19
CAPITOLO 2
RISULTATI E DISCUSSIONI
2.1 Studio della reattività di addotti di tipo inverso variamente sostituiti.
Le reazioni di riarrangiamento [3.3]-sigmatropico coinvolgenti cicloaddotti di nitroso Diels-Alder furono descritte per la prima volta da Kirby e Mackinnon. Lo stesso tipo di riarrangiamento è stato riportato successivamente anche da Streith e Knaus, in lavori indipendenti tra loro. In letteratura, questo tipo di riarrangiamento, detto anche di etero-Cope, è riportato soltanto per aroilnitroso cicloaddotti, mentre i derivati carbammici sono considerati non reattivi da Streith e da Knaus. Il fallimento di questi ultimi è stato attribuito alla riduzione dell’effetto elettron-attrattore del carbonile legato all’N3 che risulta essere non sufficiente ad indurre la rottura del legame C4-N3
necessaria per la reazione di riarrangiamento (Schema 2.1).
Schema 2.1: Trasposizione [3.3]-sigmatropica dei cicloaddotti di tipo inverso aroilnitroso e
carbammoilnitroso.
Nel laboratorio in cui ho svolto la presente Tesi sperimentale, ricercando il cleavage del legame N-O per i cicloaddotti di nitroso Diels-Alder 2.2 e 2.3 derivanti dalla 1,2-diidropiridina 2.1, in condizioni riduttive blande (per trattamento con Mo(CO)6 in una soluzione di CH3CN/H2O), erano
state ottenute le corrispondenti ene-acetammidi 2.6 e 2.7 (Schema 2.2)23. Queste necessariamente dovevano derivare da un riarrangiamento [3.3]-sigmatropico con formazione delle ipotetiche diossazine corrispondenti intermedie e non isolabili (2.4 e 2.5) che successivamente, subendo un processo di idrolisi, fornivano il composto finale.
20
Schema 2.2: Riarrangiamento [3.3]-sigmatropico seguito da idrolisi della diossazina intermedia con
formazione dell’ene-acetammide corrispondente.
Le reazioni di riarrangiamento [3.3]-sigmatropico inizialmente erano state condotte in presenza di Mo(CO)6 in una soluzione 15/1 di CH3CN/H2O a 65 °C, cioè in condizioni riduttive, dal momento
che l’obiettivo di partenza era il cleavege del legame N-O. Dato i risultati che erano stati ottenuti, è stato pensato di esaminare più in dettaglio le condizioni sperimentali in cui il riarrangiamento poteva avere luogo. Attraverso questi studi è stata chiarita l’importanza dell’acqua e della temperatura di reazione. Con temperature superiori agli 85 °C, infatti, i bicicli andavano incontro a reazioni di retro Diels-Alder con riottenimento della diidropiridina di partenza. Le temperature ottimali per avere il riarrangiamento erano state trovate tra i 65 °C e i 75 °C. D’altra parte la reazione condotta a temperatura ambiente dopo 12 ore non aveva portato alla formazione di nessun prodotto, neanche in minima quantità.
L’influenza dell’acqua in questo tipo di riarrangiamento è abbastanza documentata, e potrebbe essere attribuita alla suscettibilità e all’accelerazione dovute al legame a idrogeno quando lo stato di transizione di un processo di riarrangiamento non è perfettamente concertato. Successivi esperimenti di controllo svolti con e senza molibdeno esacarbonile avevano dimostrato che la presenza dell’acqua, a differenza del molibdeno, è essenziale nel promuovere la conversione termica dei bicicli precedentemente citata. D’altra parte quando la reazione è lasciata andare a 75 °C in acetonitrile in condizioni anidre si osserva esclusivamente prodotti di decomposizione del cicloaddotto.
21
Composti del tipo 2.6 e 2.7 risultano essere interessanti dal punto di vista sintetico dal momento che l’anello piperidinico, così come il 4-idrossipiperidinico, si ritrovano in molti composti biologicamente attivi, o possono essere utili come building blocks per la sintesi di alcaloidi. Scopo di questa Tesi sperimentale è quello di verificare la generalità del metodo o l’influenza che diversi gruppi protettivi e sostituenti possono avere sul processo di riarrangiamento.
