WASp in integrating signals critical for development and functional differentiation of iNKT cells, and suggest that defects in these cells may play a role in WAS pathology.
1. ITALIAN ABSTRACT
WASp è una proteina che regola il rimodellamento dell’actina del citoscheletro nelle cellule ematopoietiche. Mutazioni nel gene che codifica per WASp (WAS) causano la Sindrome di Wiskott-Aldrich (WAS). Sebbene WASp sia coinvolto in svariate funzioni delle cellule del sistema immunitario, il suo ruolo nei linfociti invarianti NKT (iNKT) non è mai stato investigato. Difetti delle cellule iNKT potrebbero infatti contribuire allo sviluppo di alcune caratteristiche dei pazienti WAS quali le infezioni ricorrenti e l’alta incidenza tumorale. Infatti il nostro studio ha rivelato una profonda riduzione numerica dei linfociti iNKT periferici nei pazienti WAS, direttamente correlata con la severità del fenotipo clinico dei pazienti. Per definire ulteriormente il fenotipo delle cellule iNKT prive di WASp, abbiamo esteso l’analisi ai topi was-/-. Le cellule iNKT sono significativamente ridotte anche nel timo e negli organi linfoidi periferici dei topi was-/- rispetto ai controlli wt. Inoltre l’analisi dello sviluppo delle cellule was-/- iNKT cell ha messo in luce un completo blocco maturativo allo stadio intermedio CD44+NK1.1-. In particolare, la generazione di chimere di midollo osseo, ha dimostrato un difetto maturativo intrinseco delle cellule was-/- iNKT. L’assenza di WASp non altera la stimolazione indotta dall’IL-15, che è importante nello sviluppo delle cellule iNKT. Contrariamente, WASp è coinvolto nel controllo della proliferazione omeostatica di questa tipologia cellulare. Le cellule iNKT prive di WASp presentano anche un difetto funzionale, come messo in evidenza dalla ridotta secrezione di IL-4 e IFN-γ dopo la loro attivazione in vivo. In aggiunta, saggi funzionali condotti in vitro, suggeriscono che il difetto funzionale delle cellule iNKT prive di WASp sia mediato dal TCR e che la ridotta produzione di IL-4 sia causata da un difetto funzionale intrinseco alle cellule iNKT, mentre la minor produzione di IFN-γ sembra derivare da un’interazione non efficiente tra le cellule was-/- iNKT cells e le
cellule dendritiche was-/-. Nel loro insieme questi risultati dimostrano il ruolo rilevante di WASp nell’integrare segnali critici per lo sviluppo e la funzione delle cellule iNKT, e suggeriscono che i difetti di questa popolazione linfocitaria possano contribuire alla patologia della Sindrome di Wiskott-Aldrich.
2. INTRODUCTION
2.1 THE WISKOTT-ALDRICH SYNDROME
In 1937, Alfred Wiskott, a German paediatrician, described a “familial and innate thrombopathia” affecting three brothers. The pathology was characterized by thrombocytopenia, bloody diarrhea, eczema, recurrent ear infections, and eventually led to the death of the patients because of intestinal hemorrhages and sepsis (1). In 1954, Aldrich and colleagues demonstrated the X-linked inheritance of this syndrome by analyzing the pedigree of a family in which 16 out of 40 males -but no females- were affected (2).
Nowadays, Wiskott-Aldrich Syndrome (WAS, OMIM 301000) is known as a complex and severe X-linked disorder characterized by micro-thrombocytopenia, eczema, immunodeficiency and increased risk to develop autoimmunity and lymphomas. WAS affects 1 to 10 out of a million male newborns (3), whose life expectancy is about 15 years (4).
The identification of the gene whose function is lost in WAS (WAS gene) has been achieved In 1994 through a positional cloning strategy (5). The protein encoded by the WAS gene (Wiskott-Aldrich Syndrome protein, or WASp) is a hematopoietic specific (6) regulator of actin nucleation in response to signals arising at the cell membrane (7).
The discovery of the WAS gene has allowed to link other diseases to alterations in this locus. Mutations impairing but not abolishing WASp expression, can cause X-linked thrombocytopenia (XLT). This disease can be chronic (8) or intermittent (9), and is considered an attenuated form of WAS since it is characterized by low platelet counts with minimal or no immunodeficiency. Recently, gain-of-function mutations in the
WAS gene giving rise to a constitutively active protein, were found to cause a distinct
pathology, X-linked neutropenia (XLN). It is characterized by low neutrophil counts and predisposition to myelodysplasia in the absence of thrombocytopenia and T-cell immunodeficiency (10, 11).
2.2. CLINICAL MANIFESTATIONS OF WAS
2.2.1. Hemorrhages
Hemorrhages are frequent (>80% incidence) in WAS patients, and range from non-life threatening (epistaxis, petechiae, purpura, oral bleeding) to severe manifestations such as intestine and intracranial bleeding. Death of WAS patients is caused, in 25% of the cases, by hemorrhages (12). Bleeding is due to severe thrombocytopenia (ranging from 6000 to 70000 per µl (4)) with reduced platelet size. This is the most common finding in WAS and XLT patients (100% incidence) and is due to the lack of WASp in platelets irrespectively of the severity of the mutation, possibly as a consequence of instability of the mutated protein (13).
Despite intensive research, the mechanisms underlying WASp-related thrombocytopenia and hemorrhages are incompletely understood. Indeed, defective thrombopoiesis, accelerated platelet destruction, and platelet dysfunction have been hypothesized, but their relationship with WASp deficiency is unclear.
Whether defective thrombopoiesis contributes to thrombocytopenia in WAS patients is controversial. Indeed, the number and cytological appearance of megakaryocytes in the bone marrow of XLT and WAS patients can be normal (4, 8, 14). On the other hand, defective platelet production has been suggested based on accelerated platelet turnover in WAS patients (14), and decreased in vitro proplatelet formation by megakaryocytes derived from WAS patients (15). These latter results were challenged by another study demonstrating that in vitro proplatelet formation by megakaryocytes derived from WAS patients was normal (16). An alternative hypothesis reconciling normal thrombopoiesis with decreased platelet release in the blood has recently proposed. Indeed, in vivo studies performed in was-/- mice showed that ectopic platelet
shedding within the bone marrow can occur, hampering the platelet release in the peripheral blood despite normal platelet production by megakaryocytes (17). These studies, however, await confirmation on bone marrow specimens isolated from WAS patients.
There is more general agreement on a role of peripheral platelet destruction in the pathogenesis of WAS-related thrombocytopenia. Indeed, decreased half-life of circulating platelets is a common finding in WAS patients (14, 18, 19), and can be due
to intrinsic platelet abnormalities (20) or to autoantibodies (4, 21, 22), finally leading to splenic sequestration (20). Accordingly, splenectomy can correct both platelet numbers and size (21, 23), but it is usually discouraged since it exposes the patient to a high risk of sepsis (23). Moreover, some splenectomised WAS patients suffer a thrombocytopenia relapse, which is usually immune-mediated (21) and predicts a severe prognosis (24).
Platelet dysfunction can also contribute to the hemorrhages frequently observed in WAS patients. Indeed, patients having normal number of platelets after splenectomy are not completely protected from bleeding episodes (23). The mechanisms proposed are defective platelet activation after stimulation with thrombin (22), and defective platelet adhesion to fibrinogen through the αIIbβIII integrin (25).
2.2.2. Eczema
The typical skin lesions in WAS and XLT patients resemble acute or chronic eczema in appearance and distribution. Eczema develops in 80% of the patients (4, 26), and is heterogeneous in severity and persistence. Indeed, in its most severe form, eczema is resistant to therapy, persists into adulthood, and facilitates opportunistic skin infections (molluscum contagiosum, herpes simplex, or bacteria). Severity of eczema is significantly lower in patients with residual WASp expression (4).
