!
"#!
3. M
ATERIALI E METODI.
!3.1. C
AMPIONI UTILIZZATI.
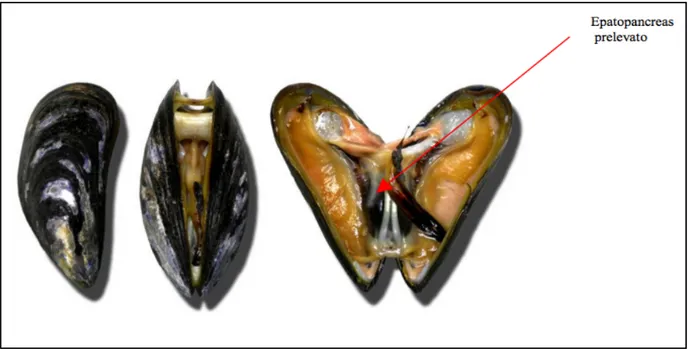
Nel presente studio sono stati utilizzati campioni di epatopancreas (stomaco ed intestino) estratti da 24 campioni di Mytilus edulis (mitilo), 24 di Donax trunculus (tellina), 24 di Crassostrea gigas (ostrica concava) e 12 di Chamelea gallina (vongola lupino).
I campioni provenivano da zone di pesca non ancora classificate per la molluschicoltura e da aree già classificate soggette ad attività di monitoraggio.
Per ogni campione è stato selezionato un numero minimo di 10 individui per le specie più grandi come le ostriche ed i mitili, 35 per le vongole e 65 per le telline. Dopo aver effettuato apertura delle valve con lama pulita, è stato asportato lʼepatopancreas (tessuto digestivo) utilizzando pinzette e forbici sterili, trasferito in piastra Petri sterile e finemente sminuzzato mediante lama di bisturi sterile. La quantità minima di epatopancreas prelevata per ogni campione è stata di 2,0 ± 0,2 g.
3.2. E
STRAZIONE DEL VIRUS.
Ogni aliquota (pari a 2,0 ± 0,2 g) è stata sottoposta alla fase di estrazione del virus mediante lʼaggiunta di 2,0 ± 0,2 ml di soluzione di Proteinasi K (0,1 mg/ml) (Sigma-Aldrich, Milano, Italia). I campioni sono stati incubati a 37,0 ± 1,0 °C per 60 ± 5,0 minuti in termostato con agitatore (circa 320 rpm) e successivamente a 60,0 ± 2,0 °C per 15 ± 1,0 minuti in bagnetto termostatico per inattivare lʼenzima. I campioni sono poi stati sottoposti a centrifugazione a 3000 x g per 5 minuti ± 30 secondi ed il surnatante è stato recuperato, misurato e portato a volume finale di 3,0 ± 0,3 ml mediante aggiunta di PBS sterile a pH 7,3. Il campione così trattato è stato conservato a -80 °C fino al momento dellʼutilizzo.
3.3. E
STRAZIONE E CONCENTRAZIONE DELLʼRNA
VIRALE.
LʼRNA virale è stato estratto utilizzando due diverse metodiche; la prima prevede lʼestrazione da 140 μl di surnatante mediante il kit QIAamp Viral RNA Mini Kit (Qiagen, Leusden, The Netherlands) mentre la seconda metodica prevede lʼutilizzo di 500 μl di surnatante per lʼestrazione dellʼRNA virale mediante il kit NucliSENS® MiniMAG® (bioMerieux, Marcy l'Etoile, France). I campioni di RNA virale ottenuti mediante le due diverse metodiche (60 μl con il kit QIAamp Viral RNA Mini Kit e 100 μl con il kit Nuclisens®Minimag®) sono stati stoccati a -80 °C fino al momento del loro utilizzo.
!
"#!
3.4. C
ONTROLLO DI PROCESSO.
Come controllo di processo è stato utilizzato un campione di Feline CaliciVirus (FCV-F9). Il virus FCV-F9 (ATCC VR-782, ceppo F9) fornito dallʼIstituto Superiore di Sanità – Dipartimento di Sanità Pubblica Veterinaria e Sicurezza Alimentare, Roma, è stato propagato su cellule Crandell-Reese Feline Kidney (CRFK) (ATCC CCL-94). Le cellule CRFK sono state mantenute nel terreno MEM/EBSS (Eagleʼs Minimum Essential Medium) con 2 mM di L-glutammina, 0,1 mM di amminoacidi non essenziali e 10% di siero di cavallo ed incubate a 37 °C con il 5% di CO2. Gli stock virali sono stati preparati attraverso congelamento e scongelamento del monostrato cellulare infettato ed incubato fino a che non è stato evidenziato lʼeffetto citopatico (sette giorni); lo stock virale è stato mantenuto a -80°C fino al momento del suo utilizzo.
