UNIVERSITÀ DI PISA
Dipartimento di Farmacia
Corso di Laurea specialistica in Farmacia
“Sintesi di ibridi strutturali di tireomimetici TRbeta
selettivi e tironamine: nuovi tools farmacologici per il
trattamento delle dislipidemie”
Relatori:
Dott.ssa Simona Rapposelli
Dott.ssa Grazia Chiellini
Correlatore:
Prof.ssa Annalina Lapucci
Candidato:
Enrico Casciotti
“Il contenuto di questa relazione è strettamente riservato, essendo presenti argomenti tutelati dalla legge come segreti. Pertanto tutti coloro che ne prendono conoscenza sono soggetti all’obbligo, sanzionato anche penalmente dagli articoli 325 e 623 del codice penale, di non divulgare e di non utilizzare le informazioni acquisite.”
Del nostro meglio Sempre pronti Servire
INDICE
INTRODUZIONE GENERALE 4
1. PREFAZIONE 5
2. ORMONI TIROIDEI 6
3. EFFETTI FISIOLOGICI E METABOLISMO DEGLI ORMONI TIROIDEI 8
4. METABOLISMO DELLE TIRONAMINE 12
La solfatazione 14
La deaminazione ossidativa 14
5. DISTRIBUZIONE TISSUTALE DELLA 3-IODOTIRONAMINA (T1AM) 15
6. EFFETTI FARMACOLOGICI DELLE TIRONAMINE 18
Attività cardiaca 18
Ipotermia 20
Neuroprotezione 22
Iperglicemia 23
Azione anoressizzante 24
Riduzione del peso corporeo 25
Riduzione del quoziente respiratorio 26
Inibizione dei trasportatori delle monoammine 27
7. RECETTORI DELLE TIRONAMINE 28
8. ANALOGHI SINTETICI DELLA T1AM 31
9. CONCLUSIONI E PROSPETTIVE 38
INTRODUZIONE ALLA PARTE SPERIMENTALE 39
PARTE SPERIMENTALE 52
1. PREFAZIONE
Le tironamine sono un insieme di composti endogeni di recente scoperta strutturalmente correlati agli ormoni tiroidei. Tra queste, la 3-iodotironamina (T1AM) e la tironamina (T0AM) che derivano rispettivamente da T4 (tiroxina) e T3 (3,3’,5-triiodotironina), sembrano avere importanti effetti funzionali e metabolici. Sia T1AM che T0AM sono prodotti per azione di enzimi quali deiodinasi e decarbossilasi su T4 e T3. A loro volta le tironamine subiscono ulteriori degradazioni metaboliche con formazione dei due cataboliti acidi TA1 (acido 3-iodotiroacetico) e TA0 (acido tiroacetico). Questi composti sono stati identificati sia nel plasma che in estratti tessutali di varie specie animali sottoposte a trattamento con dosi farmacologiche di T1AM e di T0AM, rispettivamente.
T1AM e T0AM non sono ligandi dei recettori nucleari dell’ormone tiroideo (TRs), ma si sono rivelati, in vitro, potenti agonisti del recettore associato alle amine traccia di tipo 1 (TAAR1), che è un recettore di membrana associato a proteine Gs. Oltre all’elevata affinità per il recettore TAAR1 è stato dimostrato che T1AM interagisce anche con altri recettori come il recettore adrenergico α2 (ARα2), e alcuni trasportatori delle ammine biogene (dopamina e noradrenalina).
Studi in vivo hanno evidenziato che T1AM è in grado di produrre una rapida comparsa di alcuni importanti effetti funzionali e metabolici (ipertermia, bradicardia, alterazioni metabolismo energetico) in genere opposti a quelli prodotti dall’ormone tiroideo. I rapidi e pronunciati effetti metabolici prodotti dalla somministrazione centrale di T1AM e T0AM suggeriscono che uno o più recettori medino la loro azione e che tale effetto sia il risultato di una stimolazione sia centrale che periferica.
Con lo scopo di studiare più approfonditamente il meccanismo di azione delle tironamine e i bersagli farmacologici responsabili degli effetti funzionali (cuore isolato lavorante di ratto) e metabolici (somministrazione icv a topi) indotti da T1AM in questa tesi di laurea viene descritta la sintesi di alcuni analoghi degli ormoni tiroidei, attraverso l’inserimento di specifiche modifiche strutturali in punti chiave dello scheletro molecolare “tironaminico” di T1AM.