The causes of eczema in WAS patients are currently unknown. WAS patients often have elevated IgE levels (4), therefore suggesting an atopic origin. Indeed, WAS patients often develop allergy (4). Recently, an imbalance in cytokine production towards the Th2 type has been described in WAS patients’ T cell lines (27), and might contribute to the pathogenesis of eczema and allergy. Abnormal priming of antigen-specific T cells in the skin caused by defective chemotaxis of DCs and Langerhans cells may also play a role (3).
2.2.3. Immunodeficiency
WAS patients have often symptomatic infections caused by various microorganisms including bacteria, viruses and fungi. Frequency and severity of infections are higher in WASp-negative patients, as compared to WAS patients with residual WASp
expression (4), and cause nearly 50% of death cases (12). Most common bacterial infections affect the respiratory tract, the ear, the gut, the skin, and the urinary tract. Systemic infections, such as meningitis and sepsis, are also reported (4). Bacterial infections are mainly due to encapsulated bacteria, probably because of the inability to produce antibodies against polysaccharide antigens (14). Viral infections are mostly due to Herpes simplex (can be severe and disseminated) and to
Cytomegalovirus spp. (encephalitis and hepatitis). WAS patients also develop Molluscum contagiosum and Papillomavirus infections. Fungal infections due to Candida spp. and Aspergillus spp. as well as opportunistic pulmonary infections due
to Pneumocystis carinii are also frequent in these patients (4).
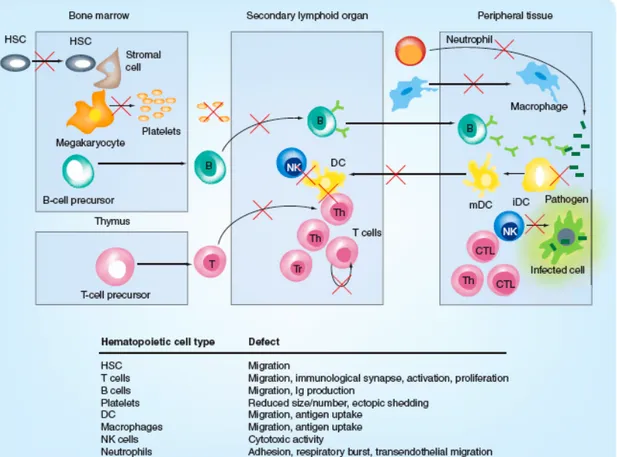
The complex immunodeficiency caused by the lack of WASp results from the dysfunction of many immune cell types. Indeed, absence of WASp impairs T cell activation (27-29), NK lytic activity (30, 31), B cell function (32), phagocytosis (33, 34) and antigen presentation by DCs (35), and microbial killing by neutrophils (36). In addition, migration of WASp-deficient hematopoietic stem cells (37) and leukocytes (38) is globally defective. Finally, WAS patients, especially young ones, can suffer from T and B lymphopenia (39). For a schematic summary of the reported cellular defects refer to Figure I.
Figure I: Cellular functions altered by WASp deficiency.
Abbreviations used: HSC: hematopoietic stem cell; B: B lymphocyte; NK: natural killer lymphocyte; T: T lymphocyte; Th: T helper lymphocyte; CTL: cytotoxic T lymphocyte; iDC: immature dendritic cell; mDC: mature dendritic cell. Modified from Trifari et al (40).
2.2.4. Autoimmunity
WAS-associated autoimmune complications are frequently observed. They have been reported to affect from 25% to 72% of the patients (4, 24, 26), irrespectively of WASp expression and overall disease severity (4). The most common manifestations are autoimmune hemolytic anemia, cutaneous vasculitis, arthritis and nephropathy. Less common autoimmune manifestations include inflammatory bowel disease, idiopathic purpura thrombocytopenia and neutropenia. Patients frequently suffer from multiple autoimmune manifestations at the same time (24).
Development of autoimmunity can have a prognostic value. Indeed, it has been reported that WAS patients who develop autoimmune hemolytic anemia or autoimmune thrombocytopenia early (<180 days) after splenectomy, have a poor prognosis (24). Moreover, autoimmunity is associated with a higher risk of a later
development of tumors and with an increased risk of mortality (26).
Until now, the mechanisms of WAS-associated autoimmunity have not been clarified. It has been proposed that autoimmunity could be the result of a bystander tissue damage originating from the chronic inflammatory state that is established after incomplete pathogen clearance (12). Another possible cause of autoimmunity is the loss of central or peripheral tolerance to self-antigens. Given the role of WASp in TCR signaling, it is possible that WASp deficiency impairs negative selection of thymocytes, leading to recirculation of abnormal proportions of potentially self-reactive T cells. It is also possible that WASp absence impairs localization or function of regulatory T cells, which can no longer suppress the activation of autoreactive T cells in the periphery. Indeed, several groups including ours have recently described a defective localization and function of naturally occurring CD4+CD25+FOXP3+ regulatory T cells in the absence of WASp (41-44).
2.2.5. Tumors
Two distinct surveys report a tumor incidence of 13% and 22% (4, 26) in WAS patients. Tumors can arise during childhood (especially myelodysplasia) but are more frequent in adolescents and young adults. WAS-associated tumors are mainly lymphoreticular malignancies, since leukemia, myelodysplasia, and lymphoma (often EBV-positive) cover 90% of the cases. WAS-associated malignancies have a poor prognosis, as less than 5% of patients survive 2 years after diagnosis (26), and cover up to the 25% of the death cases (12).
The observation that a consistent proportion of malignancies is associated to EBV infection, leads to the hypothesis that immune deficiency can contribute to the genesis of tumors. Defective NK cell functions, as well as other alterations of immune surveillance, may play a key role in the susceptibility to tumor development. However, other mechanisms can be involved, since the highest lymphoma incidence (44%) can be conferred by a single splice site mutation that is otherwise associated with a mild clinical phenotype (45). To this regard, it has been recently reported that WASp regulates cytokinesis and genomic stability in human cells (46), leading to the hypothesis that WASp mutations may directly alter cellular homeostasis.
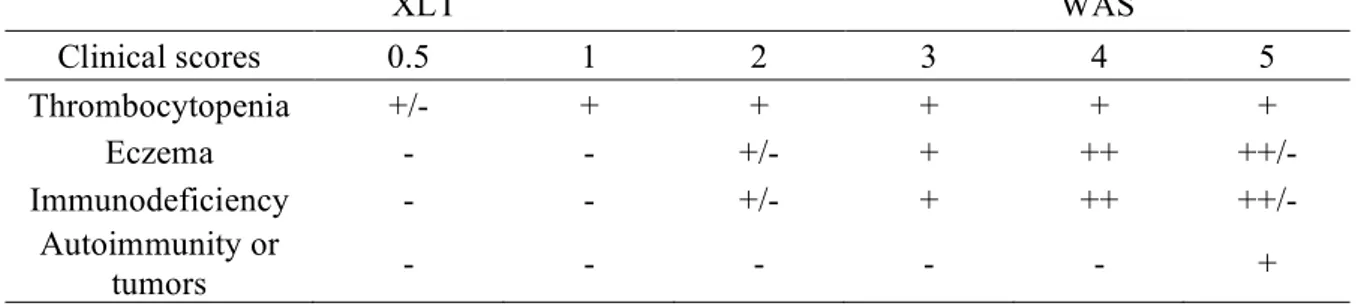
2.2.6. Scoring system
The severity of WAS-associated symptoms can be estimated through a scoring system originally developed by Zhu and colleagues (47), and slightly refined in subsequent works (4, 9).
A score of 0.5 or 1, assigned to patients with intermittent or chronic thrombocytopenia and small platelets, and a score of 2, assigned to patients with additional findings of mild, transient eczema or minor infections, identify XLT patients. Those with treatment-resistant eczema and recurrent infections in spite of optimal treatment receive a score of 3 (mild WAS) or 4 (severe WAS). Regardless of the original score, if a patient develops autoimmune disease or malignancy, a score of 5 is attributed. For a schematic summary of the scoring system, refer to Table I.