3.5. P
RIMER E SONDET
AQM
AN PER LA RILEVAZIONE DI NOROVIRUSGI,
GII
EHAV.
Per lʼamplificazione ed il rilevamento mediante Real-Time con metodica Taqman del genoma di NoV GI sono stati utilizzati il primer senso QNIF4, il primer antisenso NV1LCR e la sonda NV1LCpr (6-carboxyfluorescein/6-carboxytetramethylrhodamine) (da Silva et al., 2007).
Per lʼamplificazione ed il rilevamento del genoma di NoV GII sono stati utilizzati il primer senso QNIF2, il primer antisenso COG2R e la sonda QNIFS (6-carboxyfluorescein/6-carboxytetramethylrhodamine) (da Silva et al., 2007).
Lʼarea selezionata per la ricerca dei Norovirus è la regione fortemente conservata allʼestremità 5ʼ dellʼORF2. Lʼallineamento, utilizzando tutte le sequenze disponibili su
GenBank per la regione target, ha dimostrato che questo set di primer/probe è adeguato per la ricerca di tutti i genotipi di Norovirus GI e GII.
Per lʼamplificazione ed il rilevamento del genoma di HAV sono stati utilizzati il primer senso HAV68, il primer antisenso HAV240 e la sonda HAV150 (6-carboxyfluorescein/6-carboxytetramethylrhodamine) (Costafreda et al., 2006).
Lʼarea selezionata per la ricerca di HAV è la regione 5ʼ UTR (circa 600 nucleotidi), regione maggiormente conservata del genoma. Lʼallineamento, utilizzando tutte le sequenze disponibili in GenBank per la regione target, dimostra che questo set di primer/probe è adeguato per la rilevazione di tutti i genotipi. In aggiunta lʼanalisi di quasispecie dello spettro dei mutanti indica che questa regione non è incline alla variabilità e che questo saggio dovrebbe possedere robustezza a lungo termine. Per lʼamplificazione ed il rilevamento mediante Real Time con metodica Taqman del genoma di FCV, le sonde sono state selezionate dalla regione dellʼORF1 (nucleotidi 20-5308 sequenza GenBank M86379) (Di Pasquale et al., 2010).
Le sequenze dei primer utilizzati sono di seguito riportate:
Norovirus GI
Primer Sequenza (5ʼ!3ʼ) Amplificato (bp)
QNIF4 (FW) CGC TGG ATG CNG TTC CAT (da Silva et al., 2007) 86
NV1LCR (REV) CCT TAG ACG CCA TCA TCA TTT AC (da Silva al., 2007)
NV1LCpr (PROBE) TGG ACA GGA GAY CGC RAT CT (da Silva et al., 2007)
Probe labelled 5ʼ 6-carboxyfluorescein (FAM), 3ʼ 6-carboxy-tetramethylrhodamine (TAMRA) Y = C o T; R = A o G; N = A, T, G o C; W = A o T
!
"#!
Norovirus GII
Primer Sequenza (5ʼ!3ʼ) Amplificato (bp)
QNIF2 (FW) ATG TTC AGR TGG ATG AGR TTC TCW GA (da Silva al., 2007) 89
COG2R (REV) TCG ACG CCA TCT TCA TTC ACA (da Silva al., 2007)
QNIFS (PROBE) AGC ACG TGG GAG GGC GAT CG (da Silva al., 2007)
Probe labelled 5ʼ 6-carboxyfluorescein (FAM), 3ʼ 6-carboxy-tetramethylrhodamine (TAMRA)
Y = C o T; R = A o G; N = A, T, G o C; W = A o T
HAV
Primer Sequenza (5ʼ!3ʼ) Amplificato (bp)
HAV68 (FW) TCA CCG CCG TTT GCC TAG (Costafreda et al., 2006) 174
HAV240 (REV) GGA GAG CCC TGG AAG AAA G (Costafreda et al., 2006)
HAV150 (PROBE) CCTGAA CCT GCA GGA ATT AA (Costafreda et al., 2006)
FCV
Primer Sequenza (5ʼ!3ʼ) Amplificato (bp)
FCV (FW) ACA AGT CCG TTG GAG CAA TTG A (Di Pasquale et al., 2010) 83
FCV (REV) CCC CTG AGG TGT CCT TGT GAT (Di Pasquale et al., 2010)
FCV (PROBE) CCT ATT GAT CCT GAC TCT GTT GTT TTC TTG AAG AGA AC (Di
Pasquale et al., 2010)
Probe labelled 5ʼ 6-carboxyfluorescein (FAM), 3ʼ 6-carboxy-tetramethylrhodamine (TAMRA) !