2. ORMONI TIROIDEI
La stimolazione dell’asse ipotalamo-ipofisi (o ghiandola pituitaria) porta alla liberazione di TSH (o ormone tireotropo) da parte delle cellule basofile β dell’Adenoipofisi.
Il TSH, attraverso il circolo ematico, arriva alla tiroide, stimolando il rapido intrappolamento di ioni ioduro, la sintesi delle molecole 3,3’,5-triiodotironina (T3) e 3,3’,5,5’-tetraiodotironamina (T4 o tiroxina) sottoforma di materiale colloidale, che viene riversato nel lume dei follicoli tiroidei, e infine la loro liberazione nel circolo sanguigno.
Fig. 1
Gli ormoni della tiroide, i loro metaboliti e il TSH una volta in circolo si legano per il 70% alla TBG (Thyroxine-binding globulin), in particolare alla apolipoproteina B-100, una glicoproteina prodotta dal fegato, e per il resto alla transtiretina (prealbumina) e l’albumina.
Gli ormoni della tiroide (T4, T3 e rT3), chiamati anche tironine, si distinguono dagli altri perché in una struttura organica costituita da due molecole dell’amminoacido Tirosina incorporano un elemento inorganico, lo iodio, il quale viene fornito esclusivamente con la dieta.
O NH2 COOH I I I I O H T4 O NH2 COOH I I I O H T3 O NH2 COOH I I I O H rT3 O H COOH NH2 Tirosina Fig. 2
Il principale prodotto di secrezione della tiroide è il T4 (3,3’,5,5’-tetraiodotironina o tiroxina), una molecola fisiologicamente inattiva, che svolge la funzione di preormone circolante.
Seppur in quantità molto piccole rispetto al T4, il T3 (3,3’,5-triiodotironina) è in parte prodotto dalla tiroide, ma proviene primariamente dalla conversione del T4 a livello dei tessuti periferici. Questo processo viene svolto dalle deiodinasi (dio1/dio2), enzimi appartenenti alla classe delle ossido-riduttasi, che rimuovono l’atomo di iodio in posizione 5’ dalla molecola T4.
Un terzo ormone tiroideo, il cui ruolo biologico non è ancora stato chiarito, è il rT3 o T3 inversa (3,3’,5’-triiodotironina). Questa molecola non viene secreta direttamente dalla tiroide, ma è il risultato dell’attività di un'altra deiodinasi (dio3), che strappa l’atomo di iodio dalla posizione 5 del T4.
O NH2 COOH I I I I O H T4 O NH2 COOH I I I O H T3 O NH2 COOH I I I O H rT3 dio1 /dio2 dio3 Fig. 3
L’attività della tiroide e quindi i livelli plasmatici degli ormoni T4 e T3 sono mantenuti costanti da un sistema feedback negativo svolto dalla T3, che deprime la liberazione del TSH da parte delle cellule tireotrope adenoipofisarie.
3. EFFETTI FISIOLOGICI E METABOLISMO DEGLI ORMONI TIROIDEI La T3 e la T4 entrano nella cellula bersaglio tramite un meccanismo di trasporto che richiede energia. La T3 entra nel nucleo e, con un’affinità superiore al T4, si va a legare
ad uno specifico recettore nucleare, che è definito TR. Ne sono stati individuati due sottotipi: α e β.
Fig. 4
Il complesso T3-TR stimola e inibisce la trascrizione di numerosi mRNA. Questo meccanismo di regolazione trascrizionale si realizza in una scala di tempo relativamente lenta; tuttavia ci sono effetti a rapida insorgenza che sono causati da meccanismi d’azione diversi dal meccanismo trascrizionale, in particolare la regolazione dell’omeostasi ionica e la regolazione del consumo di ossigeno. Questo chiarisce perché l’attivazione da parte di T3 di TRα e TRβ non spiega al 100% l’azione degli ormoni tiroidei.1
I principali effetti dei THs (ormoni tiroidei) sono: l’aumento della gittata cardiaca e della ventilazione, grazie all’attivazione dei canali al Na/K ATPasi, l’aumento dell’appetito, della temperatura corporea, della sudorazione, della funzionalità sia epatica che renale. Provocano inoltre un’accelerazione della risposta metabolica al
digiuno, cioè si ha la mobilitazione di carboidrati, proteine e grassi endogeni, che determinano una riduzione della massa muscolare e del tessuto adiposo.
