61
In questo capitolo verranno descritte le tecniche di campionamento di gas e condensati e le tecniche di analisi, adottate nel presente lavoro di tesi, per la caratterizzazione di biomasse utilizzate, char, frazioni condensate e particolato prodotti durante le prove sperimentali. Particolare attenzione verrà posta sullo sviluppo del sistema di campionamento tar la cui messa a punto è stata uno degli obiettivi del presente lavoro di tesi.
3.1
Sistema di campionamento Tar
Il campionamento del tar, per le successiva analisi, è stato uno dei punti focali del presente lavoro di tesi. A tale scopo un sistema di campionamento del tar è stato progettato e costruito in collaborazione con Gamba & Botteghi S.N.C. di Guasticce (LI).
Il sistema sfrutta il metodo a condensazione, simile al Tar protocol (paragrafo 1.2.3), che si basa essenzialmente sulla continua aspirazione di syngas dal sistema in esame successivamente inviato ad un treno di separatori detti impinger. L’azione base di separazione è dovuta al raffreddamento del gas negli impinger, i quali sono mantenuti a basse temperature al fine di effettuare la condensazione del tar.
Il Tar protocol sebbene più preciso dei metodi tradizionali presenta l’inconveniente di avere un apparato sperimentale piuttosto complesso dovuto all’alto numero di impinger (6) ed alla presenza dell’isopropanolo, che può portare, oltretutto, a perdite di carico nella linea di campionamento ed alterare o impedire il flusso di campionamento.
Il sistema di campionamento sviluppato nel presente lavoro di tesi presenta notevoli vantaggi rispetto al Tar protocol dal punto di vista della complessità e della praticità d’impiego. Il sistema è, infatti, di semplice utilizzo oltre che facilmente trasportabile ed adattabile alle condizioni logistiche di campionamento. L’assenza del solvente organico consente di escludere tutte le problematiche ad esso connesse e permette un’alta ripetibilità delle prove. E’ possibile, inoltre, la determinazione diretta della quantità di tar prodotto mediante semplice analisi gravimetrica.
62
3.1.1
Punto di campionamento
Il sistema di campionamento tar viene posizionato nell’impianto di gassificazione subito dopo il ciclone accanto alla termocoppia TC-1 (Figura 2.2) e nell’impianto di pirolisi a valle del reattore 1 (Figura 2.11). Il campionatore viene collegato agli impianti connettendo l’involucro mobile contenente l’impinger 1 alle valvole di derivazione mediante clamp e guarnizione in grafite. La procedura di campionamento prevede l’accensione del sistema, il settaggio dei point, il collegamento all’impianto quando le temperature hanno raggiunto i valori di set-point, e l’accensione della pompa contestualmente all’apertura della valvola di derivazione dell’impianto.
3.1.2
Descrizione del sistema
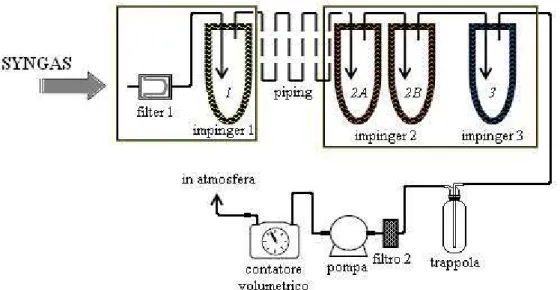
Il campionatore tar preleva una portata di gas dall’impianto a cui è collegato ed effettua un frazionamento in 3 impinger mantenuti a temperatura decrescente allo scopo di permettere la condensazione delle diverse frazioni del tar all’interno di provette in vetro da 30×150 mm. La Figura 3.1 mostra uno schema del sistema.
Fig. 3.1 – Schema Tar-sampling
Il sistema opera in depressione mediante una pompa a membrana, posta a valle, della TCR Tecora s.r.l. modello BRAVO Basic M avente campo d’impiego tra 0,1 – 35 l/min. Un vacuometro posto a pannello fornisce il valore di intasamento del dispositivo collegato e
63
permette di testare la tenuta pneumatica del sistema. Sono, inoltre, presenti un contatore volumetrico ed un termometro digitale per la misura della temperatura del gas.
Il filtro1 in acciaio sinterizzato viene mantenuto ad una temperatura di 200°C mentre l’impinger 1 (Figura 3.2 (a)) è termostatato a 120°C al fine di favorire la condensazione della frazione più pesante del tar. Entrambi sono contenuti in un involucro mobile, collegato al resto del campionatore da una tubazione di 5 m di lunghezza, coibentata e tracciata elettricamente a temperatura di 200°C, in questo modo il sistema può essere connesso anche nei punti di campionamento logisticamente più difficoltosi da raggiungere.
L’impinger 2 (Figura 3.1 (b)), a differenza degli altri, contiene due provette, anziché una, mantenute a 10°C per condensare anche le frazioni leggere, oltre al vapore acqueo. La presenza della seconda provetta serve ad incentivarne ulteriormente la condensazione, in quanto raddoppia il tempo di contatto.
L’ultimo impinger, termostatato a -15°C mediante celle Peltier, ha funzioni di guardia al fine di evitare che gocce di acqua e/o tar vengano trascinate dal flusso gassoso ed arrivino alla pompa.
All’interno degli impinger sono alloggiate delle provette in vetro su un supporto filettato inserite dal basso, la cui tenuta è assicurata da una molla a coefficiente di elasticità noto, che preme la provetta contro una guarnizione in silicone.
A valle del terzo impinger è posta una trappola in vetro sotto ghiaccio ed un ulteriore filtro per gas in materiale vetroso prima della pompa.
64
La Tabella 3.1 mostra i tempi di residenza del gas nelle provette degli impinger e nella trappola, stimati per una portata di aspirazione di 15 l/min ed in assenza di riempimenti.
Impinger Tempo di residenza (portata 15 l/min)
1 0,4 s
2 0,8 s
3 0,4 s
Trappola 2 s
Tab. 3.1 – Tempo di residenza del gas negli impinger
3.1.3
Ottimizzazione dell’impianto
Il sistema è stato consegnato in Ottobre 2010. Al fine di stimare l’efficienza di campionamento del sistema sono state effettuate 3 prove con acqua distillata che hanno fornito valori di efficienza di recupero tra 70% ÷ 80%, denotando notevoli fenomeni di trascinamento a causa della formazione di aerosol.
Al fine di aumentare la superficie specifica di contatto tra gas e provette e favorire, quindi, la condensazione del tar, sono stati inseriti anellini di ceramica all’interno della provetta 1 del primo impinger (30 anelli) e della 2A del secondo impinger (20 anelli), mentre palline di vetro sono state inserite nelle provette 2B del secondo impinger e 3 del terzo (circa 2,5 cm). Sono stati, inoltre, applicati batuffoli di cotone in cima alle provetta 2B e 3 per impedire il ritrascinamento del condensato e delle palline stesse da parte del gas.
Sono, quindi, state effettuate 4 prove con olio minerale che hanno fornito una efficienza del campionatore di oltre 87%, applicando, però, basse portate di aspirazione.