Per prima cosa ci siamo quindi preparati una serie di 1,2-diidropiridine da utilizzare nelle reazioni di nitroso Diels-Alder. Per la sintesi di queste ultime abbiamo utilizzato tre metodi generali (i dettagli delle varie procedure sono riportati nella parte sperimentale):
a) Addizione di organometalli a sali di piridinio N-protetti: l’anello piridinico viene attivato dall’attacco del cloruro acido sull’azoto, portando alla formazione del sale di piridinio corrispondente, a cui segue l’attacco dell’organometallo. Per la sintesi delle nostre diidropiridine abbiamo utilizzato, come organometalli, sia reagenti di Grignard sia organolitio derivati (Schema 2.3).
Schema 2.3: Addizione di organometalli a sali di piridinio N-protetti.
Come riportato anche in letteratura la reazione avviene con una selettività che preferisce l’attacco dell’organometallo sulla posizione 2, portando principalmente alla formazione della 1,2-diidropiridina-2-sostituita, ma si verifica in parte anche l’attacco sulla posizione 4, con l’ottenimento della 1,4-diidropiridina. Le due diidroipiridine hanno Rf diversi, solitamente la 1,4
maggiore rispetto alla 1,2. Non sempre è stato possibile separarle del tutto attraverso cromatografia flash, con l’ottenimento di una frazione in cui le due diidropiridine, risultano essere in miscela e una frazione in cui è presente esclusivamente la 1,2-diidropiridina con una buona purezza. In questi casi, dal momento che la 1,4 non interferisce sulla successiva reazione di nitroso Diels-Alder, abbiamo ritenuto opportuno utilizzare anche quella frazione di miscela, evitando così una perdita in peso del nostro composto ricercato.
22
Le 1,2-diidropiridine ottenute con questo metodo sono riportate nella Figura 2.1:
2.8a PG1= Ac, R= Ph; 2.8b PG1= COPh, R= Ph;
2.8c PG1= COOPh, R= Ph; 2.8d PG1= Cbz, R= CH3;
2.8e PG1= Ac, R=CH3; 2.8f PG1=Ac, R= C6H13.
Figura 2.1: 1,2-diidropiridine sintetizzate per addizione di organometalli a sali di piridinio
N-protetti.
b) Metodo di Fowler, per la sintesi di 1,2-diidropiridine non sostituite. L’attivazione dell’anello piridinico anche in questo caso è data dall’attacco del cloruro acido all’atomo di azoto, che in questo modo può subire l’addizione di uno ione idruro da parte del NaBH4 (Schema 2.4).
Schema 2.4: Metodo di Fowler per la sintesi delle 1,2- 1,4-diidropiridine.
Anche in questo caso si ottiene una miscela delle due diidropiridine 1,2 e 1,4, per la possibilità da parte dell’idruro di attaccare sia in posizione 2 che in posizione 4. Dalla purificazione per cromatografia si ottiene spesso una frazione in cui la 1,2-diidropiridina è in miscela con l’isomero 1,4, e una frazione “pulita”. Nel caso della diidropiridina 2.5h (vedi Figura 2.2) la procedura di letteratura prevedeva una cristallizzazione in etanolo. Purtroppo a noi la cristallizzazione ha portato a rese molto basse, ottenendo solo 2 mmoli di diidropiridina dalle 50 iniziali di piridina. Abbiamo quindi provato diversi solventi per la cristallizzazione, ma i risultati sono stati sempre deludenti. Considerando che dalla reazione si ottiene un grezzo con una purezza del 70% abbiamo infine optato per utilizzare quest’ultimo.
Le 1,2-diidropiridine ottenute con questo metodo sono riportate in Figura 2.2:
2.8g PG1= Cbz (-OCOCH2Ph);
2.8h PG1= COOPh.
23
c) Il metodo di Arndtsen16 è stato utilizzato per la sintesi di 1,2-diidropiridine-2-sostituite con alchini terminali. Questo è un metodo recente che permette di funzionalizzare le piridine anche in modo asimmetrico attraverso una reazione rame-catalizzata. La catalisi da parte del rame fa sì che le reazioni avvengano in tempi brevi e a temperatura ambiente (Schema 2.5).
Schema 2.5: Metodo di Arndtsen per la sintesi di 1,2-diidropiridine sostituite con alchini terminali.