In generale, la tiroide coordina una grande varietà di processi fisiologici, quali la crescita cellulare, il metabolismo nei mammiferi e lo sviluppo del sistema nervoso nei vertebrati.
Una volta attuata la loro funzione gli ormoni tiroidei subiscono il metabolismo, caratterizzato da reazioni di solfatazione e glucuronazione del gruppo fenolico delle iodotironine, dalla deaminazione ossidativa e decarbossilazione della catena laterale di alanina, formando derivati dell’acido iodotiroacetico, e dalla scissione del ponte etereo.
Fig. 5
La solfatazione sembra ricoprire un ruolo chiave nella regolazione generale del metabolismo delle tironine, infatti questo processo accelera notevolmente la deiodinazione e l’inattivazione.
La glucuronazione spesso precede l’escrezione bilio-fecale degli ormoni. Numerosi studi hanno dimostrato che nei ratti la stimolazione di questo processo metabolico, da parte di vari farmaci e tossine, può provocare una riduzione dei livelli di T4 e T3. Nell'uomo, la stimolazione farmaco-indotta della glucuronazione, influenza solo la concentrazione di T4, e di solito non compromette la normale funzione della tiroide.
Le tironine glucuronate e solfatate possono fungere da serbatoio di riserva delle iodotironine biologicamente attive; questo perché a livello del tratto gastrointestinale e di altri tessuti le forme glucuronate vengono idrolizzate liberando i loro precursori. Gli acidi iodotiroacetici, tetrac e triac, sono prodotti minori nella normale fisiologia tiroidea. Il triac si lega tuttavia preferenzialmente al βTR.
Fig. 6
La scissione del ponte etereo è una via metabolica minore nei soggetti normali; tuttavia sembra diventare rilevante durante le infezioni quando stimola un’attività battericida.2 Una via metabolica particolare degli ormoni tiroidei è la decarbossilazione della catena laterale dell’alanina e la deiodinazione. Tutt’oggi non è chiaro né in quali tessuti avvengano queste reazioni né quali enzimi siano coinvolti.
Il prodotto di queste modifiche strutturali è rappresentato da una varietà di composti feniletilamminici, che hanno sempre più attirato la curiosità dei ricercatori.
Un punto di svolta è stato quando alcuni gruppi di ricerca hanno individuato vie sintetiche per produrre tironamine esogene e poterle quindi somministrare alle cavie per studiarne le caratteristiche.
Da questi primi studi le tironamine si sono dimostrate potenziali ligandi di una nuova classe di recettori accoppiati a proteina G, oggi riconosciuti come TAARs, cioè recettori associati ad ammine traccia.
Gli studiosi definirono Tironamine questo gruppo di molecole endogene, derivanti dal metabolismo degli ormoni tiroidei.2
Fig. 7, Iodotironamine sintetiche.
4. METABOLISMO DELLE TIRONAMINE
Il metabolismo delle tironamine può avvenire attraverso diversi processi: mediante trasformazione della catena laterale etilaminica;
mediante deiodinazione;
mediante funzionalizzazione della molecola.
Per analogia con gli ormoni tiroidei, si assume che il gruppo amminico possa essere ossidato da aminossidasi. In particolare è stato suggerito che la T1AM venga metabolizzata da monoaminossidasi (MAO) o da una particolare aminossidasi semicarbazide-sensibile, la benzilaminossidasi (Bz-SSAO), con formazione di aldeide; che può essere ulteriormente ossidata ad acido carbossilico da un altro enzima, la aldeide deidrogenasi (ALDH) (figura8). MAO e SSAO sono enzimi con attività promiscua che prediligono come substrati le amine primarie, e sono noti per la capacità di catalizzare l’ossidazione di feniletilammine 3,4,5
ALDH è un enzima NAD-dipendente, che è universalmente distribuito nei tessuti dei mammiferi, e può ossidare un’ampia gamma di aldeidi come, ad esempio, le aldeidi derivanti dalla 5-idrossitriptammina e dalla dopamina.5 L’effetto di questi enzimi è quindi la trasformazione di tironamine in acidi tiro acetici 6
O R1 I R2 O H NH2 T3AM: R1 = R2 = I T1AM: R1 = R2 = H O R1 I R2 O H O O R1 I R2 O H OH O Triac: R1 = R2 = I TA1: R1 = R2 = H aminossidasi aldeide deidrogenasi Fig 8
È stato ipotizzato che il metabolismo delle tironamine possa avvenire anche mediante funzionalizzazione della molecola. Le reazioni di coniugazione accoppiano la specie di interesse con piccole biomolecole ionizzabili o altamente idrofile, come l’acido glucuronico o l’acido solforico, allo scopo di inattivarla o renderla nettamente idrofila per una migliore eliminazione renale; oppure con bioreagenti apolari, come nel caso delle metilazioni e acilazioni, per inattivarle e/o aumentarne il peso molecolare e facilitare l'eliminazione biliare.
