1
Summary
Facioscapulohumeral muscular dystrophy (FSHD) has been classified as an autosomal dominant myopathy, with an almost full penetrance (>95%) by age of 20 years, associated to rearrangements occurring in a 3.3 kb repeated sequence (D4Z4) located at the 4q subtelomeric region. For last 20 years, the diagnosis of FSHD in clinical practice has been performed by DNA testing, which has been considered highly sensitive and specific. It was established that normal subjects carry on alleles greater than 45 kb (≥ 11 D4Z4 repeats), whereas alleles of 35 kb, corresponding to 8 D4Z4 units, or shorter, are present in the majority of either de novo or familial FSHD patients. An inverse correlation between the number of D4Z4 repeats and the severity of FSHD has been also traditionally reported. Nevertheless, since the advent of genetic diagnosis, several reports have described FSHD families with subjects carrying D4Z4 reduce alleles and no signs of disease, these defined as non-penetrant carriers. Moreover, a growing number of evidences has emerged to complicate the evaluation of patients, reporting a wide and unexpected variability of FSHD clinical features and outcomes, also among subjects carrying the same D4Z4 allele, even within the same family. Then, it has been proposed that the reduction of D4Z4 repeats on chromosome 4q35 could be pathogenic only in a certain chromosomal backgrounds, defined as “permissive” specific haplotypes. However, the recent studies performed by the Italian Network for FHSD show that the current DNA signature of FSHD is a common polymorphism and suggest that the genetic basis of the disease should be revisited in consideration of the important implications for diagnosis and genetic counseling of at-risk families.
The present thesis is an observational cross-sectional analysis performed on the largest cohort of FSHD families described to date, accrued through the Italian National Registry for FSHD. The aim of the study has been to evaluate the clinical expression of FSHD in relation to D4Z4 reduced allele, 4q haplotype, age, gender and degree of kinship, in order to re-establish the prognostic value of D4Z4 reduced allele and identify other possible predictors of disease outcome in clinical practice.
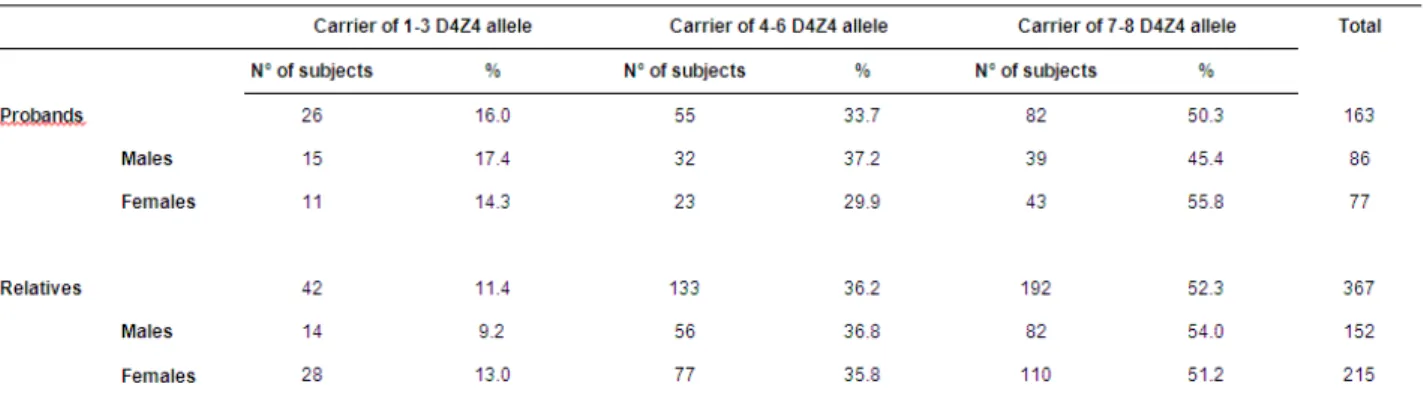
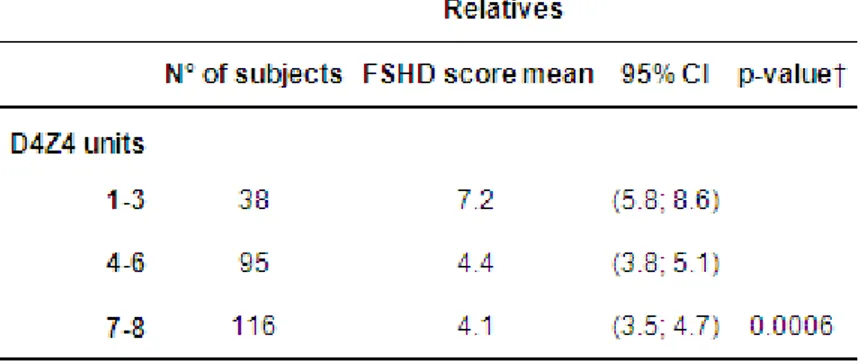
2 The analysis, performed on 367 relatives from 176 unrelated FSHD families, reveals an almost complete penetrance only in carriers on reduced allele with 1-3 D4Z4 repeats. Instead, the risk of developing FSHD is around 36% in both groups of subjects carrying 4-6 and 7-8 D4Z4 repeats by age of 30 and progressively increases with age, until 71,5% and 62,9% respectively by age of 60. The study fails to identify difference in term of penetrance and age-related risk to develop FSHD between carriers of “permissive” haplotype in comparison with the “non-permissive” haplotype, thus indicating that 4q35 haplotypes cannot have a clinical predictive value. Interestingly, a higher percentage of asymptomatic subjects between relatives with lower degree of relationship with proband is observed, suggesting a possible role of the “genetic background” and a more complex mode of inheritance in FSHD. The analysis of clinical expression on affected carriers shows that subjects carrying 1-3 D4Z4 repeats are characterized by a more precocious age at onset and by a more severe phenotype, while a wide clinical variability is observed among carriers of 4-8 D4Z4 repeats, ranging to asymptomatic-paucisymptomatic to severely affected. Finally, the discover of gender differences in term of mean age at onset and severity of disease expression proves that the female gender plays a protective effect for disease expression, suggesting variables related to gender, including hormonal and/or lifestyle factors, as possible contributing factors to be further investigated.
In conclusion, this analysis definitively confirms the previous observations about the inter- and intra-familial great variability in clinical expression, strengthening the notion that FSHD is a complex disease, whose pathogenic mechanisms, at present, are not clear. The work of the present thesis indicates that, starting from a systematic analysis of FSHD families with defined mode of inheritance and phenotypic features, it turns relevant indicators of prognostic value, these useful tools in the clinical practice, but also as incitement to further investigate the pathogenesis of FSHD.
3
Index
Introduction ... 5
Definition of diagnostic criteria for FSHD [Padberg et al, 1991] ... 6
Clinical diagnostic criteria ... 6
Genetic diagnostic criteria ... 7
Laboratory diagnostic criteria... 8
The discovery of DNA alterations associated with FSHD ... 8
The identification of specific haplotypes associated with D4Z4 reduced alleles in FSHD... 11
Molecular basis of FSHD: pathogenetic hypothesis ... 13
FRG1 as possible candidate gene for FSHD ... 13
Epigenetic hypothesis: chromatin modifications in FSHD ... 16
Hypothetical unifying pathogenic model in FSHD: the role of DUX4 gene ... 16
Genotype-phenotype correlation studies in FSHD ... 18
Size of D4Z4 allele and clinical expression ... 18
Penetrance of disease in carriers of D4Z4 reduced allele ... 21
Faciocapulohumeral muscular dystrophy without D4Z4 repeats contraction on chromosome 4q35 (FSHD2) ... 24
Atypical phenotypes associated with D4Z4 reduced alleles: clinical subtypes of FSHD or more complex myopathic conditions? ... 25
“Double trouble” conditions in FSHD families ... 29
The Italian National Registry for FSHD ... 31
The first results from the Italian National Registry for FSHD ... 33
A standardized clinical evaluation of patients affected by facioscapulohumeral muscular dystrophy: the FSHD clinical score [Lamperti et al., 2010] ... 33
New insights from compound heterozygotes and implication for prenatal genetic counseling in facioscapulohumeral dystrophy [Scionti, et al., 2012a] ... 36
Large scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy [Scionti et al., 2012b] ... 37
Genotype-phenotype correlation study from the Italian National Registry for facioscapulohumeral muscular dystrophy ... 39
Aims ... 39
Methods ... 39
Study design and subjects selection ... 39
Molecular characterization... 40
4
Results... 43
FSHD expression is not complete among carriers of D4Z4 reduced allele ... 43
Phenotypic variability in correlation with D4Z4 reduced allele size ... 44
Gender differences in disease expression ... 45
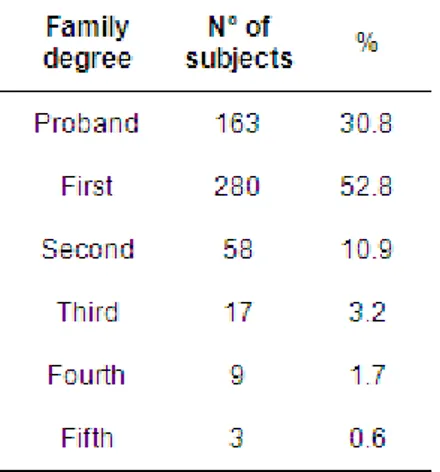
Influence of degree of kinship the disease outcome ... 45
Index cases show a more severe phenotype with gender differences ... 46
Discussion ... 46
Tables ... 52
Figures ... 59
5
Introduction
Facioscapulohumeral muscular dystrophy (FSHD, OMIM #158900) is the third most common form of hereditary myopathy with a prevalence of 1 in 20.000 [Mostacciuolo et al., 2009].