La Figura 3.3 mostra il set-up delle prove di campionamento con olio minerale. Una beuta contenente il campione, posta su una piastra riscaldante, è stata collegata all’involucro mobile del sistema di campionamento. La transfer line dalla beuta all’involucro mobile è stata riscaldata oltre i 200°C mediante fascio scaldante e la temperatura del campione è stata monitorata con una termocoppia K collegata ad un data logger portatile.
65
Fig. 3.3 – Prove di campionamento con olio minerale
L’efficienza è stata stimata rapportando il peso del condensato campionato con la perdita in peso della beuta con il campione prima e dopo la prova.
Le prove sperimentali hanno evidenziato l’esigenza di schermare ulteriormente la pompa da frazioni oleose leggere ed acqua trascinate in aerosol dal flusso di gas, specialmente ad alte portate d’aspirazione, per cui è stata inserita una trappola in vetro inserita in un bagno di ghiaccio ed un ulteriore filtro per gas in materiale vetroso (Figura 3.4) che, unitamente all’individuazione delle temperature ottimali di set-point degli impinger, hanno consentito di ottenere efficienze di campionamento fino al 98%.
66
La Tabella 3.2 mostra i parametri operativi ottimali individuati per le prove di campionamento del tar. Un parametro di fondamentale importanza da monitorare costantemente è l’andamento della depressione nel sistema visibile dal vacuometro della pompa. Esso fornisce, infatti, un indice dell’intasamento del sistema e quindi, della bontà del campionamento. Dalle prove sperimentali si è intuito che non è possibile operare con valori di
ΔP superiori a -0,5 bar. Valori di ΔP superiori a -0,25 bar sono comunque già sufficienti a far diminuire sensibilmente la portata aspirata dalla pompa.
Parametro Valore
Portata campionatore tar aspirata 15 l/min Set point Impinger 1 120°C Set point Impinger 2 10°C Set point Impinger 3 -15°C
Tab. 3.2 – Parametri operativi del sistema di campionamento tar
3.2
Campionamento ed analisi dei gas mediante micro-GC
La cromatografia comprende una serie di tecniche analitiche di separazione di una miscela nei suoi componenti, basate sulla migrazione differenziale dei vari componenti in un sistema di due fasi, di cui una fissa (detta fase stazionaria) ed una in movimento (detta fase mobile). Tutti i metodi cromatografici prevedono la presenza di queste due fasi, la fase stazionaria e la fase mobile, immiscibili fra loro e poste in contatto.
La miscela campione, introdotta nella fase mobile, subisce una serie di ripartizioni fra le due fasi, mentre viene trascinata da quella mobile. La differenza di affinità dei vari componenti per le due fasi determina la differente velocità di migrazione di questi, con conseguente separazione degli stessi.
Nella gascromatografia (GC), la fase mobile è un gas che fluisce attraverso una colonna, in cui si trova la fase stazionaria. Con questa tecnica è possibile analizzare campioni gassosi, liquidi o solidi. Le sostanze da separare devono essere portate ad una temperatura sufficiente a renderle gassose o comunque portarle allo stato di vapore. L’iniettore e la colonna devono essere dunque portati alla temperatura che rende volatili le specie da separare e termostatati.
67
La gascromatografia risulta quindi applicabile solamente a sostanze volatili e termostabili. Un dispositivo cromatografico comprende i seguenti elementi fondamentali: un sistema di alimentazione della fase mobile, un sistema di iniezione o deposizione del campione, la colonna cromatografica, ed infine, un sistema di rivelazione dei componenti eluiti.
Nella cromatografia di eluizione, il campione viene introdotto in testa alla colonna e la fase mobile, costituita dal gas di trasporto, viene fatta fluire attraverso la colonna riempita di fase stazionaria. Durante l’attraversamento della colonna, i componenti presenti nel campione si ripartiscono in modo caratteristico tra la fase mobile e quella stazionaria. Poiché un composto può muoversi attraverso la colonna solo quando è nella fase mobile, la velocità media alla quale il composto migra dipenderà dalla frazione di tempo che esso trascorre in questa fase. Questa frazione è piccola per i soluti fortemente trattenuti dalla fase stazionaria, ed è elevata quando più probabile è la ritenzione nella fase mobile. Di conseguenza, si ha una diversità nei tempi di percorrenza della colonna da parte di ciascun composto.
Se un rivelatore che risponde alla concentrazione del soluto è posto all’uscita della colonna ed il suo segnale viene riportato in funzione del tempo, si ottiene un grafico costituito da una serie di picchi, detto cromatogramma, in cui ciascun picco, avente un tempo caratteristico detto tempo di ritenzione, corrisponde ad un composto separato nella colonna. La cromatografia può, quindi, essere utilizzata per l’identificazione qualitativa dei composti analizzati, ma anche per quella quantitativa misurando l’area di ciascun picco che è proporzionale alla quantità di sostanza che dà luogo al picco stesso. In generale, i coefficienti di proporzionalità per composti diversi sono differenti, per cui è necessario ricorrere alla calibrazione della risposta dello strumento, che può essere eseguita secondo vari metodi:
• metodo della taratura diretta;
• metodo della normalizzazione interna;
• metodo della standardizzazione interna;
• metodo delle aggiunte.
Nel metodo della taratura diretta, vengono iniettate quantità note del componente da determinare, e vengono misurate le aree dei picchi; si costruisce poi una curva di calibrazione che riporta l’area del picco in funzione della quantità dello standard iniettato. Iniettando quindi un’aliquota nota di campione, si misura l’area corrispondente al picco del prodotto di interesse e, dalla curva di calibrazione, si determina la sua quantità.
68
3.2.1
Punto di campionamento
Il micro-GC (Agilent 3000) è stato posizionato nell’impianto di gassificazione dopo la soffiante vicino la termocoppia TC-3 (Figura 2.2), e nell’impianto di pirolisi a valle del reattore 3 (Figura 2.11).
La linea di campionamento comprende:
• rubinetto apri-chiudi che permette di campionare oppure di escludere l’ingresso del gas;
• filtro a cartuccia in fibra di vetro, preliminare;
• linea di introduzione campione da 1/8” in rame;
• raccordo 1/8”-1/16”;
• linea di introduzione campione da 1/16” in acciaio;
• filtro a membrana da 5 µm per eliminare H2O/particolato dal flusso di gas per non danneggiare il micro-GC;
• pompa di campionamento per l’analisi di miscele non pressurizzate in fase gassosa (la pressione massima del gas da analizzare al punto di prelievo non deve essere superiore a 2 bar); si trova all’interno del micro-GC, è programmabile e attivabile tramite il software di controllo del micro-GC.
La procedura da utilizzare per il campionamento dei gas è descritta di seguito: 1. accendere e condizionare il micro-GC;
2. controllare che l’alimentazione dei gas di trasporto del micro-GC sia sufficiente per l’esecuzione delle analisi;
3. tramite il software di controllo del micro-GC, impostare il metodo per il campionamento e l'esecuzione dell'analisi;
4. a condizioni stabilizzate, aprire il rubinetto e iniziare il campionamento: il campionamento è effettuato in maniera discontinua; il tempo minimo di campionamento è determinato dalla durata della corsa cromatografica (circa 3÷5 minuti);
69
3.2.2
Strumentazione
Lo strumento utilizzato per l’analisi dei gas è il micro-GC Agilent 3000 (Figura 3.5) il quale può eseguire una completa analisi di idrogeno, di idrocarburi saturi e olefinici (C1-C5 e C6) e di gas fissi (O2, N2, CO, CO2) in meno di 160 secondi.