La solfatazione.
T0AM, 3-T1AM e T3AM sembra vengano solfatati da una solfotrasferasi epatica umana (Sult), che presenta varie isoforme. Inoltre 3-T1AM solfatato è stato rilevato in omogenati di cervello umano e tessuto cardiaco, cioè nei principali tessuti bersaglio delle TAM. L’attività delle Sult potrebbe così servire ad attenuare e regolare l’azione delle TAM.8 Nel caso della T1AM è stato dimostrato che questa molecola è un substrato della sulfotrasferasi epatica SULT1A3 7
La deaminazione ossidativa.
Una via metabolica alternativa è rappresentata dalla deaminazione ossidativa della catena laterale etilamminica, che libera acidi tiroacetici.
3-T1AM e T3AM sono convertiti nei rispettivi acidi tiroacetici (TA1 e TA3) in colture cellulari, omogenati di tessuto, e in vivo. Queste reazioni sono state catalizzate dall’attività di un’amminossidasi e sono stati inibiti dalla monoamino ossidasi (MAO) e dall’inibitore aminico semicarbazide-sensibile (SSAO).8
L'attività biologica di TA1 è sconosciuta, finora. Nel loro insieme, ci sono prove che suggeriscono che solfatazione e deaminazione ossidativa sono rilevanti reazioni metaboliche di TAM. Finora, nessuna prova si è resa disponibile per 4’-O-metilazione o glucuronidazione di TAM.
5. DISTRIBUZIONE TISSUTALE DELLA 3-IODOTIRONAMINA (T1AM) Finora, solo due rappresentanti di TAM, vale a dire 3 -iodotironamina (3-T1AM) e tironamina (T0AM), sono stati rilevati in vivo usando il metodo della cromatografia liquida in tandem con la spettrometria di massa (LC-MS/MS). Entrambi i composti sono stati rilevati in sangue, cuore, fegato, tessuto adiposo, tiroide e nel cervello dei topi maschi adulti (di tipo C57BL), così come nel cervello di diverse altre specie; quali ratti Long-Evans e porcellini d'India. Il 3-T1AM è stato anche rilevato nel sangue e nel cervello dei Djungarian. Negli studi LC-MS/MS, i campioni sono stati simultaneamente monitorati per la presenza di T0AM, T1AM e T2AM, ma quest’ultimo isomero è mancante in tutti i tessuti.2
Dai risultati di alcuni esperimenti condotti marcando il T4AM con 131I, i ricercatori hanno rilevato la presenza di T4AM nel plasma e nella ghiandola tiroidea, ma non nelle frazioni di tireoglobulina di ratti trattati con 131I. 30
L’attenzione dei ricercatori si è sempre più rivolta al composto T1AM, poiché la sua somministrazione determina nei mammiferi significativi effetti fisiologici e comportamentali, che spesso sono opposti a quelli indotti dagli ormoni tiroidei su una scala temporale più lunga; ad esempio la riduzione della temperatura corporea, della frequenza cardiaca, la ridotta contrattilità cardiaca e la modulazione della secrezione di insulina e glucagone. 2,9
Importante in questo campo è stato lo studio condotto nel 2012 da Chiellini e coll., dove partendo dalla sintesi di [125I]-T1AM è stato esaminato, nel topo, la sua distribuzione dopo un’iniezione nella vena caudale alla concentrazione fisiologica (0,3 nM). L'espressione dei sottotipi TAAR è stata valutata con una PCR real-time quantitativa. La radioattività tissutale è diminuita esponenzialmente nel tempo, in linea con l’escrezione attraverso le vie biliari e urinarie; e, dopo 24 ore, il 75% della radioattività residua era presente nel fegato, nei muscoli e nel tessuto adiposo. I TAARs erano espressi solo in tracce nella maggior parte dei tessuti, ad eccezione dei TAAR1, rilevati nello stomaco e nei testicoli, e del TAAR8, riscontrati nell’intestino, nella milza e nei testicoli.