Duchenne de Boulogne firstly reported the FSHD syndrome: in 1862 he published one photo of an affected patient in his Album de photographies pathologiques [Duchenne, 1862] and in 1869 the photos of the same patient’s family in the Revue photographique des hopitaux de Paris [Duchenne , 1869]. He also firstly described the disease in his famous series of papers in Archives of General
Medicine in 1868, that is often cited as the earliest reference of the disease [Engel and
Franzini-Armstrong, 2004]. In 1885 the disease was called “Landouzy-Dejerine form of muscular dystrophy”, since the detailed description performed by Landouzy and Dejerine [Landouzy-Dejerine, 1885]. Subsequent reports of large pedigrees suggested an autosomal dominant inheritance of the disease [Pearson, 1933]. In 1950 Tyler and Stephens described a large family affected by FSHD (1248 relatives from six generations), identifying 159 affected individuals and reporting the neurological examination of 58 patients. In this report they firstly highlighted FSHD as a progressive disorder characterized by a precocious and pronounced involvement of the brachioradialis muscles [Tyler and Stephens, 1950]. In 1954 Walton and Natrass identified the clinical criteria for the diagnosis, showing in their analysis the slowly progressive nature of the disease and its limited and asymmetric distribution of muscle weakness.
However, the thesis of Padberg in 1982 exhaustively reviewed the literature and identified the clinical features of the diseases through the examination of a large cohort of patient. Padberg investigated a group of 107 patients, whose 73 were symptomatic. Presenting symptoms were facial weakness in 10% of cases, shoulder weakness in 82% and foot extensor weakness in 8%. None of his patients had pelvic girdle or calf muscle weakness [in Padberg G. Facioscapulohumeral disease [thesis]. University of Leiden, The Netherlands, 1982].
6 Definition of diagnostic criteria for FSHD [Padberg et al, 1991]
To date, the thesis of Padberg in 1982 remains the most detailed clinical description of FSHD patients, that give the notion of the wide clinical variability and of the degree of variation within families.
On the basis of the thesis of Padberg, the diagnostic criteria for FSHD were subsequently defined by the International Consortium in 1991, before the identification of the molecular defect associated with the disease. In particular, the International Consortium identified the clinical, laboratory and genetic criteria for FSHD diagnosis, in absence of an available diagnostic DNA test. Importantly, this work also responded to the need to select individuals that could be included in the linkage analysis [Padberg et al., 1991]. In fact, uniform and well-established clinical criteria were considered necessary to support research activities directed towards identification of FSHD gene.
Clinical diagnostic criteria
Since FSHD was defined largely on clinical grounds, the following definitions were introduced: (i) "non-penetrance" refers to an obligate gene carrier without symptoms (complaints or subjective findings) or signs (objective phenomena) relating to the disease (ii) "pre-symptomatic" indicates that a person has no complaints (symptoms) related to the disease, but has muscle atrophy and weakness demonstrable by physical examination; and (iii) "symptomatic" refers to patients with complaints and objective findings related to the weakness and muscle atrophy of FSHD. It was established that presenting symptoms had to relate to weakness and wasting in facial or shoulder girdle muscles. Onset in pelvic girdle muscles was considered suggestive of alternative diagnoses, although a subsequent pelvic girdle involvement was common during the progression of FSHD. It was reported that the clinically recognizable age of onset is often very variable. The mean age of recognizable onset, at least by clinical examination, is in the second decade. Onset before the age of 5 years are considered rare, although possible; in these precocious cases, however, the involvement
7 of facial muscles are thought necessary for diagnosis of FSHD. Facial weakness is described affecting eye closure (orbicularis oculi) and peri-oral muscles (orbicularis oris) and it is reported in the vast majority of patients. In the absence of facial weakness, a diagnosis of FSHD can be accepted only if the majority of affected family members shows facial weakness. Facial weakness can be also result very mild and noticeable by asymmetry of facial expressions. The scapular fixators are reported as the muscles most prominently involved. Also the pectoralis major muscles results early affected in most cases. The deltoid muscles remain unaffected for a long period of time and often have a particular pattern of atrophy, i.e. partial and proximal.
A typical feature of FSHD is considered the asymmetric muscle involvement. Symmetric weakness and atrophy at presentation is unusual and can necessitate caution before accepting the diagnosis as FSHD.
Progression of the disease is highly variable, involving abdominal and foot extensor muscles at an early stage. Also, the pelvic girdle weakness and upper arm weakness may occur at any time after the onset of shoulder girdle weakness. Neck extensor, intrinsic hand and triceps surae muscle weakness is described as uncommon features of disease, but occasionally observed within families. However, pre-symptomatic cases are described at any age and appear to comprise approximately 30% of all cases in large families. In the symptomatic cases, the disease is progressive in the majority of cases, the rate of progression is variable. Rarely, there can be long periods of apparent arrest of progression.
Genetic diagnostic criteria
The pattern of inheritance in familial cases is considered autosomal dominant. Sporadic cases are also reported, although their frequency is unknown. Evidence for recessive inheritance are not substantiated.
8
Laboratory diagnostic criteria
Blood creatine kinase (CK) levels can be normal, but are often elevated, though rarely exceed five times the upper limit of normal. Electromyography (EMG) shows a myopathic pattern; some neurogenic features, including positive sharp waves, are present occasionally. Motor and sensory nerve conduction velocities are normal. Muscle biopsies may exhibit nonspecific myopathic changes. Cellular infiltrates are frequently observed in FSHD and can be extensive.
In summary, there were identified four main criteria which define FSHD. These are: (1) onset of the disease in facial or shoulder girdle muscles; sparing of the extra-ocular, pharyngeal and lingual muscles and the myocardium; (2) facial weakness in more than 50% of the affected family members; (3) autosomal dominant inheritance in familial cases; and (4) evidence of myopathic disease in EMG and muscle biopsy in at least one affected member without biopsy features specific to alternative diagnoses.
Clinical and laboratory features suggestive of alternative diagnosis are following: - involvement of extra-ocular, masticatory, pharyngeal and lingual muscle; - regression of symptoms and signs;
- presence of severe and diffuse contractures;
- involvement of myocardium with presence of cardiomyopathy; - persistently high CK values above five times the upper limit.