Fig. 3.5 – Analizzatore micro-GC Agilent 3000
Il Micro-GC Agilent 3000 può simultaneamente analizzare campioni su un massimo di quattro canali indipendenti. Ogni canale (o modulo) è composto da un iniettore, da una colonna capillare ad alta risoluzione e da un rivelatore a conducibilità termica (TCD, Thermal
Conductivity Detector). Questo design offre la massima flessibilità dell'applicazione,
consentendo di sostituire i singoli moduli per adattarsi a esigenze applicative diverse.
Lo strumento, data la sua compattezza, è facile da trasportare direttamente nel luogo di campionamento. Pesa, infatti 5,1 kg ed è alto 15 cm, largo 25 cm, profondo 41 cm. Per effettuare l’analisi occorrono le bombole di gas di trasporto e un computer per il controllo strumentale e l’acquisizione dati.
Il micro-GC utilizzato nel presente lavoro di tesi è a due canali analitici. Il primo canale è una colonna a setacci molecolari Molsieve 5A (10 m x 0,32 mm x 12 micron) adatta per la separazione di H2, O2, N2, CH4, CO utilizzando argon (Ar) come gas di trasporto. Questo canale è dotato di iniettore backflush riscaldato, ovvero di sistema di introduzione completo di pre-colonna per il taglio programmato dei composti indesiderati in colonna analitica. Il secondo canale è una colonna PLOT U (8 m x 0,32 mm x 30 micron) per la separazione di CO2, C2H4, C2H6, C2H2 ed utilizza elio (He) come gas di trasporto.
Ogni canale analitico è costituito da un Modulo Plug-and-Play che integra le seguenti parti principali:
70
- micro-iniettore riscaldato dotato di controllo del volume campione variabile da 1 a 10
µL o volume fisso da 1 µL;
- colonna analitica e colonna di riferimento capillari ad elevata efficienza, entrambe inserite in un comparto termostatato a temperatura costante (temperatura massima 180°C);
- micro-rivelatore universale “Solid State Detector” con volume interno da 240 µL;
- micro-EPC (Electronic Pressure Control) per il controllo e programmazione elettronica della pressione in testa alla colonna;
- scheda elettronica di controllo ed acquisizione.
Il micro-GC comprende inoltre:
- pompa di campionamento per l’analisi di miscele non pressurizzate in fase gassosa (pressione massima del gas da analizzare al punto di prelievo non deve essere superiore a 2 bar);
- filtro a membrana da 5 µm;
- linee di introduzione campione da 1/16”;
- interfaccia LAN per il controllo dello strumento.
Le specifiche tecniche principali sono le seguenti:
- sensibilità: 1-10 ppm per la maggior parte dei componenti;
- range dinamico: 6 ordini;
- ripetibilità: ≤ 1% RSD (iniettore a volume fisso), ≤ 0.2% RSD (in volume variabile).
3.2.3
Procedura sperimentale
Il sistema SRA Soprane Networked Data System è stato realizzato per controllare tutti i parametri del Micro-GC (quali attivazione di valvole, impostazione temperatura di colonna, ingresso campione e linee di trasferimento, gestione dei segnali dei rivelatori, pneumatica elettronica) per eseguire calcoli e per archiviare risultati. I segnali digitali provenienti dalla scheda del Micro-GC sono acquisiti ad una frequenza di campionamento variabile da 20, 50 o 100 Hz.
71
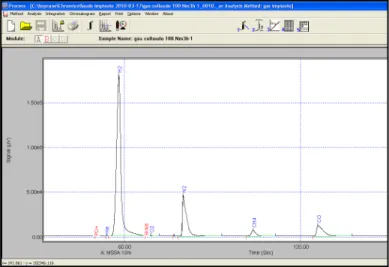
Il software Soprane può controllare il micro-GC Agilent da un PC esterno utilizzando la scheda di comunicazione LAN standard tramite protocollo (TCP/IP). È possibile infatti creare e modificare metodi e dall’analisi ottenere grafici costituiti da una serie di picchi, detti cromatogrammi, in cui ciascun picco, avente un tempo caratteristico detto tempo di ritenzione, corrisponde ad un composto separato nella colonna. In Figura 3.6 sono riportati i parametri del metodo (denominato “gas impianto”) impiegato per l’analisi del syngas.
Fig. 3.6 – Parametri del metodo impiegato per l’analisi del syngas
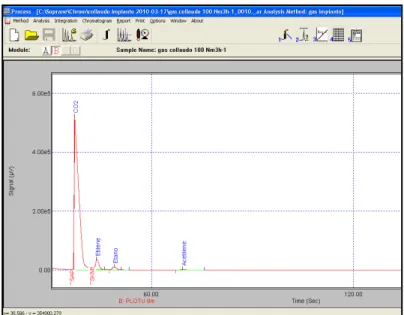
In Figure 3.7 e 3.8 sono mostrati i cromatogrammi rispettivamente per il modulo A (colonna a setacci molecolari adatta per la separazione di H2, O2, N2, CH4, CO) e per il modulo B (colonna PLOT U adatta per la separazione di CO2, C2H4, C2H6, C2H2) ottenuti in una prova di gassificazione.
72
Fig.3.8 – Esempio di cromatogramma ottenuto per il modulo B
E’ possibile, quindi, visualizzare e archiviare i risultati ottenuti (dopo calibrazione del sistema), avere un output di stampa (un esempio in Figura 3.9), leggere i dati esterni, confrontare vari cromatogrammi e trasferire i valori direttamente in un foglio di calcolo (ad esempio Microsoft Office Excel).
73
Le miscele gassose impiegate per la calibrazione sono elencate di seguito:
- H2 15,00%, N2 85,00%. - CO 0,995%, N2 99,005%. - CO 25,00%, N2 75,00%. - CO2 1,50%, N2 98,50%. - CO2 19,98%, N2 80,02%. - CH4 2,997%, N2 97,003%. - C2H4 0,500%, N2 99,500%.
- Agilent Universal Gas Calibration Standard (la cui composizione è riportata in
Tabella 3.3). Gas Concentrazioni (± mole%) Elio (He) 0,0993% (± 5%) Neon (Ne) 0,0496% (± 5%) Idrogeno (H2) 0,1019% (± 5%) Ossigeno (O2) 0,0497% (± 5%) Azoto (N2) 0,0993% (± 5%) Metano (CH4) Rimanente Etano (C2H6) 0,0500% (± 5%) Etilene (C2H4) 0,0495% (± 5%) Anidride carbonica (CO2) 0,0497% (± 5%) Monossido di carbonio (CO) 0,0998% (± 5%) Acetilene (C2H2) 0,0501% (± 5%) Propano (C3H8) 0,0504% (± 5%) Metilacetilene (C3H4) 0,0502% (± 5%) n-Butano (C4H10) 0,0497% (± 5%) n-Esano (C6H14) 0,0503% (± 5%) n-Eptano (C7H16) 0,0495% (± 5%) Tab. 3.3 – Composizione dell’Agilent Universal Gas Calibration Standard
74
3.3
Determinazione del contenuto di umidità
Per umidità si intende la quantità di acqua, libera e legata, presente nel materiale ad una data temperatura. In laboratorio il contenuto di umidità viene determinato calcolando la perdita in massa di un campione sottoposto ad essiccamento, in condizioni normalizzate, alla temperatura di 105°C.