3.6. R
EALT
IMERT-PCR
ONE-
STEP.
La Real Time RT-PCR (rRT-PCR) è stata effettuata con un Bio Rad iQ5 Multicolor Real Time 950 Detection System (Bio Rad).
3.7. P
ROTOCOLLO DIR
EALT
IMERT-PCR
ONE-
STEP.
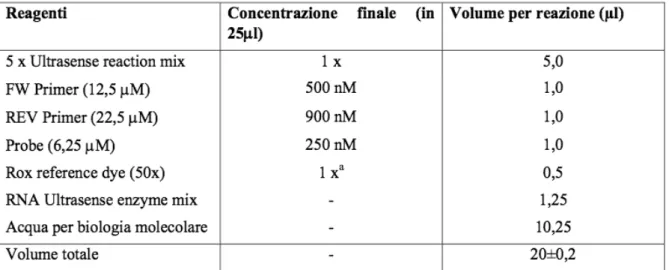
La Real Time RT-PCR (rRT-PCR) è stata effettuata utilizzando il kit commerciale “RNA UltraSense" One-Step Quantitative RT-PCR System” (Invitrogen, France). L'RNA UltraSense™ One-Step Quantitative RT-PCR System utilizza due enzimi: SuperScript™ III trascrittasi inversa (RT), che è un mutante di SuperScript II™ RT con ridotta attività RNasi H, e Platinum® TaqDNA Polymerase. Il primo presenta un tempo di dimezzamento più lungo (220 minuti a 50 °C) ed aumentata termostabilità rispetto allʼenzima non mutato. A 20,0 µl di una miscela contenente 5,0 µl di Ultrasense reaction mix 5X, 1,0 µl di Primer Forward 12,5 µM e 1,0 µl di Primer
!
""! reverse 22,5 µM, 1,0 µl di Probe 6,25 µM, 0,5 µl di Rox reference dye (50X), 1,25 μl di RNA Ultrasense enzyme mix e 10,25 µl di H2O sono stati aggiunti 5,0 µl di RNA virale estratto.
Alla reazione di retrotrascrizione, effettuata a 55 °C per 60 minuti, poi a 95 °C per 5 minuti (denaturazione), ha fatto seguito la reazione di PCR, 45 cicli di amplificazione, denaturazione a 95 °C per 15 secondi, annealing a 60 °C per 1 minuto ed estensione a 65° C per 1 minuto.
Tabella 3.3. Parametri di amplificazione delle one-step TaqMan Real-Time RT-PCR utilizzando il kit Invitrogen RNA Ultrasense One-step qRT-PCR system
Tabella 3.2. Composizione delle one-step TaqMan Real-Time RT-PCR mastermix utilizzando il kit Invitrogen RNA Ultrasense One-step qRT-PCR system
In ogni reazione di amplificazione sono stati inclusi un controllo negativo (acqua RNAsi Free) ed un controllo positivo (NoV genogruppo I e/o II e/o HAV).
La polimerasi permette lʼamplificazione attraverso PCR di un frammento di un gene specifico di Norovirus genogruppo I e II e di HAV. Unʼulteriore garanzia di specificità dellʼamplificazione è ottenuta in Real-Time attraverso lʼibridazione dellʼamplicone con una sonda TaqMan specifica per NoV genogruppo I e II e per HAV. Lʼamplicone specifico per NoV Genogruppo I e II e per HAV è misurato mediante fluorescenza FAM.
3.8. C
ALCOLO DELLʼ
EFFICIENZA DI ESTRAZIONE CONFCV.