Table I: WAS scoring system according to Zhu and colleagues (47), with subsequent refinements (4, 9). XLT WAS Clinical scores 0.5 1 2 3 4 5 Thrombocytopenia +/- + + + + + Eczema - - +/- + ++ ++/- Immunodeficiency - - +/- + ++ ++/- Autoimmunity or tumors - - - +
2.3. MOLECULAR DEFECT CAUSING WAS
2.3.1. Identification and characterization of the gene
By taking advantage of a previous mapping study which localized the WAS gene to the region Xp11.22-Xp11.3 (48), Derry and colleagues isolated the WAS gene by positional cloning and demonstrated mutations in lymphoblastoid cell lines derived from patients with WAS or XLT (5). The WAS gene encompasses 12 exons, and encodes a 502-aminoacid intracellular protein (WASp). WASp is expressed in all non-erythroid hematopoietic cells (6, 13).
The mouse was gene was identified in parallel to the human WAS gene. The mouse
was gene resides on the X chromosome, and encodes a 520-aminoacid protein which
shares 86% identity with the human counterpart (5). Two was knock-out (was-/-) mouse strains were constructed by disruption of the was gene by deletion of exons 4 to 11 (49), or by a large insertion in exon 7 (50). In both cases WASp expression was completely abrogated. These mouse strains have been validated as relevant models of WAS, since they carry many of the cellular defects originally described in WAS patients (49, 50), and are immunodeficient (51).
2.3.2. Molecular functions of WASp
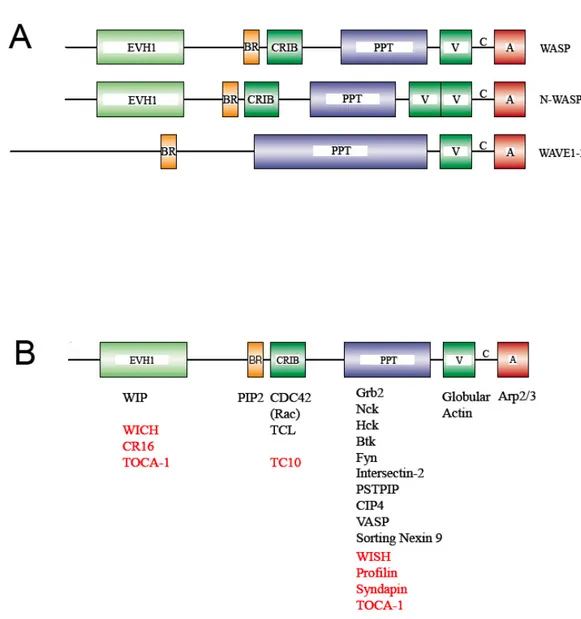
WASp is the founding member of a family of proteins whose function is to integrate the signals from many input pathways to promote actin cytoskeleton remodeling. The other members of the WASp family in mammals are the neural-WAS protein (N-WASP) and the scar/WAVE proteins (WAVE1, WAVE2, and WAVE3) (52) (Figure IIA). All these proteins share a highly similar C-terminal portion composed by a polyproline-rich region (PPT), and a Verprolin/Cofilin/Acidic (VCA) domain. In particular, the VCA domain can bind and activate the Arp2/3 complex, one of the most powerful activators of nucleation of new F-actin filaments stemming from pre-existing ones (“branching” process). On the other hand, the N-terminal portion of the WASp family proteins, which actually controls their activation, is less conserved. Indeed, WASp and N-WASp contain an EVH1 (Ena/VASP homology 1) domain, a basic region (BR), a CDC42 and Rac interactive binding domain (CRIB), while the
WAVE proteins lack such a structure, retaining the basic region only. WASp and N-WASp have a very similar domain structure, but differ for the expression pattern, since the former is hematopoietic-specific and the latter is ubiquitous, but more abundant in the nervous system. The very high structural similarities between WASp and N-WASp have led to the speculation that the molecular activation processes, as well as the binding partners, demonstrated for either of the two proteins, are actually relevant for both. This is illustrated by the recent demonstration of the redundancy of WASp and N-WASp for T cell differentiation (53). For a schematic summary of proteins interacting with WASp, see Figure IIB.
Constitutive interaction between the WASp EVH1 domain and the WASp interacting protein (WIP) stabilizes WASp through inhibition of calpain and proteasome degradation (54-56). Moreover, it could also inhibit spontaneous WASp activation (demonstrated experimentally for N-WASp (57)). It has been hypothesized that the dissociation of the WIP/WASp complex is key for WASp activation in T cells stimulated through the TCR (58). However, evidences confuting this hypothesis have been reported very recently. Indeed, Dong and colleagues demonstrated that a WIP/WASp fusion protein is functional in TCR-stimulated T cells (59).
Figure II: Structure of WASp family proteins.
A. Schematic representation of the WASp family proteins in mammals. B. Proteins
interacting with WASp. The interactions shown experimentally for N-WASp are reported in red. Modified from Thrasher et al (60).
The actin-remodeling activity of WASp is mediated by the C-terminal VCA domain, which directly binds and activates the actin nucleation complex Arp2/3. In resting conditions, WASp assumes an auto-inhibited conformation characterized by the folding of the VCA domain on the CRIB domain through hydrophobic interactions (61). As a consequence, the VCA domain of WASp is unable to bind the Arp2/3 complex and promotes actin nucleation. The molecular activation of WASp can be achieved by cooperative binding of multiple interactors. Binding of phosphatidylinositol (4,5) bi-phosphate (PIP2) to the N-terminal basic region of WASP can synergize with the binding of GTP-loaded CDC42 to the CRIB domain (62), or the binding of SH3 domain containing proteins (such as Grb2, Nck) to the PPT region (63), to release auto-inhibition and promote actin nucleation. Binding of other SH3 domain containing proteins to WASp PPT region mediates a number of other important functions. Indeed, interaction with TOCA-1 amplifies the activatory signal mediated by PIP2 and GTP-CDC42 (shown experimentally for N-WASp (64)); the Intersectin-2 (65) and Sorting Nexin 9 (66) can recruit WASp to endocytotic vesicles; binding of CIP4 could mediate interaction with the microtubule cytoskeleton (67); interaction with VASP could facilitate filopodia formation (68). In addition, several kinases and phosphatases can interact with WASp SH3 domain, and regulate its activation through phosphorylation and dephosphorylation of a single tyrosine residue (Y291). Phosphorylation of Y291 (Y293 in murine WASp), achieved by Fyn (in T cells) (69), Btk (in B cells) (70), and Hck (in phagocytes) (71), increases the basal activity of WASp conferring sensitivity to input signals through SH2-domain containing proteins (72), and leading to cell activation (69). In some circumstances tyrosine phosphorylation may occur independently of binding to GTP-bound CDC42 (69, 71). In addition, a recent finding underlines the absolute requirement for WASp phosphorilation during multiple cellular task, including migration, phagocytosis and proliferation (73). Dephosphorylation of Y291 is achieved by the PTP-PEST phosphatase, which is recruited to WASp through the adaptor protein PSTPIP (69). Moreover, constitutive phosphorylation at residues S483 and S484 by CK2 kinase is required for optimal activation of the Arp2/3 complex (74). A simplified model for WASp activation is depicted in Figure III.
Figure III: Model depicting the mechanism of WASp activation.
A. Autoinhibited conformation by hydrophobic interactions between the CRIB domain and
the VCA domain. B. By cooperative binding of PIP2 to the basic region, GTP-CDC42 to the CRIB domain, and several SH3 domain-containing proteins to the poly-proline region, autoinhibition is released. C. WASp activation is further stabilized by tyrosine 291 phosphorylation, and the VCA domain can bind the ARP2/3 complex leading to actin polymerization. Modified from Ochs et al (3).