Nel nostro caso abbiamo utilizzato il fenilacetilene che nelle condizioni di reazione va a formare il fenilacetiluro di rame, che attacca l’anello piridinico attivato dal cloruro acido.
Con questo metodo abbiamo sintetizzato due diidropiridine, ma solo una di queste, 2.8i, è stata utilizzata nei successivi studi di riarrangiamento. Le 1,2-diidropiridine ottenute con questo metodo sono riportate in Figura 2.3.
2.8i PG1= COPh, R= Ph;
2.8l PG1= COOPh, R= Ph.
Figura 2.3: 1,2-diidropiridine sintetizzate secondo il metodo di Arndtsen.
La sintesi di cicloaddotti di nitroso Diels-Alder è stata effettuata con N-idrossibenzammide (PhCONHOH), N-idrossi-2-fenil-acetammide (PhCH2CONHOH), t-butil-idrossicarbammato
(BocNHOH) e benzil-idrossicarbammato (CbzNHOH), in condizioni ossidanti per formare il nitroso derivato corrispondente in situ. Abbiamo eseguito le reazioni di nitroso Diels-Alder in diverse condizioni, che prevedono l’ossidazione in situ dell’acido idrossammico a nitroso derivato da parte di un periodato. Un metodo è condotto in diclorometano e prevede l’utilizzo di tetraetil ammonio periodato come ossidante. L’aggiunta dell’ossidante è fatta a basse temperature (-78°C), dopo di ché la reazione viene riportata a temperatura ambiente. Il secondo metodo prevede l’impiego di sodio periodato in una miscela di metanolo/acqua (2/1) a 0°C. La scelta dell’uno o dell’altro metodo varia molto a seconda della diidropiridina utilizzata. Ad esempio la diidropiridina
2.8i, nonostante risulti essere insolubile in metanolo, reagisce con PhCH2CONO (formato in situ
24
cicloaddotto di tipo inverso corrispondente, anche se in basse rese. La stessa reazione è stata ripetuta in diclorometano con tetraetil ammonio periodato, ma in questo caso, nonostante la diidropiridina sia solubile nel solvente organico, la reazione non ha portato alla formazione di alcun prodotto (Schema 2.6).
Schema 2.6: Nitroso Diels-Alder per la diidropiridina 2.8i nelle due diverse condizioni.
Al contrario, la diidropiridina 2.8b con N-idrossi-2-fenil-acetammide non dà alcun risultato in metanolo/acqua, ma reagisce nelle condizioni che prevedono l’impiego del tetraetil-ammonio periodato in diclorometano. In generale comunque possiamo dire che le reazioni di nitroso Diels-Alder avvengono con rese paragonabili nelle due condizioni appena descritte. Nella maggior parte dei casi abbiamo preferito condurre le reazioni in metanolo/acqua, sia per la maggiore facilità nell’allestimento della reazione stessa, sia perché i grezzi di reazione risultano essere più semplici, facilitando la purificazione del prodotto. Questo è in accordo con studi precedenti del laboratorio in cui ho svolto la presente Tesi sperimentale secondo cui le condizioni migliori per le reazioni di nitroso Diels-Alder coinvolgenti la diidropiridina 2.1 prevedevano l’impiego di sodio periodato in una miscela metanolo/acqua a 0 °C.
Come descritto da Streith, le reazioni di nitroso Diels-Alder coinvolgenti diidropiridine N-protette dimostrano una regioselettività in funzione del gruppo che lega il nitroso. In particolare l’effetto elettronico esercitato dal gruppo legante il nitroso modula i coefficienti orbitalici dell’ossigeno e dell’azoto a tal punto che questa differenza si trasferisce nella distribuzione dei prodotti. Infatti, dalla reazione con derivati carbammici (Boc e Cbz), in cui i coefficienti orbitalici degli atomi di ossigeno e azoto sono comparabili, abbiamo ottenuto miscele dei due cicloaddotti, di tipo diretto e di tipo inverso, anche se non in tutti i casi è stato possibile isolare il prodotto di tipo diretto, che viene degradato durante la purificazione su silice.