L’umidità delle biomasse (materiale fortemente igroscopico) può raggiungere valori superiori al 50%. L’umidità è un fattore molto importante sotto il profilo economico: viene infatti considerato un parametro del valore energetico/commerciale della biomassa e come tale può influire sul prezzo. Può essere suddivisa in umidità inerente, propria della struttura della biomassa, e umidità superficiale, fortemente influenzata dalla pezzatura e dalla condizioni ambientali. L’umidità della biomassa è una proprietà dinamica in quanto tende a portarsi in equilibrio con l’ambiente e varia in maniera consistente in funzione del tipo di biomassa, del terreno di coltivazione, delle condizioni ambientali, delle modalità e dei tempi di trasporto e stoccaggio.
Il contenuto di umidità, generalmente espresso come percentuale in massa, è un parametro importante sia sotto il profilo tecnico-operativo sia in relazione ad alcune caratteristiche qualitative del materiale, essa influisce, infatti, su numerose proprietà termiche e fisiche delle biomasse (conducibilità, permeabilità, diffusione termica, densità). Un’elevata umidità superficiale, ad esempio, è particolarmente negativa sul funzionamento dei polverizzatori (diminuisce la capacità effettiva), sui trasportatori meccanici e più in generale, può creare inconvenienti nella movimentazione della biomassa, in particolare nelle fasi di carico e scarico in caso di gelate. Per quanto riguarda altri effetti indesiderati, il contenuto di umidità influenza fortemente la densità e il potere calorifico. L’umidità infatti incide sfavorevolmente sul peso del prodotto e sui costi di trasporto e di essiccamento, e soprattutto abbassa il contenuto energetico della biomassa.
Per quanto riguarda le prestazioni in impianto, in generale si osserva un calo dell’efficienza di combustione all’aumentare dell’umidità. Dati sperimentali indicano che si può ottenere un’efficienza massima di combustione per un contenuto di umidità attorno a circa il 5%: l’acqua eserciterebbe, infatti, un’azione moderatrice nella combustione, creando condizioni più favorevoli al trasferimento di calore rispetto al brusco passaggio in presenza di legno molto secco.
75
3.3.1
Metodologia
Per la determinazione del contenuto di umidità, il campione tal quale viene pesato in idonei contenitori e messo in stufa ventilata a 105°C fino a peso costante.
Dopo raffreddamento in essiccatore, il campione viene di nuovo pesato e la differenza tra il peso del campione prima e dopo l’essiccazione fornisce il valore dell’umidità. Questo metodo è utilizzato quando è necessaria un’elevata accuratezza nella determinazione dell’umidità.
3.3.2
Strumentazione e procedura sperimentale
Per determinare il contenuto di umidità presente nella biomassa viene utilizzata una bilancia analitica (ACCULAB, Sartorius Group) per pesare i campioni e una stufa ventilata (Binder). La procedura da utilizzare per determinare il contenuto di umidità è la seguente:
1) Pesare il campione in idonei contenitori (quantità di campione: circa 5÷50 g). 2) Mettere il campione in stufa a 105°C fino a peso costante (circa 10 ore). 3) Raffreddare il campione in essiccatore.
4) Pesare di nuovo il campione.
3.4
Analisi immediata
L’analisi immediata (proximate analysis) consiste nel determinare il contenuto di umidità (Moisture), la frazione volatile (VM), il carbonio fisso (FC) e le ceneri (Ash) del campione. L’umidità determinata con questa prova rappresenta tipicamente acqua fisicamente legata. Le sostanze volatili ed il carbonio fisso rappresentano la frazione combustibile presente nel materiale, mentre umidità e ceneri, ne costituiscono la parte non combustibile. Il contenuto di ceneri di una biomassa influenza molto il processo globale. In funzione del contenuto di ceneri, l’energia disponibile in un combustibile è ridotta proporzionalmente. In processi di tipo termico, la composizione delle ceneri può creare non pochi problemi. Infatti, nelle tecnologie in cui la combustione è il cammino utilizzato per la conversione della biomassa, le ceneri sono un problema reale, poiché reagiscono per formare degli “slag”, una fase liquida che si forma ad alta temperatura, che possono ridurre il rendimento dell’impianto (ad esempio
76
per problemi di scambio termico sui fasci tubieri), con il risultato di aumentare i costi di operabilità.
3.4.1
Metodologia
Secondo i metodi di prova comunemente utilizzati, l’analisi immediata si effettua riscaldando il campione in atmosfera inerte fino ad una temperatura massima generalmente compresa tra 800 e 950°C, in condizioni normalizzate. Le sostanze volatili sono costituite dalla porzione di biomassa, esclusa l’umidità, che si sviluppa sotto forma gassosa. Il materiale rimanente, escluse le ceneri, è considerato carbonio fisso. La prova di laboratorio consente di differenziare la frazione di combustibile che brucia sotto forma gassosa (sostanze volatili) da quella che brucia sotto forma solida (carbonio fisso). In termini quantitativi, i contenuti di
sostanze volatili e di carbonio fisso sono fortemente influenzati dalla temperatura e dalle
condizioni di prova adottate dal metodo utilizzato che va dunque esplicitato.
In generale, il campione viene prima macinato e successivamente sottoposto all’analisi immediata. Semplificando il processo esso si articola attraverso tre fasi distinte:
1. Essiccamento.
2. Devolatilizzazione in atmosfera inerte (N2).
3. Ossidazione del residuo solido della devolatilizzazione (char) in atmosfera
ossidante (O2).
L’umidità viene rilasciata a bassa temperatura (intorno ai 100°C), mentre gas e tar (liquidi a temperatura ambiente, ma gassosi alla temperatura di processo, ed espulsi in fase vapore) sono rilasciati a temperature più elevate, superiori in genere ai 300°C. La devolatilizzazione controlla la distribuzione dei prodotti (gas, tar, char). Dopo il processo di devolatilizzazione rimane il char la cui ossidazione è una reazione eterogenea.
L’analisi immediata delle biomasse può essere effettuata mediante analisi termogravimetrica. La Termogravimetria (TG) o Analisi Termogravimetrica (TGA) consente di valutare la perdita in peso di un materiale sottoposto ad un programma controllato di temperatura.
È possibile controllare sia il profilo termico che l’atmosfera cui è soggetto il materiale. L’impiego di tale tecnica permette la valutazione degli intervalli di temperatura in cui il campione in esame degrada, dell’entità della formazione di prodotti volatili e residuo solido.
77
Il principale vantaggio dell’utilizzo di questa tecnica è legato alle quantità assai limitate di campione necessarie per l’effettuazione delle prove sperimentali (dell’ordine dei pochi mg). Tuttavia uno studio preliminare dovrebbe essere svolto prestando attenzione a minimizzare i problemi legati al trasferimento di calore attraverso i seguenti accorgimenti:
− limitare i gradienti di temperatura nel campione esaminato, utilizzando una piccola quantità di particelle (circa 10 mg) sufficientemente fini ed omogenee;
− alimentare il gas con una velocità relativamente alta di flussaggio (100 ml/min), per evitare reazioni secondarie.