The discovery of DNA alterations associated with FSHD
The variability in expression, severity, age at onset and need for an accurate pre-symptomatic test have given impetus to an active search for the localization, identification, and characterization of the FSHD gene [Lunt et al., 1989]. An International Consortium for FSHD linkage was organized
9 to expedite this process, by sharing DNA probes, by exchanging information on the research techniques and by pooling data. From the combined results of genetic linkage, presented in 1988 at the first meeting of the Consortium, an initial exclusion map for FSHD was constructed [Sarfarazi et al., 1989; Jacobsen et al., 1990]. In 1990 the FSHD gene was assigned to chromosome 4 by positional mapping in 10 Dutch families [Wijmenga et al., 1990]; confirmation of this location was performed in other families and with additional probes [Upadhyaya et al., 1990, 1991]. Wiimenga and coworkers in 1991 reported that D4S139, a Variable Number Tandem Repeat structure (VNTR) locus, was much more closely linked to FSHD. Two-point linkage analysis between FSHD and D4S139 in nine informative families showed a maximum combined lod score. D4S139 was mapped to chromosome 4q35-qter by in situ hybridization, thus firmly establishing the location of the FSHD gene in the subtelomeric region of chromosome 4q. In 1992, the members of the International Consortium for linkage analysis of the FSHD gene had pooled data for joint analyses, in an attempt to determine the precise location of the FSHD gene and the order of four DNA markers on 4q35 region. Six laboratories determined a total of 3078 genotypes in 65 families, consisting of a total of 504 affected subjects and 559 unaffected subjects. For each marker, a mean of 648 meioses were informative. D4S139 and D4S163 were identified as the closest linked markers to the FSHD locus As result of this effort, the assignment of the FSHD locus on region 4q35 in a total of 65 families was definitively established. The gene responsible for FSHD was refined to a position distal to the D4S139 locus. The cosmid clone 13E, isolated in search for homebox genes, was subsequently mapped to 4q35, also distal to D4S139 [Wijmenga et al., 1992]. The subclone p13E-11 resulted to detect in healthy subjects a polymorphic EcoRI fragment, usually larger than 28 kilobases (kb). The analysis performed by Wijmenga and coworkers [1992] showed that in healthy individuals the majority (72%) of EcoRI fragment detected by p13E-11 were larger than 28 kb, while in FSHD patients there was an overrepresentation of fragment smaller than 28 kb.
10 In conclusion, the hybridization of restriction enzyme EcoRI digested DNA using the p13E-11 probe identified a 3.3 kb tandemly repeated sequence (D4Z4) located at the 4q subtelomeric region, that resulted rearranged in almost all FSHD patients. Based on restriction fragment mapping and DNA sequencing, van Deutekom and coworkers [1993] confirmed that the rearrangements associated with FSHD result in deletion of central 3.2 kb repeat region of the EcoRI fragment.
Studies on the evolutionary distribution of 3.3-kb repeats revealed that in Old World monkeys 4qter contains the ancestral copy of this repeat; hybridization data have suggested that the copy number and organization of the 3.3-kb family are similar in chimpanzee and humans [Clark et al., 1996; Winokur et al., 1996].
D4Z4 is highly polymorphic with a VNTR structure [Hewitt et al., 1994] such that the variation in the size of EcoRI fragments is due to variability in the number of D4Z4 repeats [van Deutekom et al., 1993]. In humans, normal subjects carry p13E-11 EcoRI alleles usually ranges from 40 kb to approximately 200 kb (>10 D4Z4 units) originating from chromosome 4, whereas alleles of 35 kb (≤ 8 D4Z4 units) or shorter are present in the majority of either de novo or familial FSHD patients [Upadhyaya et al., 1993; Wijmenga et al., 1994; Lunt et al., 1995a].
It was also reported that the size distribution of approximately 10% of EcoRI fragments from the homologous polymorphic locus at chromosome 10q overlaps with the range seen in FSHD cases, and consequently many non-FSHD cases had shorter EcoRI fragment sizes, thus giving a reduced test specificity from EcoRI digest. In fact, it was demonstrated a 98% homology between 4q35 and 10q26 regions [Deidda et al., 1995], that is not confined to the 3.3-kb repeats but extends both proximally (42 kb) and distally to include the telomere [van Geel et al., 2002]. Despite the high homology between 4q35 and 10q26 regions, FSHD has been exclusively associated with reduction of D4Z4 repeat units on chromosome 4q [van Deutekom et al., 1996a; Matsumura et al., 2002]. Sometime 4q35 and 10qter p13E-11 alleles segregating in the same FSHD family can overlap on conventional agarose gel electrophoresis and make the interpretation of Southern blots difficult. The
11 p13E-11 probe has been hampered by the fact that it detects at least two pairs of EcoRI alleles, one derived from the 4q35 region (D4F104S1), the other from 10q26 (DlOF104S2). 4q35 and 10q26 EcoRI clones can be distinguished by restriction analysis with SfiI and Styl. The accurate comparison of nucleotide sequences between 4q35 and non-4q35 EcoRI fragments led to the identification of restriction enzymes able to cut specifically in either one of the alleles, facilitating the interpretation of the p13E-11 hybridisation patterns. The detection of sequence divergence between the KpnI tandem repeat units located at 4q and 10q showed a different distribution of restriction enzyme sites and that the restriction enzyme BlnI specifically cleaved the variable KpnI region of the 10qter p13E-11 fragments, leaving intact the tandem repeat units at 4q and thus allowing the direct identification of 4q35 alleles implicated in the disease [Deidda et al., 1996; Upadhyaya et al., 1997].
Therefore, a double restriction enzyme digest with the enzymes EcoRI and BlnI was routinely used, greatly facilitating the molecular diagnosis of FSHD.
Approximately half of new FSHD cases arise as a consequence of a postzygotic rearrangement of the repeat leading to somatic mosaicism for the D4Z4 repeat contraction [Lemmers et al., 2004a]
The identification of specific haplotypes associated with D4Z4 reduced alleles in FSHD
Since there are individuals with reduced D4Z4 alleles that do not have clinical signs of FSHD, it has been proposed that additional DNA sequences flanking the D4Z4 repeat array are necessary for disease development.
In 2002 a polymorphic segment of 10 kb directly distal to D4Z4 existing in two allelic forms, 4qA and 4qB, was identified [van Geel et al., 2002]. Although both alleles are equally common in the general population, it was reported that FSHD is solely associated with the 4qA allele. Lemmers
12 and coworkers, in 2002, analyzed 80 healthy controls and 80 unrelated individuals with FSHD for the presence of 4qA and 4qB alleles. In the controls they observed almost equal frequencies of 4qA and 4qB alleles (42% and 58% respectively) on chromosome 4, but only alleles of the 4qA type on chromosome 10. By contrast, in the 80 unrelated individuals with FSHD (44 de novo cases and 36 unrelated familial cases) they detected D4Z4 contractions exclusively in chromosomes 4 bearing the 4qA allele, and never in those with the 4qB allele [Lemmers et al. 2002]. Subsequently, three families with FSHD in which each proband carried two FSHD-sized alleles and was heterozygous for the 4qA/4qB polymorphism were identified [Lemmers et al., 2004b]. Segregation analysis demonstrated that FSHD-sized 4qB alleles were not associated with disease, since these were present in unaffected family members. Thus, the authors supposed that, in addition to a contraction of D4Z4, additional cis-acting elements on 4qA might be required for the development of FSHD. Alternatively, 4qB sub-telomeres might contain elements that prevent FSHD pathogenesis.
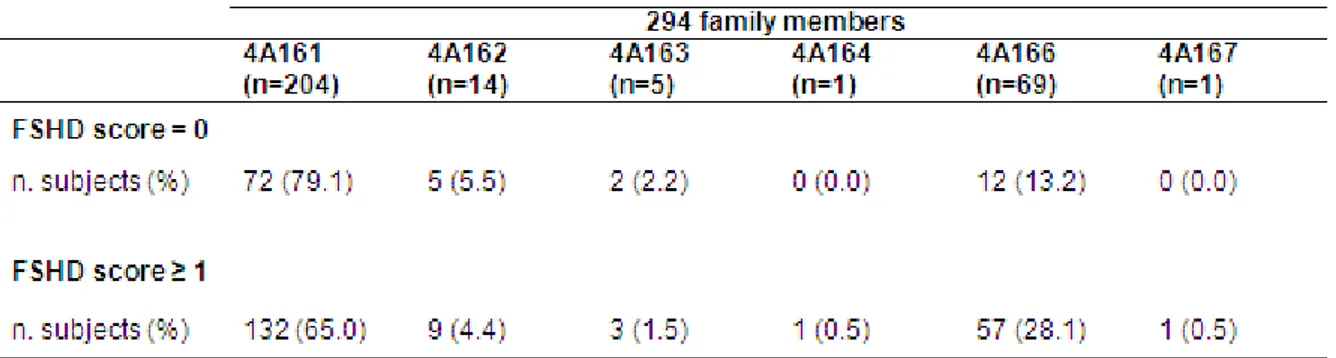
In 2007, the identification of additional sequence variations in a relatively stable Simple Sequence Length Polymorphisms (SSLP) proximal to the D4Z4 repeat was identified in the FSHD locus [Lemmers et al., 2007]. On the basis of the proximal SSLP, it was possible subdivide chromosome 4 into at least 17 genetically distinct telomeric variants and chromosome 10 into 8 telomeric variants. The contractions in only three genetically almost identical chromosome 4 sub-telomeres, the common variant 4A161 and the rare variants 4A159 and 4A168, caused FSHD, whereas contractions in other 4q sub-telomeres were not associated with disease. The authors reported the pedigrees of two FSHD families in which two different short alleles segregated, observing that the subjects carrying the D4Z4 reduce allele in association with the non-permissive 4qA166 haplotype did not manifest signs of disease.