Al fine di valutare lʼefficienza di estrazione, 24 campioni di M. edulis, 24 di D. trunculus, 24 di C. gigas e 12 di C. gallina sono stati contaminati con 10,0 μl di sospensione del controllo di processo (FCV) e sottoposti al protocollo di estrazione virale e di estrazione dellʼRNA virale mediante i due kit di estrazione, il QIAamp Viral RNA Mini Kit (Kit 1) ed il NucliSENS® MiniMAG® (Kit 2).
Lʼefficienza di estrazione viene calcolata mediante il confronto del Ct ottenuto dal campione contaminato con il Ct del controllo di processo (Feline Calicivirus).
Il controllo di processo è stato riscaldato a 99,0 ± 2,0 °C per 5 minuti ± 30 secondi in modo da rilasciare lʼRNA.
LʼRNA estratto è stato sottoposto ad amplificazione ed è stata valutata lʼefficienza di estrazione mediante la seguente formula:
!
"#! E = 2 -∆Ct d
E= Efficienza di estrazione
∆Ct = Ct campione - Ct controllo processo
Ct = ciclo soglia medio
d = diluizione
(Di Pasquale et al., 2010)!!
3.9. C
ALCOLO DELLʼ
EFFICIENZA DI AMPLIFICAZIONE DIN
OV
GII.
LʼRNA estratto dallʼepatopancreas dei molluschi bivalvi è stato sottoposto alla valutazione dellʼefficienza di amplificazione nei diversi campioni.
Per la valutazione dellʼefficienza di amplificazione per NoV GII sono stati utilizzati 24 campioni di M. edulis, 24 di D. trunculus, 24 di C. gigas e 12 di C. gallina. Cinque microlitri di RNA dei campioni sono stati contaminati con 1,0 µl di RNA di NoV GII in duplice copia nella stessa reazione. Nella stessa reazione 5,0 µl di acqua per biologia molecolare sono stati anchʼessi contaminati con 1,0 µl di RNA di NoV GII ed utilizzati come standard per il calcolo dellʼefficienza di amplificazione.
Lʼefficienza di amplificazione viene calcolata mediante il confronto del Ct ottenuto dal campione addizionato con il controllo esterno con il Ct del controllo esterno (alla medesima diluizione).
La valutazione dellʼefficienza di amplificazione è stata calcolata con la seguente formula:
E= 2 -∆Ct
E= Efficienza di amplificazione
∆Ct = Ct campione – Ct controllo esterno
Ct = ciclo soglia medio
(Di Pasquale et al., 2010)
3.10. A
NALISI STATISTICA.
Sui dati ottenuti è stata effettuata lʼanalisi della varianza (ANOVA) mediante il software statistico R (R Development Core Team, 2011): per ogni matrice sono stati analizzati i dati relativi sia allʼefficienza di estrazione che allʼefficienza di amplificazione confrontando i due diversi kit utilizzati.
Inoltre sono state confrontate lʼefficienza di estrazione e lʼefficienza di amplificazione tra le diverse matrici a seconda della metodica di estrazione utilizzata (in questo caso i campioni di C. gallina non sono stati considerati, in quanto numericamente inferiori) mediante analisi della varianza e il test per confronti multipli Student-Newman-Keuls mediante il software statistico Primer (Glantz, 2003).
!
"#! I dati ottenuti sono stati inoltre analizzati per valutare il soddisfacimento dei requisiti previsti dalla procedura utilizzata, ovvero efficienza di estrazione maggiore dellʼ1% ed efficienza di amplificazione maggiore del 50%. In questo caso è stato effettuato il test del !2 mediante il software statistico Primer (Glantz, 2003).
3.11. P
ROTOCOLLO DI TRASCRIZIONE INVERSA(RT).
La RT è stata effettuata utilizzando il kit commerciale AMV Reverse Trascriptase (Finnzymes, Vantaa, Finland).
A 10,0 µl di una miscela contenente 2,0 µl di buffer 10X, 1,0 µl di dNTPs 20 mM, 0,4 µl di Primer reverse 20 µM, 0,4 µl di enzima RT e 6,2 µl di H2O sono stati aggiunti 10,0 µl di RNA virale. LʼRNA virale di NoV GI, NoV GII e HAV è stato estratto, rispettivamente, da campioni di D. trunculus positivi per NoV GI, da sospensione virale di NoV GII derivante da materiale fecale (Laboratorio di Igiene e Virologia ambientale del Dipartimento di Biologia dellʼUniversità di Pisa) e da sospensione virale di HAV derivante da coltivazione su linee cellulari (Laboratorio di Riferimento per il Monitoraggio delle Contaminazioni Virali dei Molluschi Bivalvi dellʼIstituto Superiore di Sanità).