3.4.2
Strumentazione
L’apparecchiatura utilizzata per l’analisi termogravimetrica è la termobilancia TA-Q500, mostrata in Figura 3.10. L’analizzatore è costituito da un corpo centrale, da uno scambiatore esterno aria-acqua per il raffreddamento della fornace e da un computer che permette il controllo dell’apparecchiatura, la registrazione e l’analisi dei dati.
Il modulo centrale comprende la fornace, il sistema di flussaggio di gas e la microbilancia. Esso realizza il controllo della temperatura della fornace in modo da imporre al campione il profilo termico desiderato, acquisisce e registra i dati delle varie prove e li trasmette al computer per le successive elaborazioni.
78
Le informazioni che si rendono disponibili sono: tempo, temperatura, massa del campione e velocità di perdita in peso (dM/dt).
La Fig. 3.11 mostra uno schema della fornace in cui viene normalmente posizionato il crogiolo porta campione. Il campione è posizionato su un porta campione sostenuto da una bilancia al centro della fornace. La microbilancia è posizionata al di sopra della fornace.
Figura 3.11 – Sezione della fornace della TGA Q500 e visualizzazione dei percorsi dei gas di flussaggio.
La fornace è flussata da un gas (“furnace purge gas”) che consente il condizionamento dell’atmosfera intorno al campione e l’allontanamento dei prodotti di degradazione, mentre un secondo flusso di gas (“balance purge gas”) viene inviato alla bilancia e da questa passa nella fornace. Questo secondo flusso di gas serve ad impedire o limitare quanto meno la diffusione dei prodotti di decomposizione del campione verso la microbilancia.
Il controllo del profilo di temperatura del campione è effettuato mediante una termocoppia situata in prossimità del campione. Questa viene utilizzata per valutare la temperatura del campione e per controllare il set-point della temperatura della fornace.
L’analizzatore necessita di una calibrazione periodica del peso e della temperatura. La calibrazione della temperatura è basata sul punto di Curie (temperatura a cui si ha una transizione magnetica ovvero perdita delle proprietà magnetiche del materiale) di standard ad elevata purezza di cui è nota la temperatura di transizione.
La massima temperatura consentita dal sistema è pari a 1000°C, mentre la velocità di riscaldamento può variare da 0 a 100°C/min. La risoluzione della microbilancia è 0.1 µg.
79
A seconda del programma termico impostato è possibile effettuare diverse prove sia isoterme che in scansione variando la velocità di riscaldamento. L’atmosfera in cui vengono condotte le prove può essere scelta tra diverse possibilità: azoto, aria, miscele a diverso contenuto di ossigeno o anidride carbonica, ecc. È possibile, inoltre, cambiare il gas di flussaggio durante lo svolgimento della prova, effettuando, ad esempio, la prima parte della prova in atmosfera inerte e la successiva in ambiente ossidante.
La portata del gas di flussaggio può variare da 0 a 400 ml/min; in tutte le prove effettuate il flusso complessivo di gas è stato mantenuto costante al valore di 100 ml/min.
Il campione, che può essere sia solido che liquido, viene posto in appositi crogioli di allumina, accuratamente puliti, posizionati sul piattello della termobilancia, il quale andrà all’interno della fornace. La quantità di campione comunemente impiegata è di circa 10-15 mg.
I dati che si ottengono possono, poi, essere analizzati con l’ausilio dell’apposito software “Universal Analysis 2000”, il quale permette di riportare i segnali registrati sia in funzione del tempo che della temperatura e consente di valutarne rapidamente le caratteristiche.
3.4.3
Procedura sperimentale
La procedura seguita per l’esecuzione delle prove in termobilancia è stata la seguente: - macinazione del campione per renderlo più omogeneo possibile;
- impostazione del programma termico al processore;
- acquisizione della tara con il solo crogiolo posizionato sul piattello;
- preparazione del campione avendo cura che i campioni preparati siano di massa simile (circa 10-15 mg) per evitare possibili effetti legati alla differenza di massa;
- posizionamento del campione sul piattello; - start della prova.
Il programma di temperatura cui è stato sottoposto il campione nella termobilancia è il seguente:
1) rampa con velocità di riscaldamento pari a 20°C/min, da temperatura ambiente fino a 105°C in flusso di azoto;
2) isoterma per 10 minuti in flusso di azoto;
80
4) isoterma per 10 minuti in flusso di azoto; 5) equilibrio a 800°C in flusso di azoto;
6) cambiamento del gas di flussaggio: anziché N2, viene fornita aria al sistema; 7) isoterma per 10 minuti in flusso di aria.
Il programma termico prevede una parte iniziale di riscaldamento del campione da temperatura ambiente fino a 105°C ed un’isoterma di 10 minuti a tale temperatura, in modo da garantire la perdita totale dell’umidità contenuta nel campione. Successivamente il campione viene riscaldato in atmosfera inerte, alla velocità stabilita per il test, fino a raggiungere i 900°C. Una volta raggiunta tale temperatura il flusso del gas passa ad aria, per ottenere una fase finale di ossidazione del char prodotto, mediante isoterma di 10 min in atmosfera d’aria, per permettere di valutare il quantitativo di ceneri contenute nei campioni. In tutti i test sono state condotte almeno due prove nelle stesse condizioni, per poter confermare la ripetibilità dei risultati. Dai dati ottenuti con le prove in termobilancia, è possibile ricavare, attraverso il software “TA Universal Analysis 2000”, la curva della perdita in peso (Weight %) in funzione del tempo (o della temperatura) dei vari campioni, utile alla determinazione della composizione di ogni singolo campione, in termini di contenuto di umidità (Moisture), frazione volatile (VM), carbonio fisso (FC), ceneri (Ash).
Da questa curva si evincono, infatti, gli intervalli di tempo in cui il peso rimane costante (tratti orizzontali) e quelli in cui il peso decresce nel tempo. Misurando la differenza di peso tra due tratti orizzontali è possibile ricavare i valori cercati:
• Moisture: quantità di umidità presente nel campione ottenibile graficamente
considerando il primo gradino nella curva;
• VM: è quella porzione di solido che si trasforma in gas per riscaldamento in atmosfera
inerte, ottenibile considerando il secondo gradino nella curva;
• FC: è la massa rimanente dopo il rilascio della parte volatile, escluso le ceneri e
l’umidità (terzo gradino);
• Ash: peso del campione in %, determinato ancora una volta graficamente, come valore
nell’ultimo tratto costante della curva.
Un esempio di curva di perdita in peso è riportato in Figura 3.12. Sull’asse delle ordinate è presente il valore del peso residuo percentuale (weight %) ottenuto come rapporto tra il peso
81
attuale ad un preciso istante di tempo (W) e il peso iniziale del campione (W0); sull’asse delle
ascisse è presente il valore del tempo (time) in minuti.
Fig. 3.12 – Esempio di curva TG
3.5
Analisi elementare
Il contenuto percentuale di carbonio, idrogeno, azoto e ossigeno rappresenta la cosiddetta
analisi elementare. Tali elementi, ai quali per integrare l’informazione analitica viene spesso
aggiunta la determinazione di zolfo e cloro, costituiscono la parte organica della biomassa. Il contenuto di ossigeno viene generalmente calcolato per differenza, sottraendo a 100 i contenuti di carbonio, idrogeno, azoto, zolfo, cloro e ceneri.