Finally, it has been suggested that FSHD patients carry specific single nucleotide polymorphism (SNP) ATTAAA in the chromosomal region distal to the last D4Z4 repeat in the pLAM1 sequence of the 4qA alleles, that provides a PolyAdenylation Signal (PAS) [Lemmers et al., 2010]. Thus, the
13 molecular signature, named 4A(159,161,168)PAS, has been proposed to define alleles causally related to FSHD. This signature results from the combination of (1) a reduction in the number of D4Z4 elements, (2) the presence of the 4qA allele, and (3) the PAS in the pLAM1 sequence.
Molecular basis of FSHD: pathogenetic hypothesis
As discussed above, soon after the discovery of D4Z4 repeats on chromosome 4, it was established that the sub-telomere of chromosome 10q is almost identical to that of chromosome 4q and that it also contains a highly homologous and equally polymorphic repeat array. A considerable proportion of individuals in the population carry 4q or 10q chromosome ends with repeat arrays that have apparently been entirely or partially transferred between both chromosomes. However, the observation of linkage of the disease with chromosome 4 and the absence of linkage with chromosome 10 led to the hypothesis that the interplay between D4Z4 and other more proximal elements on chromosome 4 could explain the chromosome 4 specificity of the disease [van der Maarel et al., 2011].
Although it is thought that deletions of D4Z4 are causally related to FSHD, it is not clear how this triggers the disease. It has long been speculated that such deletions may alter the expression of genes located within or nearby the repeats.
FRG1 as possible candidate gene for FSHD
The region immediately proximal to the D4Z4 repeats harbors a number of candidate genes. This FSHD locus includes: i) FSHD-related gene 1 (FRG1), which encodes a nucleolar protein involved in RNA biogenesis [van Deutekom et al., 1996b; van Koningsbruggen et al., 2007]; ii) FSHD-related gene 2 (FRG2), a predicted transcript with no significant homology to any known protein;
14 iii) adenine nucleotide transporter 1 gene (ANT1), a gene involved in apoptosis, lying more distally from the 4qter (5.8 Mb) [Doerner et al., 1997; Bodega et al., 2009].
The overexpression of FRG1, FRG2, and ANT1 has been found in some muscles affected by FSHD [Gabellini et al., 2002; Laoudj-Chenivesse et al., 2005]. It has been shown that a transcriptional repressor complex binds D4Z4 unit, so it has been supposed that D4Z4 deletion would trigger the gene overexpression as result of the lack of repression [Gabellini et al., 2002]. Gabellini and coworkers, in 2002, found that, in FSHD muscle, 4q35 genes located upstream of D4Z4 resulted inappropriately overexpressed. In particular, it was shown that an element within D4Z4 specifically bound a multiprotein complex consisting of YY1, HMGB2 and nucleolin proteins. YY1 is a complex protein that is involved in repressing and activating a number of promoters, interacting with numerous key regulatory proteins [Thomas et al., 1999]. HMGB2 is a member of one of the three families of high mobility group (HMG) proteins, having an ‘architectural’ function and binding proteins that facilitate the assembly of multiprotein complexes on DNA [Agresti et al., 2003]. Nucleolin, which is an abundant protein of the nucleolus, has been implicated in chromatin structure, ribosomal RNA (rRNA) transcription, rRNA maturation, ribosome assembly and nucleo-cytoplasmic transport [Ginisty et al., 1999]. This multiprotein complex bound D4Z4 in vitro and in
vivo and mediated transcriptional repression of 4q35 genes. The authors hypothesized that deletion
of repeated elements in the sub-telomeric region of 4q might act on neighboring genes by derepressing their transcription and thus starting a cascade of events which ultimately lead to FSHD, also explaining the autosomal dominant transmission. This hypothesis results also consistent with the observation that haploinsufficiency of distal 4q does not cause FSHD [Tupler et al., 1996]. Interestingly, the extent of 4q35 gene overexpression in FSHD skeletal muscle resulted inversely related to the number of D4Z4 repeats, suggesting a direct correlation with disease severity. Moreover, the observation that 4q35 gene overexpression is muscle specific can explain the muscular phenotype observed in the disease. Finally, the stochastic variation in gene expression in
15 muscle cells may be responsible of the asymmetric muscle involvement and of the great clinical variability reported between and within families [Tupler and Gabellini, 2004].
Consistently with this hypothesis, interesting data come from the work of Gabellini and coworkers in 2006. In this study, a transgenic mice selectively overexpressing in skeletal muscle the 4q35
FRG1, FRG2 or ANT1 genes was generated. The authors found that FRG1 transgenic mice
developed a muscular dystrophy; by contrast, FRG2 and ANT1 transgenic mice resulted normal. The degree of mice muscle impairment appeared correlated with transgene expression levels: in particular, FRG1-low mice showed no evidence of kyphosis, whereas FRG1-intermediate and
FRG1-high mice exhibited mild and severe kyphosis respectively, due to muscle degeneration.
Skeletal muscle from FRG1 mice showed histological and ultrastructural dystrophic features characterized by the increase of fibers size variability, necrosis, nuclear centralizations and connective tissue. In the same study, the authors also found that in muscle cells from FRG1 transgenic mice and from FSHD patients, specific pre-mRNAs, such as fast skeletal muscle troponin T (Tnnt3) and myotubularin related protein 1 (Mtmr1), underwent aberrant alternative splicing. These genes resulted aberrantly spliced also in myotonic dystrophy patients and animal models [Buj-Bello et al., 2002; Kanadia et al., 2003], but not in muscle cell cultures derived from patients with Duchenne muscular dystrophy and congenital merosin deficient muscular dystrophy 1A [Gabellini et al., 2006].
Nevertheless, several follow-up studies could not reproduce the transcriptionally up-regulation of
FRG1, FRG2 and ANT1 in FSHD muscle [Winokur et al., 2003; Celegato et al., 2006; Osborne et
al., 2007]. The use of different techniques and different sources of RNA may partly explain this lack of reproducibility [de Greef et al., 2008].
16
Epigenetic hypothesis: chromatin modifications in FSHD
The D4Z4 repeat is GC-rich and contains sequences often residing in heterochromatic domains of the genome [Lyle et al., 1995]. DNA methylation analysis and studies of histone modifications has supported the hypothesis that the reduction of D4Z4 repeat, that normally is in a relatively closed chromatin configuration, causes a more open chromatin configuration facilitating the transcriptional activity of the repeat and possibly affecting the processing of the different D4Z4 transcripts [Jiang et al., 2003; van Overveld et al., 2003; Zeng et al., 2009]. In healthy subjects the D4Z4 repeats resulted densely methylated, while in FSHD subjects chromosomes presented almost 30–40% reduction of DNA methylation at specific sites tested in D4Z4 region. Moreover, chromatin immunoprecipitation studies showed that the D4Z4 repeat is normally occupied by both transcriptionally repressive as well as permissive histone modifications. In FSHD patients chromosomes, it is observed a relative loss of repressive histone modifications; these changes in chromatin structure are restricted to the D4Z4 repeat and do not seem to spread proximally. Chromatin immunoprecipitation studies also identified other chromatin factors that were lost or gained, including HP1γ, the cohesin complex, YY1 (lost) and CTCF (gained) at D4Z4 of disease alleles [Gabellini et al., 2002; Zeng et al., 2009; Ottaviani et al., 2009]. Interestingly, similar modifications of the chromatin structure in D4Z4 site were also reported in a small number of patients whose disease status could not supported by discovery of a D4Z4 reduced allele [Zeng et al., 2010; de Greef et al. 2009; de Greef et al., 2010].