Besides being key to actin cytoskeleton remodeling, WASp can also control gene transcription. Indeed, WASp N-terminal EVH-1 domain controls NFAT-mediated transcription in a way unrelated to actin polymerization, as demonstrated by studies performed on T cell lines transfected with different domains of WASp (75). Studies conducted on more relevant cell types gave further support to this hypothesis. Indeed, CD4+ and CD8+ untransformed T cell lines derived from WAS patients showed a delayed kinetic of NFA1 nuclear translocation, and a defective transcription of T-bet and Th1 cytokine genes, following TCR stimulation (27). Similarly, in primary T cells isolated from was-/- mice, a delayed kinetic of NFAT-1 nuclear translocation
after TCR engagement was described. In addition, a defective nuclear localization of p-Erk, the consequent lack of Elk-1 phosphorylation, and c-fos transcription, led to impaired AP-1 DNA-binding activity. These defects were likely causative of impaired IL-2 gene transcription (76). In line with this hypothesis, it has recently been reported that WASp deficiency in NK cells delays nuclear translocation of NFAT-2 and RelA, a subunit of NFκB, in a way independent from actin polymerization (77).
In conclusion, WASp is a molecule whose stability and function is tightly regulated, and which is able to integrate a wide array of different inputs to activate actin polymerization, and gene transcription.
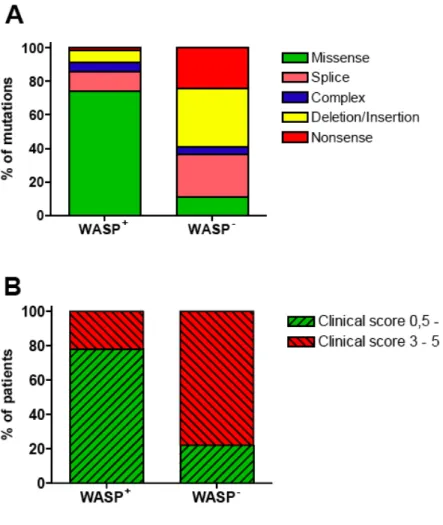
2.3.3. WAS gene mutations and genotype/phenotype correlation
Approximately 300 unique mutations have been reported in the WAS gene, spanning all 12 exons (http://homepage.mac.com/kohsukeimai/WASp/WASPbase.html). It has been demonstrated that the genotype of WAS mutations largely determines WASp expression levels (78), which in turn determines disease severity (4, 79). Indeed, missense mutations usually resulting in residual expression of a mutated protein are associated with XLT (disease score 0.5-2, see Figure IV). In contrast, nonsense mutations, deletions or insertions abolishing WASp expression are associated with a full-blown WAS phenotype (disease score 3-5, see Figure IV). However, about 20% of the WASp-expressing patients have a severe clinical phenotype (Figure IV). Moreover, it has been reported that WASp-expressing patients can progress to a score of 5 due to autoimmunity (4) or malignancy (45). Therefore, long-term prognosis
Figure IV: Genotype/phenotype correlation.
A. Correlation between mutation type and WASp expression. Data are derived from Jin et al
(79). B. Correlation between WASp expression and clinical score. Data are derived from Notarangelo et al (80).
2.4. CELLULAR FUNCTIONS REGULATED BY WASp
2.4.1. Hematopoietic stem cells
Early studies conducted on human samples have demonstrated that WAS RNA is transcribed already at the stage of CD34+ hematopoietic stem cells (HSC), and its expression is kept along hematopoietic differentiation (81). Despite this, the cytological appearance of bone marrow of WAS patients is often normal (14), and CD34+ cells isolated from a WAS patient can differentiate into a normal number of myeloid colonies in vitro (82). Therefore, WASp might be dispensable in early haematopoiesis.
The study of obligate female carriers of a mutated WAS allele shed light on the function of WASp in hematopoietic stem cells. Indeed, a non-random X-inactivation in CD34+ cells and in mature hematopoietic cells was observed in these individuals, leading to the hypothesis that WASp can play a role in the lyonization process (83). However, a direct role for WASp in X-chromosome methylation has never been demonstrated. Alternatively, non-random X-inactivation in the bone marrow can be explained by a migratory defect of WASp-null HSC. Analysis of heterozygous was
+/-mice, and competitive transplantation experiments, demonstrated that WASp is crucial for transition of hematopoietic stem cells from the fetal liver to the bone marrow, and for their engraftment (37). Besides providing a possible explanation to the non-random X-inactivation, these findings are highly relevant for WAS gene therapy, as they predict the preferential engraftment of gene corrected hematopoietic stem cells.
2.4.2. T lymphocytes
T cell defects, affecting effector, helper and regulatoryfunctions, are thought to play a key role in WAS-associated immunodeficiency.
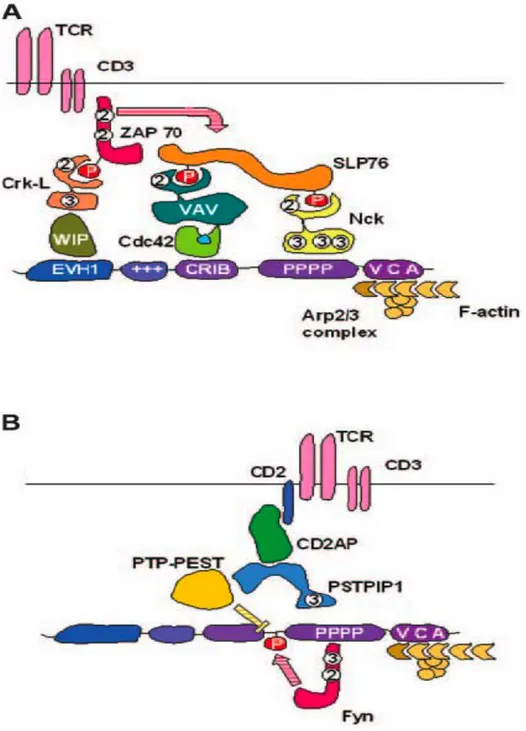
WASp plays a key role in T cell activation and actin cytoskeleton remodeling after the engagement of the T cell receptor (TCR) (29, 49, 50, 84), and the co-stimulatory molecules CD28 (66) and CD2 (85). T cell activation is regulated by the formation of the immunological synapse (IS), a polarized cluster of the TCR, co-stimulatory
molecules, signaling molecules, and integrins to the T cell:APC interface. The IS is a symmetrical structure organized in concentric rings, with the TCR, the TCR- associated molecules, and co-stimulatory molecules residing in the centre, while integrins are localized in a concentric ring. Larger molecules such as CD45 and CD43, which may interfere with synapse assembly through steric hindrance, are actively excluded from the IS. To promote their lateral movement on the plasma membrane, the molecules being recruited to the IS are associated with specific cholesterol-enriched membrane microdomains, called lipid rafts. Following TCR engagement, WASp is promptly recruited to the lipid rafts and the IS through Nck (86), which can directly interact with WASp, and/or by the adaptor protein CrkL, which binds WIP and recruits the WIP-WASp complex (58). In addition, WASp recruitment to the IS could be mediated by CD2 through the adaptor molecules CD2AP and PSTPIP1(85). TCR ligation also causes PKCθ-dependent phosphorylation of WIP and disengagement of WASp from the WIP-WASp complex (58). In parallel the Tec tyrosine kinase Itk mediates the TCR-induced recruitment of Vav to the IS (87, 88), which in turn activates CDC42. GTP-CDC42 finally activates WASp in situ (86, 89). Alternatively, as mentioned above, Fyn-mediated tyrosine phosphorylation might activate WASp independently of binding to GTP-bound CDC42 (69, 71). For a schematic representation of the mechanisms leading to the recruitment of WASp to the IS, refer to Figure V. In the absence of WASp, IS can be formed only after strong TCR stimulation (90). In addition, lipid raft dynamics during IS formation (28), and IS stability (91) are compromised. Another level of regulation of T cell activation is achieved by prompt internalization of the TCR and the CD28 co-stimulatory molecule after specific engagement, functions that are defective in WASp deficient cells (65, 66). As a consequence of impaired signaling through the TCR and co-stimulatory molecules, T cells from WAS patients and was-/- mice show
defective proliferation as well as impaired production of IL-2 (27, 29, 49, 50). The inability of was-/- cells to proliferate upon initial stimulation precludes the ability in the following downstream events such as Th differentiation and the production of effector cytokines. Indeed human WASp deficient T cells display an impaired production of IFN-γ, while the murine counterpart can produce Th1 cytokines but are defective in their secretion (27, 29, 49, 50, 92). These defects are associated with delayed NFAT-1 nuclear translocation and defective T-bet induction (27, 76). Very
recently WASp was also shown, in mice but not in humans, to control the Th2 effector function of CD4 T cells. Morales-Tirado et al. in fact demonstrated a post-transcriptional requirement for WASp in IL-4 production (93).