Le informazioni che è possibile trarre dall’analisi elementare sono molteplici e si basano sostanzialmente sul rapporto percentuale tra i principali elementi, contribuendo a definire il più corretto utilizzo della biomassa. Un elevato rapporto carbonio/azoto (C/N >30), ad esempio, è indice di un materiale con elevata attitudine alla combustione e come tale idoneo alle conversioni termochimiche.
L’analisi elementare viene, inoltre, utilizzata come supporto per valutare la qualità energetica della biomassa. Come andamento generale, ad alti contenuti di carbonio ed idrogeno corrispondono elevati poteri calorifici mentre elevate concentrazioni di ossigeno ed azoto hanno un effetto opposto.
82
Azoto, zolfo e cloro sono particolarmente importanti ai fini della valutazione delle emissioni inquinanti, delle prestazioni in impianto (con particolare riferimento a fenomeni corrosivi) e delle problematiche a loro connesse. Il cloro in particolare, oltre ad essere un precursore per la formazione di diossina durante la combustione, svolge un ruolo primario anche nella formazione di ceneri particolarmente aggressive.
3.5.1
Metodologia
I metodi impiegati per l’analisi elementare si basano sul principio della combustione in corrente di ossigeno (metodo Dumas), seguita dalla determinazione quantitativa dei prodotti di reazione.
In generale è possibile distinguere le strumentazioni per l'analisi elementare in due categorie: strumenti per la microanalisi, spesso utilizzati per la determinazione della formula bruta di composti organici incogniti, che analizzano campioni fino a 2 mg circa e strumenti per la macroanalisi utilizzati per il controllo di qualità, specie in campo industriale, che operano con quantità più elevate (fino a 1 g circa) e quindi più rappresentative di miscele o materiali non omogenei.
Gli strumenti CHNS per microanalisi prevedono comunemente una combustione dinamica “flash”, seguita da riduzione, separazione cromatografica dei gas di combustione e rivelazione con rivelatore a termo conducibilità (TCD). I campioni pesati in contenitori di stagno, ad intervalli prestabiliti sono introdotti in un tubo di quarzo verticale mantenuto intorno a 1000°C, attraverso il quale fluisce una corrente costante d'elio. Dopo l'introduzione dei campioni, la corrente d'elio è temporaneamente arricchita con ossigeno puro che dà luogo ad una combustione flash. La combustione completa è garantita dal controllo dell'iniezione dell'ossigeno durante la combustione ad alta temperatura. La miscela di gas ottenuta viene fatta passare sopra una colonna di riduzione di rame a 650°C, per rimuovere l'eccesso di ossigeno e per ridurre gli ossidi di azoto ad N2 e successivamente, attraverso una colonna cromatografica. I singoli componenti (N2, CO2, H2O) sono separati per cromatografia ed inviati ad un rivelatore TCD.
Un altro sistema per la misura di C, H, N e S, in campioni fino a 2 mg, opera con un sistema a combustione in flusso di ossigeno su CuO a 900°C, riduzione su catalizzatore a base di rame a 650°C, determinazione di H2O, SO2 e CO2 mediante celle di rivelazione IR e di N2 mediante cella TCD, previa purificazione del gas da umidità e da CO2.
83
Alcuni strumenti effettuano la determinazione dell'ossigeno in composti organici mediante pirolisi del campione, conversione a CO mediante carbone metallizzato, separazione cromatografica e rivelazione con TCD; altri strumenti, dopo la fase di pirolisi, utilizzano celle di rivelazione IR.
Gli strumenti che possono essere utilizzati per la macroanalisi comprendono l’analizzatore
LECO TruSpec CHN e l’analizzatore Elementar vario Macro CHNS. Il primo si dedica
all’analisi elementare operando con un forno a resistenza a temperatura di circa 1000°C in corrente di ossigeno e permette l’analisi di macrocampioni fino a 1 g circa. È dotato di quattro rilevatori, ciascuno dei quali dedicato ad un elemento. Il metodo di rilevazione è ad assorbimento all’infrarosso per carbonio e idrogeno e a conducibilità termica per azoto. Lo strumento è espandibile all’analisi dello zolfo, con modulo e rilevatore IR dedicato. L’analizzatore Elementar vario Macro CHNS è un analizzatore elementare per la determinazione simultanea di C, H, N e S. Il principio di funzionamento è basato sulla tecnica di combustione di Dumas e di separazione dei gas con la tecnica “Purge-and-Trap” ovvero con un sistema di separazione dei gas in colonne differenti. Tutti i gas prodotti dalla combustione (N2, CO2, H2O e SO2), dopo la separazione con la tecnica “purge-and-trap”, vengono direttamente letti da un detector a termoconducibilità (TCD) senza separazione cromatografica. Il sistema è configurabile per l’analisi di O2 con la sostituzione della colonna di combustione con la colonna di pirolisi catalitica ed il set-up dedicato delle colonne di purge-and-trap.
3.5.2
Strumentazione
Per la macroanalisi è stato utilizzato l’analizzatore elementare LECO TruSpec CHN (Figura 3.13). Lo strumento ha tempi di analisi molto brevi: per una determinazione occorrono tipicamente circa 4 minuti e permette l’analisi di macrocampioni, fino ad 1 g circa. Il campo di misura e la precisione sono le seguenti:
Campo di misura*
- Carbonio da 0.005% a 50% - Idrogeno da 0.02% a 50% - Azoto da 0.008% a 100%
84 Precisione*
- Carbonio 0.5% RSD (deviazione standard relativa) - Idrogeno 1% RSD
- Azoto 0.5% RSD
* per un campione di 500 mg
Fig. 3.13 – Analizzatore elementare LECO TruSpec CHN
Il principio di funzionamento di questo strumento prevede una combustione rapida e completa del campione all’interno di una fornace, alla temperatura di 950°C in eccesso di ossigeno. I prodotti della combustione sono fatti passare attraverso un forno di post-combustione a 850°C per una ulteriore ossidazione.
La Figura 3.14 mostra il tubo ad U nel quale avviene la combustione. Quest’ultima avviene nel lato primario del tubo (nella parte destra della figura), mentre nel lato secondario si ha una post-combustione che garantisce la completa ossidazione. I crogioli porosi (porous crucible) raccolgono le ceneri formate durante l’analisi.
I gas di combustione (CO2, H2O ed NOx) sono poi raccolti in un contenitore detto ballast, di volume pari a 4,5 litri, all’interno del quale sono omogeneizzati ed inviati alle rispettive unità di rivelazione. I gas sono inviati a due rivelatori ad assorbimento infrarosso per la misura dell’anidride carbonica e dell’acqua; il carbonio è, infatti, misurato sotto forma di CO2 e l’idrogeno sotto forma di H2O.
Un’aliquota dei gas di combustione, pari a 3cc è, invece, trasferita in un flusso di elio e fatta passare attraverso un catalizzatore di rame caldo per la rimozione dell’ossigeno e la conversione degli NOx ad azoto elementare ed attraverso ulteriori filtri per la rimozione dell’anidride carbonica e dell’acqua. Infine un rivelatore TCD è usato per la determinazione del contenuto di azoto.