I cicloaddotti di tipo inverso ottenuti con t-butil-idrossicarbammato e successivamente utilizzati nel riarrangiamento [3.3]-sigmatropico sono riportati nello Schema 2.7.
25 2.9a PG1= COPh, R= Ph; 2.9b PG1= Ac, R= Ph; 2.9c PG1= COOPh, R= Ph; 2.9d PG1= Cbz, R= CH3; 2.9e PG1= Cbz, R= H; 2.9f PG1= Ac, R= C6H13.
Schema 2.7: Cicloaddotti di tipo inverso ottenuti con t-butil-idrossicarbammato utilizzati nel
riarrangiamento.
I cicloaddotti di tipo inverso ottenuti con benzil-idrossicarbammato e successivamente utilizzati nel riarrangiamento sono riportati nello Schema 2.8.
2.10a PG1= COOPh, R= Ph;
2.10b PG1= Ac, R= CH2COOCH3;
2.10c PG1= Cbz, R= H.
Schema 2.8: Cicloaddotti ottenuti con benzil-idrossicarbammato.
Con i derivati nitroso di natura ammidica (PhCH2CONHOH e PhCONHOH), in accordo con quanto
riportato in letteratura, abbiamo ottenuto esclusivamente il cicloaddotto di tipo inverso. Nel LUMO degli acil-nitroso derivati, i coefficienti orbitalici degli atomi di azoto e ossigeno sono diversi, e in questo caso sono maggiori sull’atomo di ossigeno e questo dirige l’attacco verso la formazione del cicloaddotto di tipo inverso.
26
I cicloaddotti ottenuti con N-idrossi-2-fenil-acetammide e sottoposti a riarrangiamento sono indicati nello Schema 2.9. 2.11a PG1= COOPh, R= H; 2.11b PG1= COOPh, R= Ph; 2.11c PG1= COPh, R= Ph; 2.11d PG1= COPh, R= CCPh; 2.11e PG1= Cbz, R= H; 2.11f PG1= Cbz, R= CH3; 2.11g PG1= Ac, R= C6H13; 2.11h PG1= Ac, R= Ph.
Schema 2.9: Cicloaddotti ottenuti con N-idrossi-2-fenil-acetammide .
I cicloaddotti ottenuti con N-idrossibenzammide e sottoposti a riarrangiamento [3.3]-sigmatropico sono indicati nello schema 2.10.
2.12a PG1= COOPh, R= H;
2.12b PG1= Ac, R= Ph;
2.12c PG1= Ac, R= CH2COOCH3.
Schema 2.10: Cicloaddotti ottenuti con N-idrossibenzammide.
Abbiamo già parlato dell’importanza dell’acqua nelle reazioni di riarrangiamento [3.3]-sigmatropico. All’inizio di questo lavoro di Tesi era già stato stabilito che il molibdeno esacarbonile non fosse essenziale e che il riarrangiamento avvenisse in condizioni di acqua/acetonitrile 1/15 a caldo. Nel nostro primo esperimento utilizziamo quindi queste stesse condizioni sul biciclo 2.11e (Schema 2.9) e dopo 40 ore a 65 °C la reazione non è ancora finita, con una conversione del 75%. A questo punto abbiamo provato a cambiare le condizioni di reazione per vedere se fosse possibile aumentare la conversione e diminuire i tempi. Per prima cosa abbiamo aumentato la temperatura portandola a 75 °C: dopo 40 ore ci sono solo minime tracce del biciclo di partenza (Schema 2.11 es. 1).
27
La stessa reazione viene ripetuta a 70 °C nel tubicino, in CD3CN/D2O (15/1) e seguita via 1H
NMR. Già dopo 30 minuti si cominciano a vedere nuovi segnali dovuti alla formazione del prodotto riarrangiato a 6.90 ppm circa e a 3.94 ppm dello spettro protonico. Dopo 80 minuti i segnali sono diventati più evidenti, ma rimangono ancora importanti quelli del biciclo. Dopo 19 ore sono ancora visibili i segnali caratteristici del biciclo che cadono intorno a 6.50 ppm.