Notably even natural occurring regulatory T cells (nTreg), a key population in the control of autoimmunity, are dysfunctional in the absence of WASp (42-44). Indeed, similar as for the other T cell subsets, WASp is required for the TCR/CD28-triggered proliferation, for TGF-β production and for the suppressive function of nTreg.
In addition to its role in T cell activation and cytokine production, WASp is also critical for T cell chemotaxis in vitro in response to SDF-1α (94) and in vivo homing to secondary lymphoid organs (95).
A reduction in the numbers of circulating naïve CD4+ and CD8+ T cells may be present in WAS patients, especially at young age, contributing to the immunodeficiency (39). A decreased thymic output has been hypothesized based on the description of abnormal thymic involution in a limited number of histological studies (96, 97). On the other hand, recent studies have highlighted that WASp is dispensable for thymic generation of T cells in mice. Indeed, was-/- mice have a
relatively normal thymic development, but abrogation of both WASp and N-WASp function through a dominant negative portion of WASp (98), or simultaneous knock-out of N-Was (53), cause the block of thymocyte maturation at the DN3 stage. Thus, N-WASp can complement WASp deficiency to promote the generation of normal numbers of T cells. In addition, the observation that the TCR Vβ repertoire is normal in young WAS patients, indicates that WASP absence does not impair thymopoiesis qualitatively (99). In the same study, it has been observed that the TCR Vβ repertoire of WAS patients becomes skewed after 15 years of age (99). This finding supports the hypothesis of defective T cell survival in the periphery. Accordingly, T lymphocytes isolated from the blood of WAS patients are abnormally prone to spontaneous in vitro apoptosis due to decreased Bcl-2 (100), or increased Fas levels (101).
Despite the above information, the precise relationship between T cell abnormalities and WAS-associated immune deficiency, autoimmunity and cancer remains to be elucidated.
Figure V: Mechanisms of WASp recruitment to the IS in T lymphocytes.
A. Recruitment through WIP and Nck (58, 86, 89). B. Recruitment through PSTPIP1, CD2AP
and CD26 (69, 85). SH2 and SH3 domains are indicated by “2” and “3”, respectively. P indicates phosphorylation. Modified from Burns et al (102).
2.4.3. B Lymphocytes
WAS is also characterized by impaired humoral immunity. Indeed, WAS patients often have elevated IgA and IgE titers. Serum IgM levels are decreased in some patients, but in other ones they are elevated and predict a poor prognosis (24). In addition, WAS patients present a specific defect in the production of Abs against polysaccharide antigens (14).
In contrast to what observed in T lymphocytes, activation of WASp-deficient B lymphocytes can occur normally. Indeed, measurements of calcium fluxes after BCR engagement highlighted a mild defect in WASp-deficient EBV-transformed B cell lines (103), but not in primary B cells isolated from WAS patients (104). Moreover, B cells isolated from was-/- mice did not show any activation defect (49, 50). In addition, in vitro immunoglobulin class-switching (32, 105) and presentation of soluble
antigens (35) do not appear to depend on WASp.
On the other hand, WASp-deficient B cells show alterations of the cytoskeleton (32, 106) likely due to defective F-actin nucleation (107), impaired cell polarization (32, 106), decreased chemotaxis in vitro (32), and decreased homing to B cell areas in the spleen in vivo (32). In addition, an abnormal splenic architecture characterized by involution of T and B cell areas, and marginal zone, has been described in both was-/- mice (32) and WAS patients (108). Germinal centre reaction can also be impaired (32). This finding is in agreement with the observation that WAS patients may have an increased proportion of circulating germinal centre precursors, and a decreased percentage of CD27+ post-germinal centre B cells (105).
WAS patients may also suffer from B cell lymphopenia (105). The underlying causes are unknown, but increased spontaneous apoptosis of WASp-deficient B lymphocytes might cause peripheral destruction (100). In contrast with this hypothesis, two recent studies performed in was-/- mice clearly indicate a role for WASp in peripheral homeostasis of mature B cell subsets rather than in apoptosis (109).Alternatively, B cell ontogenesis might be affected by the absence of WASp.
In conclusion, B cell intrinsic defects may act in concert with defective T cell help to generate the impaired humoral immunity observed in WAS patients.
2.4.4. Natural Killer lymphocytes
The high susceptibility of WAS patients to viral infections and lympho-reticular malignancies suggests an impairment of NK cell-mediated cytotoxicity and immune surveillance.
Even though WAS patients have a normal or increased percentage of circulating NK cells (30, 31), these cells have a reduced ability to form conjugates, and fail to polarize actin towards the target cells in vitro. Consequently, both direct and CD16-mediated cytolysis (a model of ADCC) are impaired (30, 31). In addition, secretion of GM-CSF is defective in WASp-deficient NK cells (77).
WASp is physiologically recruited to the contact area between NK cells and their targets (the lytic synapse) (31), and is activated by GTP-CDC42 and by tyrosine phosphorylation after CD16 or β2-integrin engagement (30). WASp deficiency in NK cells alters significantly the molecular composition of the lytic synapse, and delays the nuclear translocation of NFκB and NFAT-2 (77).
In addition, defects in other cell types can contribute to the dysfunction of NK cells in WAS patients. To this regard, it has been demonstrated that in vitro lytic activity of NK cells isolated from WAS patients can be rescued by treatment with exogenous IL-2 (30), leading to the hypothesis that impaired IL-IL-2 production by WASp-deficient T cells might play a role in overall NK cell dysfunction in WAS. Moreover, it has been shown that was-/- mature DCs are unable to activate NK cells in vitro (110).
Therefore, WASp deficiency triggers mechanisms intrinsic or extrinsic to NK cells, which can cooperate in causing the profound impairment of NK lymphocyte function observed in WAS patients.
2.4.5. Myeloid cells
Defects in myeloid cell functions, such as pathogen clearance and antigen presentation, can contribute to the immunodeficiency in WAS patients.
WASp absence impairs F-actin reorganization and polarization of monocytes (111), macrophages (112), and DCs (113), leading to defects in directional chemotaxis in
vitro. In macrophages and DCs, migration depends on the formation of podosomes,
peculiar cell adhesion structures characterized by an actin core surrounded by an integrin ring. WASp localizes to podosomes through WIP (54), and is absolutely
required for their formation (114).
WASp is also key for efficient phagocytic cup formation in human macrophages, and its deficiency impairs the uptake of opsonised bacteria and apoptotic cells (33, 34, 115).
Impaired DC function in the absence of WASp can result in defective priming of T cells. Indeed, studies on was-/- mice have shown that was-/- DCs home inefficiently to
secondary lymphoid organs (116) and cause defective in vivo priming of CD4 and CD8 T cells (117, 118). Moreover, was-/- DCs are impaired in presenting particulate
antigens to T cells in vitro (35). In addition to the above findings, in vitro activation of NK lymphocytes by was-/- DCs is inefficient (110).