85
Fig. 3.14 – Tubo di combustione dell’analizzatore Leco
Un’aliquota dei gas di combustione, pari a 3cc è, invece, trasferita in un flusso di elio e fatta passare attraverso un catalizzatore di rame caldo per la rimozione dell’ossigeno e la conversione degli NOx ad azoto elementare ed attraverso ulteriori filtri per la rimozione dell’anidride carbonica e dell’acqua. Infine un rivelatore TCD è usato per la determinazione del contenuto di azoto.
Sulla base dei dati di calibrazione ottenuti con materiali di riferimento standard di composizione nota (comunemente viene impiegato l’acido etilendiamminotetraacetico o ETDA), lo strumento fornisce la percentuale in peso dei vari elementi presenti nel campione.
3.5.3
Procedura sperimentale
Le procedure da utilizzare per un corretto funzionamento dell’analizzatore CHN sono elencate di seguito:
86
2) Preparare lo strumento per il funzionamento, come indicato nel manuale di istruzione fornito all’operatore.
3) Impostare il metodo.
4) Effettuare la prova di bianco. 5) Calibrare con EDTA.
6) Pesare circa 0,10-0,15 g di campione e inserire il valore del peso e l’identificazione del campione che vogliamo analizzare nel programma di acquisizione dati.
7) Mettere nella posizione appropriata del carosello il campione e procedere con l’analisi. 8) Effettuare almeno due repliche per ogni campione.
I parametri di combustione sono i seguenti:
− temperatura della fornace: 950°C;
− temperatura del forno di post-combustione: 850°C.
Il profilo della combustione relativo ai due metodi utilizzati nel presente lavoro di tesi è riportato in Tabella 3.4:
Metodo Tempo (s) Flusso O2
Biomassa 40 alto 30 medio 30 alto Carbone 30 alto 180 medio 30 alto
87
3.6
Analisi TG-FTIR
L’accoppiamento della termogravimetria (TG) con la spettrometria infrarossa in Trasformata di Fourier (FTIR) per l’analisi simultanea dei composti gassosi emessi è una tecnica che si è rapidamente diffusa negli ultimi anni. Il sistema sperimentale è costituito da un analizzatore termogravimetrico interfacciato, tramite una linea di collegamento termostatabile, ad uno spettrometro FTIR per l’analisi on-line dei prodotti rilasciati in fase gas. Tale accoppiamento permette il monitoraggio simultaneo della perdita in peso di un campione sottoposto ad un definito programma termico in una specifica atmosfera di reazione, e della composizione dei prodotti volatili originati da questo.
La tecnica combinata TG-FTIR è largamente impiegata per l’analisi di prodotti gassosi di pirolisi o di combustione. L’analizzatore TG permette di simulare le condizioni di temperatura, tipo di atmosfera e velocità di riscaldamento in cui avviene un processo di degradazione termica; dall’analisi dei dati IR è possibile identificare le specie chimiche generate e monitorare la loro evoluzione in funzione del tempo o della temperatura dell’analizzatore termico.
I dati TG-FTIR vengono utilizzati principalmente per l’analisi qualitativa di prodotti di decomposizione o di reazione, anche se l’impiego del sistema TG-FTIR può essere utilizzato per determinazioni quantitative dei composti generati nelle prove TG.
I vantaggi principali della tecnica TG-FTIR sono legati alle quantità limitate di campione richieste (dell’ordine dei pochi milligrammi) ed alla rapidità dell’analisi.
Limitazioni nell’uso della tecnica possono derivare da difficoltà nell’interpretazione dei risultati IR, legate all’eventuale formazione contemporanea di un elevato numero di composti volatili durante i processi indagati. Deve inoltre essere ricordato che le molecole biatomiche omodinucleari non assorbono nell’infrarosso e quindi non possono essere individuate[42].
Nel presente lavoro di tesi le prove di analisi simultanea TG-FTIR hanno consentito la caratterizzazione del contenuto d’acqua del condensato campionato con il sistema di campionamento tar, nelle prove di pirolisi e gassificazione.
88
3.6.1
Strumentazione
Le prove di analisi simultanea TG-FTIR condotte nel presente lavoro di tesi sono state effettuate utilizzando un sistema combinato costituito dall’analizzatore termico simultaneo TG/DSC Netzsch STA 409 C e da uno spettrometro FTIR Bruker Equinox 55.
Lo spettrometro FTIR è caratterizzato dalla presenza di due distinti vani di misura. Il vano principale, dotato di un detector di tipo DTGS (Deuterate Triglycine Sulphate), permette l’analisi di campioni solidi, liquidi o gassosi. Il vano ausiliario, munito di un detector di tipo MCT (Mercury-Cadmium-Telluride), è fornito di una cella termostatabile per l’analisi on-line di composti gassosi. La cella, con temperatura programmabile tra quella ambiente ed una massima di 250°C, ha un cammino ottico di 12.3 cm ed un volume di 8.7 ml.
L’accoppiamento tra l’analizzatore termico e la cella termostatabile per l’analisi on-line dei composti gassosi dello spettrometro FTIR è realizzato con una transfer line riscaldata (Figura 3.15). La transfer line è costituita da un tubo in teflon di lunghezza pari a 800mm e con diametro interno di 2mm, inserito all’interno di una guaina coibentata riscaldata tramite una resistenza elettrica.
Fig. 3.15 – Schema dell’accoppiamento TG-FTIR
Uno specifico software (OPUS) consente il controllo dello spettrometro, l’acquisizione e l’analisi degli spettri.
89
3.6.2
Determinazioni quantitative FTIR
Grandezze fondamentali per l’analisi FTIR quantitativa sono la trasmittanza (T), definita come rapporto tra la potenza radiante trasmessa dal campione e quella incidente, e l’assorbanza (A), legata alla trasmittanza dalla relazione:
) T log(
A=− (3.1)
La legge di Lambert-Beer offre un’espressione che lega l’assorbanza ad una specifica lunghezza d’onda alla concentrazione della specie chimica responsabile dell’assorbimento:
C l
A=ε⋅ ⋅ (3.2)
dove
ε
è il coefficiente di estinzione (coefficiente di assorbimento molare o assorbanza specifica molare), l il cammino ottico attraverso il campione e C la concentrazione. Il valore diε
è funzione della specie chimica analizzata, della lunghezza d’onda e della temperatura. La legge di Lambert-Beer stabilisce dunque una relazione lineare tra assorbanza e concentrazione. Possono comunque presentarsi deviazioni di vario tipo: reali, chimiche, fisiche, e strumentali, dovute al fatto che spesso si è costretti ad operare con bande passanti larghe rispetto all’ampiezza del picco. Di conseguenza ci si trova ad operare su valori medi anziché puntuali come previsto dalla legge la quale è rigorosamente valida solo per radiazione monocromatica.Quest’ultimo fattore rende le misure strettamente dipendenti dal potere risolutivo dello strumento. La risoluzione spettrale degli spettrometri IR è in genere bassa, ciò comporta necessariamente che il valore del coefficiente di assorbimento molare sia dipendente dalla risoluzione scelta per le misure.