A questo punto del mio lavoro di Tesi abbiamo eseguito quindi una serie di esperimenti sul biciclo
2.11e (0.2 mmol) in presenza di diverse quantità di acqua, lasciando invariata la quantità di
acetonitrile (1.23 mL) e mantenendo la temperatura a 75 °C: le prove sono state fatte, rispettivamente, con 40 μL di acqua (acetonitrile/acqua 30/1), pari a 2.2 mmol di acqua, e con 18 μL di acqua, corrispondenti ad una mmole di acqua. Come abbiamo già visto quando il biciclo è lasciato sotto agitazione magnetica in una miscela acetonitrile/acqua 15/1, in cui il rapporto biciclo/acqua è di 1/22 , a 75 °C dopo 40 ore la reazione non è ancora andata a completezza. La reazione condotta in una miscela acetonitrile/acqua 30/1, in cui abbiamo quindi dimezzato la quantità di acqua (biciclo/acqua 1/11), nello stesso arco di tempo, va a completezza (conversione 100 %), con una resa isolata del 45 %. Diminuendo la quantità di acqua abbiamo ottenuto un leggero miglioramento nell’andamento della reazione, che avviene con una conversione completa, anche se le rese rimangono basse (Schema 2.11 es.2). Abbiamo provato a diminuire ulteriormente la quantità di acqua per vedere l’effetto sulla reazione Purtroppo dopo 24 ore a 75 °C con 1 mmole di acqua (biciclo/acqua 1/5) la reazione, seguita via TLC, è ancora ferma ed è ancora presente molto materiale di partenza (Schema 2.11 es. 3).
Schema 2.11: Riarrangiamento [3.3]-sigmatropico del biciclo 2.11e nelle diverse condizioni di
reazione.
Quindi le condizioni trovate ottimali sono quelle che prevedono una temperatura di 75 °C e un rapporto acetonitrile/acqua 30/1 (biciclo/acqua 1/11).
Una volta ottimizzate le condizioni di reazione per quanto riguarda la temperatura e la quantità di acqua, siamo passati ad esaminare i processi di riarrangiamento di cicloaddotti nitroso diversamente
28
sostituiti. La discussione sarà articolata considerando separatamente i bicicli di natura carbammica (con Boc e Cbz) e quelli di natura ammidica (con PhCH2CONHOH e PhCONHOH).
Per i bicicli con il Boc, 2.9a-f, nella maggior parte dei casi si ha o la formazione di una miscela complessa (2.9d-f) o una conversione molto bassa (2.9b) (Schema 2.12).
Schema 2.12: Risultati del riarrangiamento dei cicloaddotti Boc-protetti 2.9b e 2.9d-f.
Solo per i bicicli 2.9a e 2.9c abbiamo ottenuto dei prodotti interessanti, anche se molto particolari in quanto privi della protezione Boc. Questi sono rispettivamente il biciclo 1,4,2-diossazin-3-one
2.13 e il corrispondente monociclo decarbonilato 2.15. È stato curioso vedere che questi composti
sono caratterizzati da un Rf particolarmente basso [2.9a Rf 0.12 (esano/acetato di etile 8:2); 2.9c Rf
0.12 (esano/acetato di etile 7:3)] e che da esperimenti 1H- e 13C-NMR in entrambi i casi si verifica la perdita del gruppo protettivo Boc come t-butanolo. La struttura dei prodotti è stata confermata dalla spettrometria di massa [ESI-MS= 358.96 (M+Na+) per il composto 2.13; ESI-MS= 331.14 (M+Na+) per il composto 2.15].
Ipotizzando un plausibile meccanismo di reazione si è pensato che l’attacco da parte dell’acqua sulla diossazina intermedia non isolabile, in questo caso, sia seguito dalla perdita di t-butanolo come gruppo uscente, portando alla formazione del biciclo 2.13. Abbiamo inoltre ipotizzato che un composto simile si possa formare anche nel caso del composto 2.9c, come intermedio di reazione non isolabile 2.14, nella sintesi del monociclo 2.15, che potrebbe subire una reazione di idrolisi a carico del legame C-O estereo (Schema 2.13). Si tratta, quindi, di un processo di addizione-eliminazione di acqua che procede in maniera diversa rispetto a ciò che succedeva nel caso dei bicicli 2.2 e 2.3 (Schema 2.2).
29
Schema 2.13: Meccanismo di reazione ipotizzato.