La reazione di retrotrascrizione è stata effettuata a 65 °C per 5 minuti, 42° C per 60 minuti ed a 95° C per 5 minuti.
3.12. P
ROTOCOLLO DIPCR.
DNA Polymerase (Invitrogen, France).
A 45,0 µl di una miscela contenente 5,0 µl di buffer 10X, 4,0 µl di dNTPs 2,5 mM, 1,5 µl di MgCl2 50 mM, 1,0 µl di Primer forward 12,5 µM e 1,0 µl di Primer reverse 22,5 µM, 0,2 µl di Taq Platinum e 32,3 µl di H2O sono stati aggiunti 5,0 µl del prodotto di RT (precedente reazione).
La reazione di PCR è stata effettuata con un protocollo termico che ha previsto, dopo 5 minuti a 95 °C necessari per la denaturazione, 45 cicli di amplificazione, con denaturazione a 95 °C per 15 secondi, annealing a 60 °C per 1 minuto ed estensione a 65° C per 1 minuto, a cui è seguita unʼestensione finale a 65° C per 10 minuti. I prodotti di PCR sono poi stati controllati su gel dʼagarosio 1% mediante corsa elettroforetica.
3.13. P
URIFICAZIONE CDNA
DA UTILIZZARSI PER COSTRUZIONE PLASMIDE.
Il frammento di nostro interesse, retrotrascritto a cDNA dalla reazione di RT ed amplificato dalla reazione di PCR, è stato controllato su gel dʼagarosio 2,5% per valutare che le dimensioni dellʼamplificato corrispondessero a quelle del frammento target. La banda di cDNA prelevata per mezzo di un bisturi sterile è stata purificata mediante il kit NucleoSpin®Extract II (MACHEREY-NAGEL, Duren, Germania). Il cDNA così purificato è stato conservato a -20 °C fino al momento dellʼutilizzo.!
"#!
3.14. C
OSTRUZIONE PLASMIDE.
Sia per NoV GI e GII che per HAV è stato costruito il plasmide contenente il retrotrascritto. Il cDNA purificato è stato clonato nel vettore plasmidico pCR®II TA Cloning Kit Dual Promoter (Invitrogen, France) mediante ligation a 14 °C over night, come da indicazioni fornite dal produttore.
Il prodotto di ligation è stato quindi inserito in cellule di Escherichia coli competenti (INVαFʼ) mediante trasformazione per shock termico (30 secondi a 42 °C).
Le cellule sono state trasferite in ghiaccio per 2 minuti e successivamente è stato aggiunto 1 ml di S.O.C. Medium (Super Optimal broth with Catabolic repressor) stabilizzato a temperatura ambiente. La provetta contenente le cellule trasformate è stata poi incubata a 37 °C per 1 ora in agitazione a 225 rpm in posizione orizzontale. In seguito la provetta è stata centrifugata a 3000 rpm per 5 minuti, eliminati 850 μl di surnatante e risospeso il pellet nei 150 μl di terreno rimasti. La sospensione è stata seminata su piastra Petri contente LB-K Agar (Luria Bertani Agar Medium + Kanamicina, 1000X) e X-Gal (40 mg/ml) e la piastra è stata incubata a 37 °C overnight.
Lʼavvenuta trasformazione delle cellule competenti INVαFʼ con il vettore pCR®II è stata verificata mediante la crescita delle cellule su LB-K. Il vettore pCR®II presenta, infatti, il gene per la kanamicina resistenza (posizione 1359-2153 basi), che permette di effettuare la selezione ed il mantenimento di E. coli. La crescita su LB-K Agar è quindi indice di avvenuta trasformazione delle cellule.
Lʼavvenuto inserimento dellʼamplificato nel plasmide è stato verificato mediante lo screening della colorazione delle colonie. Le colonie batteriche con un gene lacZ funzionale, ovvero non interrotto dallʼinserto, presentano una colorazione blu; lʼX-Gal
infatti viene scisso dalla !-galattosidasi producendo galattosio e 5-bromo-4-cloro-3-idrossindolo, che viene ossidato in 5,5ʼ-dibromo-4,4ʼ-dicloro-indaco, un composto blu insolubile. Lʼavvenuta ligation del prodotto di PCR nel vettore pCR®II allʼinterno del gene lacZ" non permette lʼespressione della !-galattosidasi e quindi la manifestazione della colorazione blu delle colonie in presenza di X-Gal. Sono quindi state selezionate le colonie bianche.