La limitata risoluzione degli spettrometri IR comporta che l’equazione (3.2) sia generalmente utilizzata su un intervallo di lunghezze d’onda o di numeri d’onda ( ):
(3.3) dove:
(3.4)
K è una costante di proporzionalità incognita il cui valore dipende dal composto in esame,
dall’intervallo di numero d’onda, dalla temperatura del gas e dalla risoluzione dello strumento. C K Cd l d A I =
∫
ν
ν
=∫
ε
ν
⋅ ⋅ν
= ⋅ ν ν ν ν ~ ) ~ ( ) ~ ( 2 1 2 1 ~ ~ ~ ~ 2 1,~ ~ ν νν
ν
ε
ν ν ~ ) ~ ( 2 1 ~ ~ d l K =∫
⋅ ⋅90
Per questi motivi, valori attendibili di K possono essere determinati solo mediante calibrazione sperimentale. Inoltre K dipende anche dalla concentrazione, a meno che siano trascurabili le deviazioni della legge di Lambert-Beer (ad esempio, qualora si operi su intervalli di concentrazione limitati).
In merito alla dipendenza dalla temperatura, nel caso di analisi on-line di gas, data la piccola inerzia termica di questi ultimi, la temperatura di ingresso del gas nella cella è in genere relativamente poco influente, ed il gas può essere considerato alla temperatura della cella durante la misura.
Per la scelta dell’intervallo di numeri d’onda su cui calcolare l’integrale riportato nell’equazione (3.3), è necessario individuare una zona spettrale in cui il composto in esame presenti una banda di assorbimento priva di interferenze dovute all’assorbimento da parte di altre specie chimiche. Questo aspetto costituisce una delle limitazioni più importanti all’impiego della spettrometria IR per analisi quantitative di miscele.
La tecnica convenzionale usata per determinazioni FTIR quantitative di composti gassosi è basata su una calibrazione realizzata mediante l’impiego di diverse miscele gassose a concentrazione nota del composto di interesse. Tuttavia ciò richiede la disponibilità di un elevato numero di standard a diversa concentrazione.
Al fine di limitare i problemi citati, sono stati proposti metodi di calibrazione a impulso. Tali metodi prevedono l’invio allo spettrometro di un impulso contenente una quantità nota del composto di interesse. Nel presente lavoro tale impulso è stato effettuato tramite vaporizzazione, in cui la quantità nota di sostanza da inviare allo spettrometro è ottenuta mediante vaporizzazione nell’analizzatore TG di campioni di composizione nota.
Le misure TG-FTIR sono stare effettuate vaporizzando campioni di acqua nella fornace della termobilancia. Mediante l’uso di una siringa gascromatografica, quantità di soluzione variabili tra 5 e 15 μl sono state poste all’interno di un crogiolo di alluminio sul quale è stato posto un coperchio forato. Il coperchio ha lo scopo di ridurre l’evaporazione, determinata da fenomeni diffusivi, all’inizio della prova TG. Dal peso iniziale del campione è possibile ricavare l’esatta quantità vaporizzata durante la prova. L’uso di campioni a diverso peso ha permesso di coprire un intervallo piuttosto ampio di quantità del composto di calibrazione inviate allo spettrometro.
Le prove sono state eseguite a 10°C/min, impiegando azoto come gas di trasporto (60 ml/min). Il metodo è stato applicato alla determinazione dell’acqua.
91
(3.5) L’intervallo di numero d’onda selezionato per l’acqua è 3792-4025 cm-1.
L’analisi dei dati ottenuti dai metodi di calibrazione ad impulso richiede l’integrazione dell’equazione (3.5) nel tempo:
(3.6) dove l’intervallo temporale (t1,t2) è quello durante il quale il composto di interesse passa attraverso la cella di campionamento.
La quantità totale n del composto sviluppata nell’intervallo di tempo (t1,t2) può essere
espressa come:
(3.7) dove F è la portata volumetrica totale di gas alla temperatura della cella di misura.
Il valore dell’integrale D può essere correlato a quello di n attraverso le equazioni (3.6) e (3.7):
(3.8)
dove K' è un fattore di correlazione dipendente da n e dalle condizione operative adottate nella prova. Una volta selezionato il numero d’onda d’interesse, il valore di D può essere misurato in prove di calibrazione a impulso, permettendo quindi la stima del valore di K' in funzione di
n.
Dall’equazione (3.8) è evidente che i valori di K' risultano essere dipendenti dall’entità della portata gassosa F nella cella IR. Questo non comporta una forte limitazione, purché venga impiegata la medesima portata gassosa in tutte le prove. Inoltre, comunemente solo un intervallo limitato di valori di portata è di interesse nelle prove TG-FTIR, dato che tale parametro in genere è stabilito dalle calibrazioni di flusso termico o di temperatura dell’analizzatore termico.
Nelle prove di calibrazione per l’acqua, i valori di I sono stati integrati nel tempo, calcolando il corrispondente valore di D secondo l’equazione (3.6). I risultati ottenuti sono stati riportati in funzione della quantità di sostanza n, consentendo la valutazione della costante K'.
( )
t d K C( )
t C l d A I =∫
ν
ν
=∫
ε
ν
⋅ ⋅ ⋅ν
= ⋅ ν ν ~ ) ~ ( ) ~ ( 2 1 2 1 ~ ~∫
∫ ∫
= = 2 1 2 1 2 1 ~ ) ~ ( ~ ~ t t t t Kcdt dt d A Dν
ν
ν ν∫
⋅ = 2 1 t t cdt F n n K n Fcdt Kcdt D t t t t ⋅ = ⋅ =∫
∫
' 2 1 2 192
3.7
Analisi GC-MS
L’accoppiamento della gascromatografia con la spettrometria di massa (GC/MS) costituisce un potente metodo di indagine per l’analisi qualitativa di miscele complesse in quanto combina le grandi capacità di separazione della GC con la capacità di identificazione e caratterizzazione della struttura delle molecole tipica della spettrometria di massa.
Nel presente lavoro di tesi è stata effettuata una caratterizzazione qualitativa delle frazioni del condensato campionato con il sistema di campionamento tar nelle prove di pirolisi e gassificazione, mediante gascromatografia-spettrometria di massa (GC-MS).
3.7.1
Strumentazione
Le analisi GC-MS sono state condotte con un gascromatografo Fisons GC 8060 interfacciato con uno spettrometro di massa a quadrupolo Fisons MD 800. Per la separazione cromatografica è stata utilizzata una colonna capillare in silice fusa a fasi legate con fase stazionaria SE30 (100% polidimetilsilossano) (lunghezza 25m, diametro interno 0.32mm, spessore film 0.25µm), impiegando elio come gas di trasporto. Le iniezioni sono state effettuate in modalità splittless, con temperatura dell’iniettore pari a 250°C. Il programma trmico impiegato è il seguente:
- isoterma iniziale a 40°C di 5 min; - riscaldamento a 6°C / min fino a 250°C; - isoterma finale a 250°C di 30 min.
L’acquisizione MS è stata effettuata in scansione sull’intervallo m/z 10-819, impiegando la modalità di ionizzazione per impatto elettronico.
L’analisi degli spettri di massa ed il loro confronto con gli spettri presenti in libreria ha consentito l’identificazione di vari composti presenti nei tar analizzati.