Per i bicicli di natura carbammica con il Cbz come gruppo protettivo, 2.9a-c, la reazione di riarrangiamento [3.3]-sigmatropico non ha portato a risultati interessanti, ma soltanto a miscele complesse e conversioni molto basse (Schema 2.14). Di fatti i carbobenzilossi carbammoil-derivati risultano essere non efficaci. Soprattutto nel caso del biciclo 2.10a ci aspettavamo un risultato diverso, vista la somiglianza chimica con il biciclo 2.9c (uno dei due casi di bicicli Boc-protetti che riarrangia), derivando dalla stessa diidropiridina di partenza, 2.8c, ed essendo anche questo di natura carbammica.
Schema 2.14: Risultati del riarrangiamento dei cicloaddotti Cbz-protetti 2.10a-c.
Per i composti di natura ammidica, invece, le reazioni di riarrangiamento [3.3]-sigmatropico portano alla formazione di composti caratterizzati da un Rf elevato, maggiore rispetto al composto di
partenza e ai casi visti in precedenza. Analizzando i prodotti ottenuti attraverso studi 1H e 13C NMR, 2D-COSY e ESI-MS, è stato visto che dal riarrangiamento dei bicicli di natura ammidica si ottengono le corrispondenti diossazine 2.16 e 2.17 (Schema 2.15 e 2.16) considerate in precedenza
30
come intermedi di reazione non isolabili. Infatti da studi 1H NMR, si poteva già supporre che questi prodotti avessero una struttura chiusa, mancando il segnale dell’NH presente invece per i composti
2.6 e 2.7 (che cade rispettivamente a 9.46 ppm e 8.10 ppm), scambiabile con D2O. Questo è stato
poi confermato da analisi con ESI-MS (la descrizione dettagliata dei vari prodotti è riportata nella parte sperimentale). Infatti la massa di questi prodotti varia di una molecola di acqua rispetto a quella dei composti sopra citati. In questo caso tali composti possono essere isolati e sembrano non subire facilmente processi di idrolisi.
2.16a PG1= COOPh, R= H; 2.16b PG1= COOPh, R= Ph; 2.16c PG1= Cbz, R= H; 2.16d PG1= Ac, R= C6H13; 2.16e PG1= Ac, R= Ph; 2.16f PG1= Cbz, R= CH3.
Schema 2.15: Risultati del riarrangiamento dei cicloaddotti fenil-acetil-protetti.
2.17a PG1= COOPh, R= H;
2.17b PG1= Ac, R= Ph;
2.17c PG1= Ac, R= CH2COOCH3.
Schema 2.16: Risultati del riarrangiamento dei cicloaddotti fenil-acil-protetti.
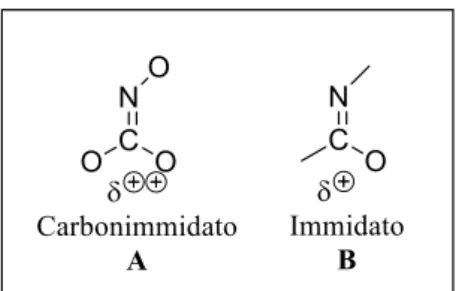
Probabilmente, la loro stabilità molto maggiore rispetto alle diossazine ottenibili dalle protezioni di natura carbammica dell’acido idrossammico, è imputabile alla ridotta elettrofilicità del carbonio di tipo immidato B, rispetto alla struttura di tipo A (Figura 2.4).
31
Figura 2.4: Strutture carbonimmidati e immidati.
Questo potrebbe fare la differenza e causare il processo di idrolisi o reazioni di degradazione per i composti aventi le protezioni Boc e Cbz. Questo tipo di reazioni non avviene per i derivati di tipo
2.16 e 2.17, su cui la natura del carbonio di tipo immidato conferisce una minor reattività al
composto, particolarmente evidente nelle strutture 2.17 in cui è presente anche una coniugazione del doppio legame dell’immidato con un sistema aromatico (Schema 2.16).