WASp is also key for neutrophil function, as testified by defective cell adhesion, migration, and oxidative burst in WASp-deficient neutrophils activated through the β1, β2, and β3 integrins (36). In a separate study, defective phagocytosis of yeast zymosan particles by was-/- granulocytes has been observed (49). WASp is also
required for mast cell degranulation and cytokine production after FcεRI engagement (119).
In conclusion, the defects described above are likely to impair pathogen recognition and clearance, which in turn may contribute to WAS-associated immunodeficiency, chronic inflammation, and development of autoimmunity.
2.4.6. Megakaryocytes and platelets
Platelet defects are observed in all WAS patients irrespectively of the severity of the mutation, likely as a consequence of specific protein instability and degradation (13). Whether thrombopoiesis is defective in WAS patients is still a matter of debate. Indeed, the number of megakaryocytes in the bone marrow of WAS patients is generally normal (4, 14), and defective proplatelet production by megakaryocytes derived from WAS patients was reported in one study(15), but excluded by another one (16). To be efficiently released into the blood stream, platelets must be generated in close proximity to, or even within, the blood vessels in the bone marrow. In contrast, a recent study reported that platelets are ectopically released and accumulate within the bone marrow stroma of was-/- mice, because was-/- megakaryocytes lack
(17). This aspect might contribute to defective platelet release in the circulation, but awaits confirmation on human samples.
Peripheral destruction of platelets by the reticulo-endothelial system is thought to play a major role in the pathogenesis of the thrombocytopenia. Indeed, the half-life of WASp-negative platelets is decreased (14, 18, 19, 42), the frequency of platelets co-localizing with macrophages in the spleen of WAS patients is abnormally high (20), and splenectomy can ameliorate WAS-related thrombocytopenia (23). However, how WASp regulates platelet lifespan is largely unknown. Since platelet-associated IgG have been found in some but not all patients (4), it is likely that the general cause of thrombocytopenia is not related to autoreactive antibodies-dependent elimination. To this regard, it has been reported that platelets isolated from WAS patients abnormally expose at their surface the phospholipid phosphatidylserine (PS), a signal for engulfment by macrophages. The underlying mechanism has not been investigated yet (20).
Platelet function is also dependent on WASp. Indeed, WAS patients with normal platelet counts after splenectomy are not completely protected from hemorrhages (23). In platelets, WASp is activated by phosphorylation after engagement of the collagen receptor GPVI (120). In addition, WASp-null platelets show impaired activation after thrombin stimulation (22), and decreased adhesion to fibrinogen through the αIIbβIII integrin (25). Nonetheless, defective in vitro aggregation of platelets isolated from WAS patients is an inconsistent finding (14, 18, 120, 121). Therefore, despite intensive research, the cellular mechanisms causing thrombocytopenia and platelet dysfunction in WAS patients are still largely unknown.
2.5 INVARIANT NATURAL KILLER T (iNKT) CELLS
2.5.1 Identification of invariant Natural Killer T (iNKT) cells
Invariant Natural Killer T (iNKT) cells are narrowly defined as a T cell lineage expressing NK lineage receptors, including CD161 in human and its murine homologue NK1.1 in the C57BL/6 background, in addition to semi-invariant CD1d-restricted αβ T cell receptors (TCRs) (122).
Several lines of research indicated NKT cells as a separate lineage of T lymphocytes. The first observation included the identification of a canonical Vα14-Jα18 rearrangement in a set of hybridoma derived from mouse KLH (keyhole limpet hemocyanin)-specific suppressor T cells (123-125), and later in cDNA extracted from lymphoid organs of unimmunized mice (126, 127). In parallel, other independent studies leaded to the identification of a murine CD4-CD8- double negative T cell subset with a Vβ8 usage bias (128, 129) and of a recurrent Vα24Jα18 rearrangement in human DN peripheral blood lymphocytes (130, 131). These observations were pieced together when a subset of CD4 and DN IL-4 producing thymocytes co-expressing NK lineage receptors was independently identified and shown to express a biased set of Vβ8, Vβ7 and Vβ2 TCR β chains (132-135) combined with a canonical Vα14-Jα18 chain in mouse (136), and with the homologous Vα24-Jα18/Vβ11 pair in human (136, 137).
The finding that mouse and human NKT cells were autoreactive to cells expressing CD1d (137-140), a member of the CD1 family of MHC-like molecules, completed the initial characterization of this lineage and raised new question regarding their specificity, development and function.
2.5.2 Distinctive features of iNKT cells
One of the most important features distinguishing iNKT cells from conventional T cells is the TCR composition, which is semi-invariant in mouse and invariant in human (122). More than 80% of these TCR are Vα14-Jα18/Vβ8, Vβ7 and Vβ2 in mouse (or Vα24-Jα18/Vβ11 in human), while the remaining 20% represents a
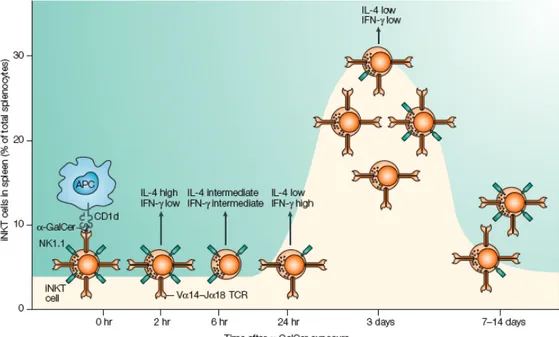
collection of rare Vα3.2-Jα9/Vβ8, Vα8/Vβ8 and other TCRs (141, 142). Although both the Vα14 and the non-Vα14 iNKT cells exhibit autoreactivity to CD1d expressing cells, particularly thymocytes, their antigen specificity does not overlap. Thus, mVα14 and hVα24 iNKT cells, irrespective of their Vβ-Dβ-Jβ chain usage, recognize a marine sponge-derived α−GalactosylCeramide (αGalCer) (143, 144) and closely relate microbial α-glycuronylceramides (145-147), as well as the self-antigen isoglobotrihexosylceramide (iGb3) (148). In contrast, the self and foreign antigens recognized by non-Vα14 NKT cells remain to be identified. Thereon I will focus on the canonical mVα14 and hVα24 iNKT cells, which I will simply refer as iNKT cells, because they are the best-characterized elements of the NKT cell family. Indeed the generation of αGalCer loaded CD1d tetramers (149), the golden tool for iNKT cell detection, allowed the achievement of new important findings in iNKT cell biology. Many iNKT cells in human and mice express CD161, a cell surface molecule usually observed on NK cells that correspond to the NK1.1 antigen, and that is the reason why they are referred as Natural Killer T cells. Nevertheless not all αGalCer/CD1d tetramers+ T cells express CD161 (150). Murine and human iNKT cells express additional receptor commonly found in NK cells. iNKT cells in C57/BL6 mice express intermediate level of IL-2 receptor β (CD122). Moreover the expression of the inhibitory NK receptor Ly49A and Ly49G2 has been reported on iNKT cells (151), as well as the expression of other NK markers as NKG2D and DX5 (CD49b) (122, 152).
A striking feature of most murine and human iNKT cells is their expression of marker associated with recently activated or memory T cells. In mice, most iNKT cells are CD44hiCD69intCD45RhiBCD62LloCCR7neg, while in humans CD45RO+CD45RA -CD25+CD62L-CCR7-, and only 5%-15% express CD69 (153-155). Interestingly, iNKT cells in human cord blood and in germ-free mice display this activated/memory surface phenotype, suggesting that previous exposure to foreign microbial antigens is not the reason for this phenotype and that stimulation by CD1d-presented self ligands is likely to be sufficient (154-156).
In addition iNKT cells in mice, and to a lesser extent in humans, express intermediate levels of TCR at the cell surface, which may be the consequence of continuous
low-level TCR stimulation provided by recognition of self antigens that are constitutively presented by CD1d (150).