Hypothetical unifying pathogenic model in FSHD: the role of DUX4 gene
Detailed sequence analysis revealed that the D4Z4 repeat contains the open reading frame (ORF) of a double-homeobox transcription factor, DUX4 [Hewitt et al., 1994], a 424-aa protein. Homeobox sequences, which are highly conserved during evolution [Gehring et al., 1990], encode homeodomains, that are characteristic domains of some gene regulatory proteins, homeobox proteins, coordinating the expression of sets of genes during development.
17 The DUX4 ORF is in a single exon, whereas other members of the double-homeobox family have multiple introns, indicating that DUX4 was inserted into the genome as a retrotransposed mRNA from an intron containing the DUX gene [Leidenroth et al., 2010]. In contrast to the many pseudogenes retrotransposed to our genome, the DUX4 retrogene maintains a conserved ORF [Clapp et al., 2007]. It was proposed that the contraction of the D4Z4 array results in the transcription of the DUX4 retrogene, although the abundance of the DUX4 mRNA and protein results extremely low [Dixit et al., 2007]. However, the study by Lemmers and coworkers [2010], reporting the requirement of DUX4 polyadenylation site for develop FSHD, have suggested a new developmental model for the disease that is consistent with the extremely low abundance of the mRNA and protein, providing support for the expression of DUX4 as a major cause of FSHD. The distal end of the repeat array and flanking pLAM1 sequences are thought to be crucially important for the development of FSHD. This hypothesis has been further corroborated by the finding of a FSHD family in which the disease segregated with a contracted D4Z4 allele of chromosome 10: importantly, the last part of this disease-associated repeat array was replaced by permissive chromosome 4 sequences. The identification of this family in which FSHD segregates with chromosome 10 has suggested the importance of the distal end of the repeat and pLAM1 sequences, apparently precluding a prominent role for other proximal candidate genes on chromosome 4. According to this hypothesis, the major transcript in each unit, the DUX4 gene, is not stable, probably due to the absence of a polyadenylation signal in internal D4Z4 units. Spliced and unspliced transcripts of the DUX4 gene in the last unit, however, use a unique 3′ untranslated region (UTR) in the pLAM1 region which is immediately distal to this last unit and which contains a poly(A) signal that presumably stabilizes this distal transcript. Consistently, by transfecting this crucial region in murine C2C12 muscle cells, stable DUX4 transcripts were identified when the constructs derived from permissive chromosomes with the polyadenylation signal. Therefore, these genetic studies seem to demonstrate the requirement for the polyadenylation site utilized by DUX4 mRNA and to implicate DUX4 protein as a cause of FSHD. However, as noted above, although
18
DUX4 mRNA was detected in FSHD muscle, it was still at extremely low abundance. It was
supposed that low abundance mRNA in a population of cells could reflect either a small amount of mRNA in all cells or an abundant amount of mRNA in just a few cells. RT-PCR amplification of
DUX4 mRNA in small pools of 100 or 600 differentiated FSHD muscle cells identified relatively
abundant transcripts in a subset of the pools [Snider et al., 2010]. Approximately 1 in 1000 FSHD muscle cell nuclei were detected with an abundant amount of DUX4 mRNA. The DUX4-expressing FSHD muscle nuclei had characteristics consistent with DUX4 induced toxicity, including an aggregation of nuclear DUX4 protein that occurs coincident with DUX4-induced apoptosis. Therefore, the very low abundance of DUX4 mRNA in FSHD muscle represented relatively abundant amounts of DUX4 mRNA and protein in a small subset of the nuclei, probably leading to dysfunction or death of those DUX4-expressing nuclei [van der Maarel et al., 2011].
In conclusion, in this unifying pathogenic model of FSHD, the inefficient chromatin-mediated repression, either related to the contraction of the array, may result in the occasional escape from repression in muscle cells, and possibly other somatic cells, with a consequently inappropriate expression of DUX4 protein [van der Maarel et al, 2011]. On this basis, healthy subjects carrying reduced D4Z4 alleles would be explained by the absence of the 4A(159,161,168)PAS [Lemmers et al., 2010].
Genotype-phenotype correlation studies in FSHD
Size of D4Z4 allele and clinical expression
Since the discovery of the FSHD molecular defect, genotype-phenotype studies have conducted in order to evaluate if the size of the EcoRI fragment could be correlated with the clinical manifestations and to assess the impact of the molecular defect on the phenotypic expression.
19 Lunt and coworkers in 1995 [Lunt et al., 1995b] reported the analysis performed on 14 FSHD families and 25 clinically isolated cases, presumed to be due to new mutation, associated with D4Z4 reduced allele (respectively in two groups the range in allele size was 19-30 and 13-24 Kb). The study revealed a clear correlation between smaller fragment sizes and earlier age at onset. The median age at onset on sporadic cases resulted 6.9 years (range <1-16 years) and on familial cases 18 years (range 8-23). Interestingly, the authors also observed within families a difference between generations in reported onset ages, with onset age becoming younger in successive generations, although it was hypothesized that this trend might be more a reflection of ascertainment bias than a biological anticipation. A similar correlation with fragment size was also observed for age to loss of ambulation in 16 subjects using a wheelchair [Lunt et al., 1995a]. The authors proposed that FSHD families could be divided broadly into three groups, necessarily with some overlap: i) new mutation cases with early onset (range <1-16 years), severe presentation, and small fragment size ≤18 kb; ii) large 'typical' families with median onset age ranging from 8-22 years associated with fragment size is 19-30 kb; iii) small families, often with a later onset presentation (median 15-23 years), or scapulohumeral presentation, in which a 4q35-cosegregating fragment of size 30-38 kb was present and non-penetrance may still be observed above 20 years of age.
The subsequent study of Tawil and coworkers in 1996 [Tawil et al., 1996] confirmed the same results, by examining the genotype in a clinically and genetically well-defined 157 FSHD subjects. In particular, this analysis showed the presence of anticipation and that the size of the deletion and the disease severity were closely related.
Three years later, Ricci et al. [1999], on a cohort of 165 patients with FSHD (range in size 10-27 Kb), further reported the inverse correlation between fragment size and clinical severity. The probability of developing a severe form of disease resulted 100% in the presence of very short fragment (1-2 D4Z4 repeats), decreased to 54% in patients carrying fragments of 16 to 20 kb (3-4 D4Z4 repeats) and dropped to 21% or less in patients carrying fragment larger than 20 kb (>4 D4Z4
20 repeats). The severe form of FSHD was defined when it was present: severe weakness of pelvic and proximal leg muscle or both (strength<3 in at least one of these muscle) with inability to stand up from a chair without support or to walk unaided, or wheelchair use.
In the genotype-phenotype correlation study performed by Tonini et al. in 2004 on 238 subjects from 106 unrelated families, it was observed that individuals with larger fragments showed a milder course while those who have the smaller ones were more severely affected. However, when genders are analyzed separately, this correlation was significant for females but not for males.
In 2003, Butz and coworkers conducted a systematic study of 39 unrelated FSHD patients with borderline D4Z4 repeat numbers and 102 healthy controls, in order to identify the molecular diagnostic cut-off point between FSHD cases and the control population and describe the phenotype in patients with borderline D4Z4 repeat numbers. The results indicated that there was not a definite D4Z4 diagnostic cut-off point separating FSHD, FSHD-like myopathies and healthy controls, without the expected correlation of D4Z4 repeat number and clinical severity. Therefore the authors suggested the D4Z4 cut off of 8 repeats [Butz et al., 2003].
In summary, the above studies showed an inverse correlation between the number of D4Z4 repeats and the severity of the disease. Alleles with 1-3 D4Z4 repeats are generally associated with a severe form of disease that presents in childhood, 4-7 D4Z4 repeats with the classical form of FSHD, and 8-10 D4Z4 repeats with a milder disease [Lunt et al., 1995a; Tawil et al., 1996; Ricci et al., 1999]. In addition, D4Z4 alleles between 38-45 kb in size (9-11 D4Z4 repeats) have been described both in normal and affected individuals and are considered as borderline [Butz et al., 2003; Vitelli et al., 1999].