Questo tipo di composti viene ottenuto con rese da moderate a buone (40-90 %) potrebbe risultare comunque interessante in quanto possibile precursore di 4-idrossipiperidine enecarbammati. In particolare, la possibilità di effettuare il cleavage del legame N-O in questi derivati permetterebbe l’ottenimento del diolo corrispondente. Dall’esame della letteratura questo è stato ottenuto solo in un caso isolato usando Raney-nichel e con bassissime rese19. Sarebbe stato interessante approfondire questo argomento, utilizzando agenti riducenti di elaborazione più recente, come SmI2, Cp2Ti(III)Cl o TiCl3, con cui probabilmente questo cleavage è possibile, ma per ragioni di
tempo questa tematica non è stata affrontata.
Nell’ultimo periodo della presente Tesi sperimentale abbiamo esaminato condizioni di reazione alternative rispetto al riarrangiamento [3.3]-sigmatropico termico, per verificare se si potesse ottenere anche in condizioni più blande. Abbiamo trovato che Yb(OTf)3, un acido di Lewis
tollerante all’acqua, è in grado di catalizzare il processo di riarrangiamento a temperatura ambiente in poche ore, con aumento sia della resa che della conversione, anche se nei casi (Schema 2.13 e 2.14) in cui in condizioni termiche si ottenevano miscele complesse o conversioni molto basse, il risultato non cambia (Tab. 2.1, es. 1, 3 e 4). Nel caso del biciclo 2.9c, ad esempio, con la catalisi da parte dell’itterbio si ottiene il composto 2.15 con una resa isolata del 52 % in 12h a temperatura ambiente (Schema 2.17), mentre nelle precedenti condizioni termiche si aveva una resa isolata del prodotto del 30 % dopo 24h.
32
Schema 2.17: Riarrangiamento [3.3]-sigmatropico catalizzato.
Tabella 2.1: Risultati ottenuti dal riarrangiamento[3.3]-sigmatropico in condizioni catalizzate
[Yb(OTf)3 (10 mol%)].
Es. Substrato PG1 R PG2 Condizioni Conversione Prodotto
1 2.9b Ac Ph Boc Rt, 12h >95% Miscela complessa 2 2.9c COOPh Ph Boc Rt, 12h >95% 2.15 3 2.10a COOPh Ph Cbz Rt, 12h - N.D. 4 2.10b Ac CH2COOCH3 Cbz Rt, 12h - N.D. 5 2.11b COOPh Ph COCH2Ph Rt, 12h >95% 2.16b 6 2.12b Ac Ph COPh Rt, 12h >95% 2.17b
In conclusione abbiamo ottimizzato le condizioni di reazione per il riarrangiamento [3.3]-sigmatropico sui bicicli di tipo inverso derivanti da nitroso Diels-Alder, trovando che un acido di Lewis, come il Yb(OTf)3, è in grado di catalizzare la reazione a temperatura ambiente, portando
anche ad un miglioramento delle rese.
In conclusione, i risultati ottenuti dal riarrangiamento [3.3]-sigmatropico con bicicli diversamente sostituiti, sia di natura carbammica che di natura ammidica, si sono rivelati abbastanza discordanti. Nel caso di bicicli di natura carbammica (protetti con Boc e Cbz) nella maggior parte dei casi si ottengono miscele complesse o conversioni molto basse. Solo in due casi abbiamo isolato composti che derivano da un riarrangiamento seguito da idrolisi, quali il biciclo 1,4,2-diossazin-3-one 2.13 e il corrispondente monociclo decarbonilato 2.15 (Vedi Schema 2.13), in cui si osserva la perdita del gruppo protettivo Boc come t-butanolo in seguito all’attacco dell’acqua sulla diossazina intermedia. Nel caso di bicicli di natura ammidica, invece, dai risultati ottenuti, possiamo generalizzare dicendo che il riarrangiamento [3.3]-sigmatropico porta alla formazione delle [1,4,2]-diossazine
33
corrispondenti 2.16 e 2.17, considerate in precedenza intermedi di reazione non isolabili. In questo caso tali composti possono essere isolati e sembrano non subire facilmente processi di idrolisi.
![Tabella 2.1: Risultati ottenuti dal riarrangiamento[3.3]-sigmatropico in condizioni catalizzate [Yb(OTf) 3 (10 mol%)]](https://thumb-eu.123doks.com/thumbv2/123dokorg/8015181.121722/14.892.216.687.106.235/tabella-risultati-ottenuti-riarrangiamento-sigmatropico-condizioni-catalizzate-otf.webp)