Based on CD4 expression, two different iNKT cell subset, with different functional properties, surface markers expression and tissue distribution, can be distinguished: the CD4+CD8- (CD4+) and the CD4-CD8- (double negative, DN) iNKT cells (157). In general, in mice the majority of iNKT cells is CD4+, while in humans a mean of 50% of αGalCer/CD1d tetramers+ T cells is CD4+, with high donor-to-donor variability (150). However a depth analysis of different subsets revealed marked differences in tissue distribution and cytokine production. For instance whereas CD4+ iNKT cells represent the major subset in thymus, spleen and liver, they are a relatively minor subset in lymph nodes and bone marrow (157). In human, but less clearly in mice, this classification provides an important functional distinction, since CD4+ iNKT cell make both Th1 and Th2 cytokines, whereas CD4- iNKT cells mainly make Th1 cytokines (158).
2.5.3 Tissue Distribution and Localization of iNKT cells
In mice, αGalCer/CD1d tetramers+ T cells are approximately 0.5-1% in thymus, 1-2% in spleen, 0.5% in lymph nodes, 10-50% in liver, 0.5% in bone marrow and 1% of intestinal intraepithelial lymphocytes (IEL) of the total lymphocytes per organ (149, 159). In humans, iNKT cells account for a mean of 0.2% of peripheral blood T cells, as determined by the use of both Vα24Vβ11 antibodies and αGalCer/CD1d tetramers (131, 158). Moreover the number of iNKT cells in human liver is lower than in the murine counterpart.
Many iNKT cells display chemokine receptor and homing molecule profiles which are more similar to those of effector (memory) T cells than naïve T cells. For instance in humans the majority of iNKT cells express CCR5, CXCR3 and CXCR6, chemokine receptors associated with Th1 responses and migration to sites of inflammation. Only a small fraction of human iNKT cells express CCR7 and no circulating iNKT cells express CXCR5, receptors required for migration into T and B cell zones of lymphoid organs (158, 160, 161). In mice most iNKT cells express CXCR3 and migrate robustly to MIG/CXCL9. Although a large proportion of murine iNKT cells express also the chemokine receptor CXCR6, they weakly migrate to the
CXCR6 ligand (CXCL16) (162). Their expression of CXCR6 matches the expression of CXCL16 on the endothelial cells lining the liver sinusoids and appears to be important for survival rather than for migration to the liver (163).
Only murine splenic NK1.1- iNKT cells, considered immature cells recently released from the thymus (164, 165), migrate in response to the CCR7 ligand SLC/CCL21(162). Interestingly only iNKT cells from the spleen, but not liver, migrate in response to the CXCR5 ligand BCA-1/CXCL13, suggesting that these cells could be recruited to B cell-rich areas in the spleen and thus modulating the follicular B cell responses (162).
Only a small fraction of human and murine iNKT cells expresses high level of L-selectin (CD62L), that would allow entry into secondary lymphoid organs via high endothelial venules (HEVs), suggesting that iNKT cells recirculate through peripheral tissue and enter lymph nodes most likely through the afferent lymphatics rather than HEVs (150).
Moreover CD4+ and DN iNKT cell subsets can present differences in chemokine receptor expression (160, 161) and in chemotactic responses (162), indicating that they may be recruited to different sites.
2.5.4 Ligands of iNKT cells
There is nowadays a general consensus that CD1d, like other CD1 family members, evolved to present lipid to T cells (150). However the nature and the source of the various lipids naturally binding to CD1d remain poorly elucidated.
Kirin Pharmaceuticals identified the first ligand of iNKT cells during a screening for the identification of antitumoral compounds. During this study, they found that extracts from the marine sponge Agelas mauritianus prolonged the survival of mice bearing B16 melanoma (166). The active principle was an α-branched galactosylceramide and it was slightly modified to produce the compound commonly known as αGalCer (Figure VI) (167). The lipid nature of this compound was demonstrated to induce a strong CD1d-restricted and TCR-dependent activation of iNKT cells (143).
The αGalCer structure resembles mammalian ceramides since it contains a sphingosine-like base, an amide linked acyl chain and an O-linked pyranose (Figure VI). However the anomeric carbon of the sugar is in the α-linkage to the oxygen, whereas in mammals the corresponding linkage is of the β-anomeric type (150). Although αGalCer has no physiological function in mammalian immunity, it has been broadly used as a tool to study iNKT cell activation. Irrespective of their variable CDR3 β sequence, most of mouse and human iNKT cells recognize αGalCer, and the mouse CD1d-αGalCer tetramers stain mouse, human and non human primate iNKT cells as well, providing a clear evidence of the high degree of conservation of this recognition system (144).
Several independent groups reported that also some microbial glycolipid antigens could directly activate iNKT cells by engaging their invariant TCR. Indeed in 2005 three reports described glycosphingolipid antigens from Sphingomonas spp, activating essentially all iNKT cells (figure VI) (145-147). Glycosylceramides from the cell wall of Sphingomonas spp display structural features similar to αGalCer, including the unusual α-linkage of the sugar to the sphingosine-containing lipid. They can bind CD1d and specifically activate mouse and human iNKT cells (145-147). Moreover direct recognition by the invariant TCR was confirmed by staining with CD1d tetramers loaded with glycosphingolipid from Sphingomonas spp.
Glycosphingolipids expressed by another bacterium, the causative agent of Lyme disease Borrelia burgdorferi, can directly stimulate iNKT cells (168). However recognition of intact or heat-killed bacteria could not be demonstrated and only one isolate report has suggested defective bacterial clearance in vivo in mice lacking iNKT cells (169).
In addition, various CD1d-expressing cell types can stimulate mouse and human iNKT cells at low-level, in the absence of foreign microbial antigens (137, 139, 170). This autoreactivity, together with the presence of IL-12 induced by TLR engagement, was shown to be required for iNKT cell activation during immune responses against Gram negative, LPS positive bacteria (145, 171). Moreover autoreactivity may explain the thymic development of iNKT cells (140), which includes an expansion phase following the positive selection (164) and the acquisition of a memory phenotype independent of microbial exposure or TLR signaling (156). The
and human iNKT cells upon presentation by DC or plastic-bound CD1d/iGb3 preformed complexes (148, 172). iGb3 was hypothesized to be the endogenous ligand essential for the positive selection of mouse iNKT cells in the thymus. Indeed mice deficient in the β-subunit of β-hexosaminidase, an enzyme involved in the metabolism of lysosomal glycosphingolipid including iGb3, exhibit greatly reduced frequency of iNKT cells (148). However a normal iNKT cell frequency was found in mice lacking iGb3 synthase (iGb3S), which had no detectable isogloboseries (iGb3, -4, and-5) (173), suggesting that the loss of iNKT cells in the previous model probably results from lipid storage alterations per se rather than the specific absence of iGb3. Further studies will be required to unravel the identity of the endogenous ligand/s involved in iNKT cell positive selection and peripheral activation.
Figure VI: Self and microbial glycosphingolipid ligands (GSL) of NKT cells.
A. Marine sponge αGalCer (KRN7000) with carbon atom number assignments on sphingosine (C), acyl (C’), and carbohydrate (C’’). B. Sphingomonas GSL-1 through GSL-4. C. Mammalian isoglobotrihexosylceramide (iGb3), or Galα1,3Galβ1,4Glcβ1,1Cer. Note that the proximal glucose of the mammalian glycosphingolipid has a β-anomeric linkage to ceramide, in contrast with the α-branched galactose of αGalCer or glucuronyl of Sphingomonas GSLs. Modified from Bendelac et al (122).
2.5.5 Cell biology of lipid presentation by CD1d
CD1d is constitutively expressed by APCs. In fact DCs, macrophages and B cells (in particular marginal zone B cells) express CD1d at high level (174, 175).