Per verificare la presenza dellʼinserto allʼinterno del plasmide è stata effettuata inoltre una PCR di screening utilizzando i primer SP6 e T7 del plasmide esterni allʼinserto. Con un puntale sterile si preleva una piccolissima quantità delle colonie da testare e si inserisce nei tubi contenenti la mix. A 25,0 μl di una miscela contenente 5,0 μl di Buffer Mix 10X, 4,0 μl di dNTP 2,5 mM, 1,5 μl di MgCl2 50 mM, 1,25 μl di Primer Forward SP6 12,5 μM, 1,25 μl di Primer Reverse T7 1,5 μM, 0,5 μl di Platinum® Taq DNA Polymerase (Invitrogen, France) e 11,5 μl di H2O è stata aggiunta una piccolissima quantità delle colonie da testare. Il protocollo termico prevede, dopo un primo ciclo di 10 minuti a 94 °C per favorire lʼestrazione del DNA dalle colonie, 25 cicli di amplificazione con 1 minuto di denaturazione a 94 °C, 1 minuto di annealing a 46 °C (temperatura di annealing necessaria per questi primer) ed 1 minuto di estensione a 72 °C.
Lʼamplificazione del frammento SP6-T7 (senza inserto) presenta una lunghezza di 184 bp, ed è indice di mancata ligation dellʼinserto nel plasmide. Lʼamplificazione di un frammento più lungo (184 + numero di basi del prodotto di PCR utilizzato per effettuare la ligation) indica lʼavvenuta ligation allʼinterno del plasmide.
!
"#!
Amplificazione frammento SP6-T7 senza inserto 184 bp
Amplificazione frammento SP6-T7 con inserto NoV GI 184 + 86 = 270 bp
Amplificazione frammento SP6-T7 con inserto NoV GII 184 + 89 = 273 bp
Amplificazione frammento SP6-T7 con inserto HAV 184 + 174 = 358 bp
Lʼassenza di amplificazione indica la mancata presenza del plasmide nelle cellule. In ogni reazione effettuata è stata testata anche una colonia blu come controllo negativo di ligation.
In seguito le colonie bianche risultate positive alla PCR di screening per la presenza dellʼinserto sono state prelevate e poste in beute contenenti 50 ml di LB Medium in agitatore overnight a 37 °C.
Il plasmide dalle colonie risultate positive è stato purificato mediante il Pureyield Plasmid Midiprep System (Promega, Milano, Italia) secondo le indicazioni fornite dal produttore.
3.15. T
RASCRIZIONE DELLʼ
INSERTO.
Il DNA plasmidico contenente lʼinserto, dopo quantificazione allo spettrofotometro, è stato linearizzato mediante taglio enzimatico a 37 °C per 2 ore utilizzando lʼenzima di restrizione SpeI (M•MEDICAL, Milano, Italia) seguendo le indicazioni fornite dalla ditta produttrice. Lʼenzima è stato disattivato mediante trattamento a 70 °C per 20 minuti. Il DNA plasmidico così linearizzato è stato purificato mediante il kit NucleoSpin®Extract II (MACHEREY-NAGEL, Duren, Germania), trascritto in RNA
mediante il kit T7 RibomaxTM Express Large Scale RNA Production System (Promega, Milano, Italia) per 4 ore a 37 °C.
LʼRNA del trascritto è stato poi estratto mediante la metodica fenolo/cloroformio/alcool isoamilico; dopo lʼaggiunta di 1 volume di fenolo/cloroformio/alcool isoamilico (rapporto 125:24:1) la provetta è stata centrifugata a 13˙000 rpm per 2 minuti. Il surnatante è stato trasferito in una nuova provetta ed è stato aggiunto 1 volume di cloroformio/alcool isoamilico (24:1) e centrifugato nuovamente a 13˙000 rpm per 2 minuti. La fase acquosa è stata trasferita in una nuova provetta ed il cloroformio è stato rimosso con brevi centrifugazioni seguite dalla rimozione della fase sedimentata. Sono stati poi aggiunti 0,1 volumi di sodio acetato 3 M pH 5,12 e 2,5 volumi di alcool etilico al 95%, mixati e posti in ghiaccio per 5 minuti. In seguito la provetta è stata centrifugata a 13˙000 rpm per 10 minuti e, una volta eliminato il surnatante, il pellet è stato lavato con 1 ml di alcool etilico al 70%. Infine il pellet è stato risospeso in 100 μl di buffer TE e stoccato a -80 °C.