Nevertheless, over the years since the advent of molecular diagnosis for FSHD, a growing number of evidences have emerged to complicate the evaluation of patients, reporting a wide and unexpected variability of FSHD clinical outcomes, also among subjects carrying the same D4Z4
21 allele, even within the same family [Fitzsimons, 1999; Galluzzi et al., 1999; Felice et al., 2000]. Interestingly, the marked phenotypic discordance observed was further supported by the observation of two sets of monozygous twins that, although carrying identical de novo small EcoRI fragments, presented a different clinical expression [Tupler et al., 1998; Sakellariou et al., 2012].
Penetrance of disease in carriers of D4Z4 reduced allele
In pre-genetic era, the first observations performed on large FSHD families suggested an almost complete penetrance of the disease [Bailey et al., 1986; Becker 1953, Tyler and Stevens 1950, Chung and Morton 1959]. However, the analysis of Padberg in 1982 revealed several mildly affected subjects who are unaware of symptoms, showing that the presence of recognizable characteristic clinical signs may precede the onset of symptoms in these 'abortive' cases for several years. Two main studies regarding the FSHD penetrance were performed by Lunt and coworkers respectively in 1989 and 1991. The estimation of age dependent penetrance based on the presence of the characteristic clinical signs was >95% by age 20, without difference between the families [Lunt et al 1989]. In particular, the penetrance of disease has been estimated <5% for ages 0 to 4 years, 21% for ages 5 to 9, 58% for ages 10 to 14, 86% for ages 15 to 19, and 95% penetrance for age 20 years and over at 95% for patients aged ≥ 20 years [Lunt et al., 1989]. The mode of inheritance was observed to be autosomal dominant.
FSHD: clinical criteria for affected status [Lunt et al.,
22 Nevertheless, over the years since the advent of molecular diagnosis for FSHD, subjects carrying D4Z4 reduced alleles without signs of disease have been reported, have suggesting a lower penetrance than expected. FSHD families including subjects carrying D4Z4 reduced alleles and no signs of the disease, defined as non-penetrant carriers, have been also reported [Griggs et al., 1993; Zatz et al., 1998; Ricci et al., 1999; Tonini et al., 2004; Scionti et al., 2012a; Scionti et al., 2012b], doubting the notion of almost full penetrance of disease. More interestingly, a gender difference in term of penetrance and severity of disease [Zatz et al., 1998; Tonini et al., 2004; Sakellariou et al., 2012] has been also reported, observing a more mild phenotype in females than males.
Lunt et al. [1995a] from analysis of 30 FSHD families (carriers of 19 to 38 kb fragment size), observed small families with 4q35 cosegregating fragments of 30 kb and 34 kb in which subjects who inherit this fragment resulted clinically unaffected above age 20 years, supporting a lower penetrance in these families. The authors hypothesized that in families with a larger fragment size, >30 Kb, penetrance of the FSHD gene was lower than previous estimated value of 95% above age 20 years.
In the study of Zatz et al. [1995] on 34 Brazilian FSHD families, two unrelated multigenerational families with multiple affected patients were described, where the parents of affected patients (a female aged 46 years and a male aged 79 years) resulted asymptomatic carriers of FSHD allele; the author did not report the correlation with the fragment size.
In the subsequent study of Zatz et al. [1998], the estimated penetrance for FSHD allele on 52 families with fragment smaller than 35 kb was 85% for patients until age 30; furthermore, when the authors considered the sexes separately, the estimated penetrance of the FSHD allele resulted significantly greater for males (95%) than for females (69%). Interestingly, among 27 families with at least two clinically affected patients it was observed that in 21 the pattern of inheritance was autosomal dominant (four of them with incomplete penetrance). In three pedigree the pattern of
23 inheritance was compatible with autosomal recessive since there were at least two affected sibs born from asymptomatic parents.
Ricci et al. [1999] reported 7 subjects, age 20 to 69 years, with fragment ranging in size between 21 and 37 kb, without symptoms or signs of FSHD, classified as non-penetrant carriers. In this study, unaffected individuals belonging to families with D4Z4 allele smaller than 20 kb were non observed.
Tonini et al. [2004], analyzing 238 subjects with reduced D4Z4 allele <35 kb from 106 unrelated families, observed that about 20% of individuals related to FSHD patients who carried a deleted EcoRI fragment remained asymptomatic or was minimally affected with a significantly higher proportion of females than males; asymptomatic carriers were found in about 30% of the families.
Finally, the most recent work of Sakellariou and coworkers [2012] reports the clinical and genetic analysis of 133 individuals carrying D4Z4 reduced allele (71 probands and 62 relatives) from 71 unrelated Greek families, revealing a high percentage (almost 50%) of asymptomatic relatives carrying a contracted 4q allele, older than 30 years. The percentage of unaffected carriers was lower between males. It is also noteworthy that 16 among the 38 multiple-case families (42%) were found to have at least one symptom-free individual, with a greater proportion of asymptomatic or minimally affected gene carriers concentrating in some pedigrees. A significant statistical association between the genders and the clinical manifestation of the disease was observed: in particular, among the females the percentage of the symptomatic patients was found to be 66.7% whereas among the males the same percentage was 86.6%.
24 Faciocapulohumeral muscular dystrophy without D4Z4 repeats contraction on chromosome 4q35 (FSHD2)
A subgroup of FSHD subjects, termed patients with FSHD2 (almost 5%), have no contraction of the D4Z4 repeat on chromosome 4q35 [Tawil et al., 2010], although they result clinically indistinguishable from patients with FSHD associated with D4Z4 reduced allele (also defined FSHD1). Recently, de Greef and coworkers [2010] have performed a cross-sectional study on 33 patients with FSHD2 from 27 families, the largest cohort described to date. In the above analysis, FSHD2 patients appeared identical to FSHD1 in clinical presentation. Of the 33 patients with FSHD2, 20 (61%) were male. The average age at symptom onset was 26 years (range 0–60), which is almost 10 years later than in FSHD1. The initial symptom was scapular weakness in 61%, foot dorsiflexor weakness in 27%, facial weakness in 10%, and hip girdle weakness in 3%. A gender differences in disease severity in FSHD2 was not observed. Interestingly, notable difference between FSHD1 and FSHD2 is the mode of inheritance. In fact, the analysis showed that he majority (20/33, 67%) were sporadic, 11 were familial, and in 2 the inheritance pattern was uncertain, suggesting that the familial to sporadic ratio in FSHD2 is inverse to the ratio in FSHD1. Of the familial cases, 3 resulted dominant in inheritance (parent-child pairs) and 2 seemed recessive in inheritance (sibling pairs). It has been suggested that similar epigenetic and molecular mechanisms, also supposed to be pathogenic in FSHD associated with D4Z4 repeat contraction, are involved. In particular, it was observed that, unlike FSHD1, patients with FSHD2 showed loss of DNA methylation on both chromosome 4q and 10q D4Z4 repeats, suggesting that a defect in establishing or maintaining the D4Z4 repeat chromatin structure may cause FSHD2 [de Greef et al., 2009]. In the study performed by de Greef and coworker in 2010, all patients with FSHD2 (33 subjects) carried at least 1 D4Z4 repeat on the permissive haplotype 4A161. The authors studied DNA methylation levels of the D4Z4 repeat at 4 methylation-sensitive restriction sites in patients
25 with FSHD2 and their unaffected relatives. The analysis showed that at the 4 sites tested, a significant D4Z4 hypomethylation was present in patients with FSHD2 as well as in patients with FSHD1. In particular, patients with FSHD2 showed significant D4Z4 hypomethylation on chromosomes 4q and 10q, while in patients with FSHD1 significant loss of D4Z4 methylation is restricted to the contracted chromosome 4q. The levels of D4Z4 hypomethylation in patients with FSHD2 were not correlated with phenotype severity. To explain the high percentage of FSHD2 sporadic cases, it was hypothesized that FSHD2 development may result from two independent events: D4Z4 hypomethylation by an unknown mechanism and presence of at least 1 repeat on the permissive 4A161 haplotype.