Also cortical thymocytes express CD1d, and this expression is mandatory for the development of iNKT cells (140). Moreover CD1d is expressed on hepatocytes, Kupffer and endothelial cells lining liver sinusoids, where iNKT cells are most abundant in mice (163). However in the liver CD1d expression is not required for iNKT cell homing (176). In addition, similar to the MHC class II molecules, most other solid tissue cells and non-antigen-presenting hematopoietic cells express low or undetectable levels of CD1d.
Within the lumen of the endoplasmic reticulum (ER), newly synthesized CD1d molecules interact with the chaperone proteins calnexin, calreticulin and the thiol oxidoreductase ERp57 (177), which are involved in the association of CD1d with β2-microglobulin (β2-m). CD1d molecules can traffic to the cell surface in the absence of β2-m. Mutation analysis revealed that the loss of glycan-2, at the interface between CD1d and β2-m, results in faster CD1d egress from the ER and reduced stability at the cell surface (178). Supporting the hypothesis that lipid ligand binding to CD1d can occur in the ER, it was shown that CD1d molecules engineered to be retained in the ER bound phosphatidylinositol (179).
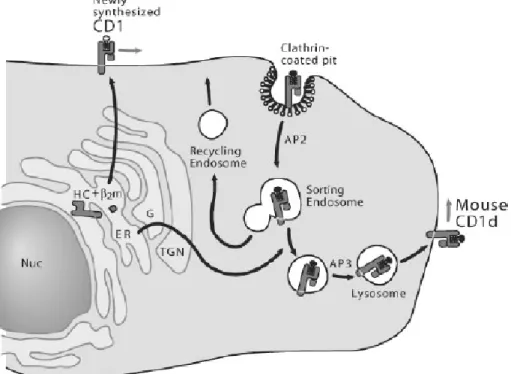
From the ER, CD1d molecules traffic to the cell surface through the secretory pathway before being reinternalized into the endo-lysosomal compartment. This
recycling between the plasma membrane and the endo-lysosomal compartment is dependent on a tyrosine motif encoded in the CD1d cytoplasmic tail (180, 181). The tyrosine motifs allow CD1d to bind to the specific adaptors protein (AP) complexes AP-2 and AP-3, which in turn direct CD1d trafficking. The intracellular localization of human CD1d is different in mice and in humans: although both are found within early and late endosome as a result of binding AP-2, murine CD1d also binds AP-3, explaining why most murine CD1d molecules are found within lysosome (182-184). Mutations or deletions of the cytoplasmic tail motif and modifications of CD1d trafficking to acidified lysosomes in mice with a defect in AP-3 impair antigen presentation and iNKT cell development (180, 185, 186), indicating the importance of trafficking within endosome and lysosome for the presentation of endogenous ligand
and, consequently, the thymic generation of iNKT cells. See figure VII for an overview of CD1d trafficking.
A number of endosomal proteins are involved in the processing and loading of glycolipids onto CD1d molecules (187). These proteins include saposins, Niemann-Pick type C2 (NPC2) protein, and microsomal triglyceride transfer protein (MTP). Saposins A, B, C, and D are produced from the proteolytic cleavage of prosaposin within the endosome. Prosaposin-deficient mice have defective iNKT cell development and reduced ability to process and present glycolipid antigens (188). Loading of human CD1d with αGalCer in the absence of saposins is similarly impaired (189). Individual saposins differ in their ability to load particular lipid, indeed saposin B is the most efficient of the saposins in transferring glycolipids onto CD1d for presentation. Mice lacking the NPC2 protein, a lysosomal lipid transfer protein, have impaired presentation of glycolipids and impaired selection of iNKT cells in the thymus (190). It was shown that NPC2 dimers can bind iGb3 and facilitate its loading onto CD1d molecules. Finally MTP, another lipid transfer protein expressed in the ER was found to co-precipitate with CD1d and genetic or drug-induced inhibition of MTP was associated with defect in lipid antigen presentation. Interestingly a recent report from Im J.S. and colleagues demonstrated that not all the glycosphingolipid antigens display the same requirement for endosomal loading (191). Indeed they found that αGalCer analogues, able to induce biased Th2 cell type responses, were presented with rapid kinetics and without a requirement for intracellular loading of CD1d, apparently as a result of their ability to rapidly associate with CD1d molecules directly at the cell surface. This was in marked contrast to classical αGalCer, which underwent intracellular loading and was presented more slowly.
Figure VII: Intracellular trafficking of CD1d.
Newly synthesized CD1d molecules may or may not assemble with β2-microglobulin in the ER, acquire self-lipids (open circle with zigzag tail), and traffic to the plasma membrane along the secretory route. CD1d molecules are internalized from the plasma membrane and traffic through the early and late endosomal compartment and are delivered to lysosomes in an AP3-dependent manner. CD1d then acquires distinct self-lipid antigens (filled circle and zigzag tail) in late endosomal/lysosomal compartments. Human CD1d traffics mainly in early and late endosomes but not in recycling endosomes and only partially localizes in lysosomes. Moreover human CD1d does not associate with the adaptor protein AP3. Modified from Brigl and Brenner (150).
2.5.6 iNKT cell development
For many years the developmental origin of iNKT cells was a matter of debate (152). Although iNKT cells are present in the thymus, their origin has remained controversial mostly because these cells were first identified by using surrogate markers such as NK1.1, which are also expressed by other cells.
Some studies suggested that iNKT cells develop very early in ontogeny, independently from the thymus, and before the appearance of conventional T cells (192, 193). However, there are now compelling evidences that iNKT cells are a thymus-dependent population. Indeed they are absent in nude mice (165, 194); do not develop in thymectomized mice (195-197) and are detectable in the thymus shortly later than other T-cell subsets (164, 165, 198-200).
Different models that explained the basis of the iNKT cell lineage were initially hypothesized (150). The “committed precursor” model suggests that iNKT cells originate from precursors committed prior to T cell receptor (TCR) expression. On the contrary the “selective” model proposes that the development of iNKT cells is a selective event stemming from the random production of a TCR that recognize CD1d (152). Convincing evidences support the last model. Indeed although iNKT cells always express the invariant TCR Vα14-Jα18 chain (136), their non-transcribed TCRα allele shows clear evidence of random recombination (201). Furthermore, although the complementarity-determining region 3 (CDR3) of the α-chain is invariant at the aminoacid level, this is not the case at the nucleotide level, in which non-templated nucleotide additions contribute to the invariant amino-acid sequence (136).
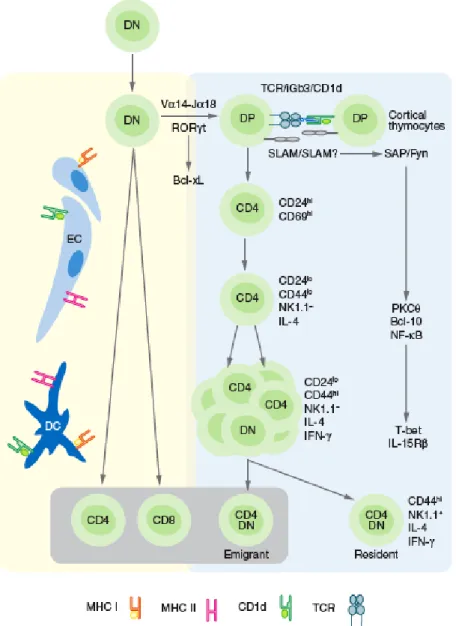
It is so far commonly accepted that iNKT cell lineage in mice branches from the developmental pathway of conventional T cells at the CD4+CD8+ DP stage (200, 202, 203) (Figure VIII). This event occurs when DP thymocytes, bearing the randomly generated TCR of iNKT cells, undergo to positive selection (152). The positive selection of iNKT cells requires their ligation to endogenous glycolipid antigens presented by CD1d on DP cortical thymocytes. This was shown using bone marrow chimeras or transgenic restoration of CD1d expression in CD1d-/- mice, to restrict the
expression of CD1d to the hematopoietic or stromal compartment of mice. iNKT cells were selected only when CD1d was expressed by hematopoietic cells, and more