LʼRNA del trascritto così purificato è stato utilizzato come controllo positivo nella reazione di Real Time RT-PCR e per la valutazione dellʼefficienza di amplificazione come previsto dal protocollo adottato.
3.16. Q
UANTIFICAZIONE E TITOLAZIONEDNA
PLASMIDICO.
In seguito alla purificazione del DNA plasmidico contenente lʼinserto sia per NoV GI e GII che per HAV si è proceduto alla quantificazione mediante lettura spettrofotometrica alla lunghezza dʼonda di 260 nm. I tre diversi plasmidi sono stati
!
"#! quantificati mediante diluizione 1:50 e si è potuto così calcolare la loro concentrazione (ng/μl) mediante la seguente formula:
Concentrazione DNAplasmide (ng/μl) = A 260 nm x fd x fc
Dove fd è il fattore di diluizione e fc è il coefficiente di estinzione molare medio, che dipende dalla natura dellʼacido nucleico:
Acido nucleico fc
(ng/μl)
DNA ds 50
DNA ss 33
RNA ss 40
Successivamente è stato calcolato il peso di un singolo plasmide in Dalton (Da) mediante la seguente formula:
Peso singolo plasmide (Da) = n° bp plasmide x peso molecolare medio Deossiribonucleotidi monofosfati (Da) x 2
Dove il peso molecolare medio dei Deossiribonucleotidi monofosfati è 330 Da e il n° bp plasmide è 3971 + bp inserto.
In seguito, mediante il programma Open Access UnitConversion (www.unitconversion.org), è stato calcolato il peso di un singolo plasmide da Dalton
(Da) in nanogrammi (ng) ed è stato calcolato il numero di copie plasmide/μl mediante la seguente formula:
N° copie plasmide/μl = concentrazione DNA plasmide (ng/μl) / peso singolo plasmide (ng)
I tre diversi plasmidi sono poi stati diluiti con diluizioni scalari 1:10 e sottoposti a Real Time PCR utilizzando il kit commerciale Platinum! qPCR SuperMix-UDG (Invitrogen, France) mediante un protocollo termico che ha previsto, dopo 5 minuti a 95 °C necessari per la denaturazione, 45 cicli di amplificazione, con denaturazione a 95 °C per 15 secondi, annealing a 60 °C per 1 minuto ed estensione a 65° C per 1 minuto. A 20,0 µl di una miscela contenente 12,5 µl di Platinum! qPCR SuperMix-UDG, 1,0 µl di Primer Forward 12,5 µM e 1,0 µl di Primer reverse 22,5 µM, 1,0 µl di Probe 6,25 µM e 4,5 µl di H2O sono stati aggiunti 5,0 µl del DNA plasmidico diluito in serie 1:10.
In questo modo è stato possibile calcolare una retta di taratura mediante i Ct medi delle varie diluizioni dei diversi plasmidi.
3.17. Q
UANTIFICAZIONE E TITOLAZIONE DELLʼRNA
TRASCRITTO.
Successivamente il DNA plasmidico contenente lʼinserto di NoV GI e GII è stato trascritto in RNA (come descritto precedentemente) ed anche lʼRNA è stato quantificato mediante lettura spettrofotometrica alla lunghezza dʼonda di 260 nm. I trascritti sono stati quantificati mediante diluizione 1:50 e si è potuto così calcolare la loro concentrazione (ng/μl) mediante la seguente formula:
!
"#! Concentrazione RNA trascritto (ng/μl) = A 260 nm x fd x fc
Dove fd è il fattore di diluizione e fc è il coefficiente di estinzione molare medio.
In seguito i trascritti di NoV GI e GII sono stati diluiti con diluizioni scalari 1:10 e sottoposti a Real Time RT-PCR utilizzando il kit commerciale “RNA UltraSense One-Step Quantitative RT-PCR System” (Invitrogen, France) seguendo il protocollo termico di RT-PCR precedentemente descritto. In questo modo è stato possibile quindi anche calcolare una retta di taratura mediante i Ct medi delle varie diluizioni dei diversi trascritti.