Atypical phenotypes associated with D4Z4 reduced alleles: clinical subtypes of FSHD or more complex myopathic conditions?
In last years, the use of the molecular analysis as diagnostic test has led to the identification of different phenotypes of the disease, expanding the clinical pattern associated with D4Z4 reduced allele. Several subtypes of FSHD with atypical clinical presentation have been described.
Firstly, van der Kooi and coworkers [2000] described six sporadic cases that did not meet most of the diagnostic criteria defined in 1991 but were diagnosed as FSHD by DNA testing, which showed small EcoRI fragments on chromosome 4q (range 26 to 38 kb). The predominant clinical features was in three patients the foot drop, in the others inability to walk on toes, shoulder pain and pelvic limb weakness with difficulty in walking respectively. None of them had apparent facial weakness, only one complained of weakness in the shoulders. Interestingly, none had a positive family history, however if all patients were really de novo mutations remained unclear because the parents had not undergo to DNA analysis.
26 In 2000, Felice and coworkers described 10 of the 14 patients with facial sparing scapular myopathy associated with restriction fragments consistent with the 4q35 deletion (range 20 to 39 kb). Except for the absence of facial weakness, most patients had clinical and laboratory features otherwise consistent with FSHD. Five patients referred also a positive family history of similar weakness, although the DNA analysis was not performed on other family members [Felice et al., 2000].
Felice and Moore [2001] described four patients, each harboring 4q35 deletions, presented with atypical phenotypes including facial-sparing scapular myopathy (25kb), limb-girdle muscular dystrophy (LGMD) (34kb), distal myopathy (30kb), and asymmetric brachial weakness (34 kb). Only the first two patients had been also performed muscle biopsy that showed unspecific dystrophic features, while the patients with LGMD-like asymmetric brachial weakness had not performed muscle biopsy. However, none of these patients had been subjected to other molecular investigations for differential diagnosis. Interestingly, the patients with LGMD phenotype and asymmetric brachial weakness did not report a positive family history for neuromuscular diseases, although his parents had not been investigated. In this work, the authors concluded that the availability of the DNA test allowed to definitively establish the diagnosis without the need for the more invasive and less specific muscle biopsy, considering DNA test highly sensible and specific.
Krasnianski et al. [2003] described three patients from a single family (father and two sons) in which a D4Z4 reduced allele of 20 kb segregated, showing signs consistent with diagnosis of typical FSHD phenotype (including facial weakness, symmetric-predominant proximal arm and shoulder girdle paresis and atrophy, bilateral scapular winging, lumbar hyperlordosis and prominent foot drop), also associated with chronic progressive external ophthalmoplegia. The oculomotor impairment was reported as the initial manifestation of disease since infancy. Only the father, 50 years old, fulfilled the diagnostic criteria of FSHD, while the two son at time of examination (age of 15) presented scapular winging and hyperlordosis. The muscle biopsy demonstrated prominent myopathic changes without ragged red fibers or histopathological features of other neuromuscular
27 diseases. The absence of singular or multiple deletions of mitochondrial DNA apparently excluded a coincidental diagnosis of CPEO of mitochondrial origin. On the other hand, the classic FSHD distribution of the muscle weakness had been never described in patients with CPEO. In the same paper [Krasnianski et al., 2003], the authors further described other two familial cases and one sporadic case with facial sparing FSHD syndrome associated with D4Z4 reduced allele (34 and 30 kb allele respectively).
More recently, five unrelated cases carrying D4Z4 reduced alleles presenting with an unusual phenotype and vacuolar myopathy with rimmed vacuoles were described [Reilich et al., 2010]. The atypical clinical features included a form of LGMD phenotype with facial sparing, a form of distal and proximal weakness, in one patient also associated with dysphagia, and a form of a prevalent asymmetric lower limb distal weakness. Other signs such as scapular winging or facial weakness were also reported, suggesting an overlapping FSHD syndrome. The family history was negative for neuromuscular disorders or motor impairment, although genetic analysis were not performed in other family members. Only the mother and two sisters of one proband underwent to genetic test, resulting carriers of the same short D4Z4 allele and presenting a mild facial involvement. The five muscle biopsies of the above unrelated cases showed a pattern of degenerative myopathy with rimmed vacuoles and inflammatory infiltrates. Immunohistochemistry did not detect abnormal desmin, myotilin or alphaB-crystallin deposits, excluding the diagnosis of myofibrillar myopathies. Electron microscopy revealed autophagic vacuoles containing myelin-like material and filamentous nuclear inclusions. Interestingly, MRI imaging did not reveal the typical muscle lower limbs involvement reported in FSHD patients [Olsen et al., 2006].
Finally, it has been described an atypical FSHD phenotype with bent spine syndrome, a clinical condition characterized by a stooped posture in the standing position, which is exaggerated in walking or in exercise and disappears in the supine position, sometimes associated with a dropped head [Umapathi et al., 2002; Wood-Allum et al., 2004; Kottlors et al., 2010; Jordan et al., 2010;
28 Papadopoulos et al., 2011]. The first case report [Umapathi et al., 2002] was about a 59-years-old woman presenting with an overlapping condition with camptocormia, scapular winging and mild facial and proximal weakness. Kottlors et al. [2010] described the case of a 65-years-old man complaining lower back pain and progressive bent spine syndrome, since the age of 60, carrying 31 kb D4Z4 reduced allele. The patient recalled that his mother had a similar posture that began at age of 80. The genetic analysis performed on the available family members revealed the short fragment in the two daughters. The physical examination of the daughters showed signs of myopathic facies and, in one, slight weakness of foot extensors. Nevertheless, none in the family presented a typical FHSD phenotype. Jordan and coworkers [2010] reported six sporadic cases with prevalent axial weakness carrying a D4Z4 reduced allele (range 21-34 kb). All patients referred a late age at onset (IV-VI decades). Muscle MRI imaging revealed that in all six patients the most severely affected muscles were the thoracic and lumbar spinal tract together with hamstrings. Hamstrings, semimembranosus and gastrocnemius muscles resulted to be involved, as previously described in FSHD patients [Olsen et al., 2006].
Often in these articles, the conclusion of the authors is that the extensive use of the genetic analysis has expanded the clinical and morphological spectrum of FSHD, considering the discover of the short D4Z4 allele highly sensitive and specific for the diagnosis of the disease [Tawil et al., 2010]. On the other hand, the wide heterogeneity associated with alterations on chromosome 4q35 can suggest that other factors/pathologic conditions influence and modulate the disease expression, such as epigenetic or environmental factors, concomitant inflammatory disease, or even another hereditary myopathy. Interestingly, the atypical phenotypic cases are often sporadic, without a clear autosomal dominant inheritance. It may be supposed that in these cases the shorter D4Z4 fragment is not per se sufficient to trigger the disease and that other genetic or environmental factors play a role in reaching the disease threshold and justify the overlapping clinical features.
29 “Double trouble” conditions in FSHD families
In 2002, Tonini and coworkers [Tonini et al., 2002] published the article: “Facioscapulohumeral
(FSHD1) and other forms of muscular dystrophy in the same family: is there more in muscular dystrophy than meets the eye?”. The authors reported two unrelated Brazilian families with
members affected by two different forms of muscular dystrophy. In the first one, the 35-years-old male proband showed limb-girdle muscular dystrophy with proximal weakness, elevated CK (16-fold above normal) and a myopathic muscle biopsy. Muscle protein immunohistochemical and western blot analysis revealed a normal pattern for dystrophin, the four sarcoglycans, calpain, dysferlin and telethonin; DNA analysis for caveolin-3 gene was negative. Two of his sisters also complained of muscle weakness. The oldest sister showed clinical signs consistent with FSHD, confirmed through the molecular analysis (30 kb EcoRI/BlnI fragment). The short fragment was also found in another six relatives: four of them (aged 72, 45, 36 and 22 years) were asymptomatic and two (aged 19 and 16 years) showed only mild facial hypostenia. Surprisingly the short allele did not detect in the affected proband. In the second family, a 57-years-old male with a typical FSHD phenotype was carrier of a 17 kb EcoRI/BlnI fragment, which was also present in other affected relatives. However, in the 14-year-old severely affected male cousin, confined to a wheelchair since age 12, but without facial weakness, the small fragment was not found; the patient refused to underwent muscle biopsy and other investigations were not reported. The present families illustrate complicated situations that may occur in the diagnosis and genetic counseling of neuromuscular disorders. Considering that the prevalence of hereditary neuromuscular disorders is very approximately 1/1000, Tonini and coworkers highlighted in their discussion that the finding of two families with an additional neuromuscular disorder were about three times higher than expected. Therefore, although the presence of different neuromuscular disorders in the same genealogy could be only a coincidence, they speculated that some epigenetic mechanisms might turn individuals more prone to pathological mutations.
30 However in FSHD, more than in other neuromuscular disorders, several “double trouble” conditions, in which the D4Z4 reduced allele is associated with well-known pathogenic mutations of other genes in the same subject, are described, causing complex and overlapping phenotypes. In particular, it has been reported cases of patients with CMT1A/ FSHD [Palmucci et al., 2001], mitochondrial myopathy/FSHD [Filosto et al., 2008], Becker dystrophy/FSHD [Rudnik-Schöneborn et al., 2008], Duchenne dystrophy/FSHD [Lecky et al., 1991; Korngut et al., 2008], Leber’s hereditary optic neuropathy/FSHD [Chuenkongkaew et al., 2005], LGMD1C with rippling disease/FSHD [Ricci et al., 2012], suggesting a synergistic effect of those simultaneous mutations in reaching and in modulating the clinical expression.
The extensive use over the past 20 years of DNA analysis for studying Mendelian disorders has revealed many complex mechanisms in addition to single mutant genes that cause disease. Identical phenotypes may be produced by mutations in different genes, the same mutation can cause different phenotypes, and distinct mutations in the same gene may result in different disorders that segregate with diverse Mendelian or even multifactorial patterns. In addition, the incomplete penetrance of certain mutations argues for the importance of modifying loci or epigenetic mechanisms influencing the clinical expression in many Mendelian disorders. Thus, establishing the value of mutational events underlying genetic diseases may be complex even when there are simple patterns of inheritance in diseases with a well-characterized pathologic course [Kanagawa and Toda, 2006; Chahwan et al., 2011]. FSHD seems to fall in this complex pattern even though it is currently considered a fully penetrant disease with a wide variability in clinical spectrum, ranging from subjects with very mild muscle weakness to severe motor impairment, with loss of gait ability.
31 The Italian National Registry for FSHD
Despite the significant breakthroughs made, FSHD pathophysiology remains elusive. To date, several questions must be addressed: 1) Are the diagnostic criteria previously established for FSHD still valid? 2) Can prognostic tools be developed? 3) Which factors may influence the FSHD clinical phenotype? 4) Is FSHD a genetically heterogeneous disorder?
In the last decade, difficulty in patients recruitment has been the greatest obstacle in conduction of successful clinical trials and genotype-phenotype correlation study. Patients registries which facilitate access to patients interested in participating in clinical trials, therefore, represent an important aspect of clinical trial readiness [Tawil et al., 2010]. The National Registry for Myotonic Dystrophy and FSHD Patients in the United States represents the most well-established of such registries. In Europe, there is the large Italian National Registry (www.fshd.it). Other smaller informal FSHD registries exists in other countries or are in the development phase.
The Italian National Registry has been developed since 2007 by Italian Clinical Network for FSHD. The project, entitled “Clinical and laboratory criteria for FSHD diagnosis in view of a national
registry for the disease”, has been supported by Telethon Foundation (Grant GUP07001 and Grant
GUP08004). The FSHD Italian Network is composed by i) two diagnostic laboratories directed by Prof. Rossella Tupler at the Università di Modena e Reggio Emilia and by Dr. Giuliana Galluzzi at the Fondazione Santa Lucia, Rome, and ii) fourteen Clinical Centers with expertise in diagnosis and management of neuromuscular disorders. The neuromuscular Clinical Centers are directed by Dr. Maurizio Moggio (Department of Neurology, IRCCS Fondazione Ospedale Maggiore Policlinico, University of Milan) and Dr. Lucia Morandi (IRCCS Foundation, C. Besta Neurological Institute) at Milan, Dr. Tiziana Mongini (Department of Neuroscience, University of Turin) at Turin, Prof. Corrado Angelini and Prof. Carlo Pietro Trevisan (Department of Neurosciences, University of Padova) at Padua, Dr Angela Berardinelli (IRCCS “C.Modino” Foundation, University of Pavia) at Pavia, Dr. Giuliano Tomelleri (Department of Neurological Sciences and Vision, University of
32 Verona) at Verona, Dr. Massimiliano Filosto (University Hospital "Spedali Civili” of Brescia) at Brescia, Prof. Gabriele Siciliano (Department of Neuroscience, University of Pisa) at Pisa, Prof. Enzo Ricci (Department of Neurosciences, Policlinico A. Gemelli, Università Cattolica of Rome) and Prof. Giovanni Antonini (Department of Neurology, S. Andrea Hospital, "Sapienza" University of Rome) at Roma, Dr. Antonio Di Muzio (Center for Neuromuscular Disease, University "G. d'Annunzio" of Chieti) at Chieti, Prof. Lucio Santoro (Department of Neurological Sciences, “Federico II” University of Naples) at Naples, Dr. Carmelo Rodolico (Department of Neurosciences, Psychiatry and Anaesthesiology University of Messina) at Messina,.
The aims of the projects have been the following: 1) to allow the classification of FSHD clinical features in view of patients molecular characterization, 2) to identify factors influencing the disease clinical expression; 3) to verify the presence of genetic heterogeneity in FSHD. All these data can provide key clinical and basic research tools useful to identify prognostic predictors of clinical impairment and measures of outcome, with further relevant repercussions for genetic counseling and possible implications in improving the understanding of the molecular pathogenetic aspects of FSHD. In the funding period from 2008 to 2012, a solid clinical network has been established, allowing to collect the majority of the FSHD Italian family and to create the National Registry for FSHD (to date, 223 families, 557 index cases and 1161 relatives). In the Clinical Centers, FSHD subjects have been recruited and clinically evaluated; the neurological examination has been also extended to all available FSHD family members, also addressing relatives to further diagnostic analysis. The diagnostic laboratories have been providing molecular characterization of index cases and their family members. A dedicated specific software and a website for data management has been designed. The dedicated website for data management, description of the project and participating groups are available on-line at www.fshd.it.
33 The first results from the Italian National Registry for FSHD
A standardized clinical evaluation of patients affected by facioscapulohumeral muscular dystrophy: the FSHD clinical score [Lamperti et al., 2010]
To define numerically the clinical severity of the disease, the Italian Network for FSHD has developed a standardized protocol that allows the quantification of muscle weakness in FSHD patients through the functional evaluation of six muscle groups specifically affected in FSHD. To this purpose, a new protocol that examines muscle groups specifically affected in FSHD by using functional criteria, allowing expression of clinical severity in quantitative terms easily in the medical office, was generated. To validate reproducibility of the protocol, 69 patients were randomly selected from the registry of FSHD patients at the University of Modena and Reggio Emilia and at the Molecular Genetic Laboratory of UILDM, Rome. The group was composed of 39 females and 30 males. The study included 56 individuals carrying the D4Z4 FSHD-sized alleles and 13 subjects carrying D4Z4 alleles within the normal range. The clinical form consists of three parts, named a, b, and c, that examine three aspects of the disease and have been designed to facilitate accurate study of molecularly defined FSHD subjects. Part a investigates the patient’s clinical history, focusing on medical conditions and particular habits. Part b evaluates the patient’s disability. Part c assesses muscle segmental involvement by using the Medical Research Council (MRC) scale. The evaluation procedure allows to assess the strength and the function of muscular groups belonging to I) face (score from 0 to 2); II) shoulder girdle (score from 0 to 3); III) upper limbs (score from 0 to 2); IV) distal legs (0-2); V) pelvic girdle (score from 0 to 5) VI) abdominal muscles (0-1). More detailed information such as asymmetry of presentation or any observed peculiarity can be added in the section “others”. The functional examination of six different groups of muscles, as described in part b of the clinical form integrated with the results of part c, generates the FSHD clinical score. The total score can range from 0, when no signs of muscle weakness are present, to 15, when all muscle groups tested are severely impaired (Figure 1). The FSHD clinical