Mentre per T1AM è stata osservata una distribuzione sistemica, i TAARs sono espressi solo in determinati tessuti, suggerendo l’esistenza di altri bersagli molecolari ad alta affinità per le tironamine oltre ai TAARs.
Fig. 9: distribuzione di [125I]-T1AM nei tessuti e nei 16 organi che sono stati considerati in cinque momenti diversi.11
Gli alti livelli rilevati nella cistifellea e nell’intestino fino a 30-60 min dopo l'iniezione potrebbero avvalorare l’ipotesi di un’escrezione biliare e di un eventuale riassorbimento enterico, mentre l'elevata concentrazione renale nello stesso periodo di tempo dimostrerebbe un’escrezione urinaria, che a quanto pare rappresenta la frazione più grande dell’eliminazione della radioattività di tutto il corpo.
Un altro organo in cui [125I]-T1AM era presente in concentrazioni significativamente alte rispetto alla concentrazione in circolo al 30’ minuto è lo stomaco. Una possibile spiegazione è che la secrezione gastrica sia un altra via di escrezione di T1AM, oppure lo stomaco sia un altro sito di stoccaggio in cui si ha una breve permanenza di T1AM. Nel fegato, la concentrazione di [125I]-T1AM è risultata significativamente superiore a quella presente nel sangue in tutte le fasi di valutazione; e dopo 24 h i due terzi della radioattività residua sono stati rilevati nel fegato, nei muscoli e nel tessuto adiposo, suggerendo che questi tessuti potrebbero rappresentare i siti di stoccaggio di T1AM.
Un’altra parte dello studio consisteva nella contemporanea somministrazione di una dose oltre 2000 volte superiore di T1AM non marcato e [125I]-T1AM marcato. Nella maggior parte degli organi i livelli di radioattività diminuiscono più del 90 % rispetto alla riduzione del composto non marcato; l’unica eccezione è stata la pelle. Questa osservazione suggerisce che la maggior parte della radioattività era situata in siti di legame specifici e saturabili. I bassi livelli di radioattività della tiroide, che nel corso del tempo non hanno mostrato alcuna tendenza ad aumentare, suggeriscono che la deiodinazione (ottenuta liberando ioni ioduro libero radiomarcato) è stata scarsa. I presunti cataboliti di [125I]-T1AM contenenti [125I]-I (come acido [125 I]-3-iodotiroacetico) non potrebbero essere dosati specificamente. Tuttavia, in alcuni esperimenti, al 30’ minuto sono stati raccolti campioni di urina e la cromatografia su strato sottile ha dimostrato che la maggior parte della radioattività (70 %) era associata a [125I]-T1AM.
Sapendo che T1AM è il più potente attivatore del TAAR1, in questo studio è stata confrontata la distribuzione di [125I]-T1AM con quella del TAAR, valutando l'espressione di ciascun gene TAAR a livello di mRNA mediante PCR quantitativa assoluta.
Fig. 9
I livelli di espressione sono risultati in generale molto bassi. L’unica eccezione è risultata essere l’espressione significativa di TAAR1 e TAAR8 in stomaco, intestino, milza e testicoli. Al contrario, nonostante l'elevato assorbimento di [125I]-T1AM, l'espressione di TAAR a livello del fegato e dei reni è risultata nulla. Questi risultati
confermano l'assenza di un correlazione tra le concentrazioni tissutali endogene di T1AM e l’espressione genica dei sottotipi TAAR.10,11
Lo studio suggerisce che T1AM potrebbe agire come un vero ormone, cioè un messaggero chimico con una distribuzione sistemica in grado di produrre particolari effetti funzionali legandosi a specifici bersagli molecolari presenti in diversi organi; e il legame ai TAARs non può essere l'unico fattore per spiegare la distribuzione di [125 I]-T1AM nei tessuti del topo.11
6. EFFETTI FARMACOLOGICI DELLE TIRONAMINE
Come già discusso in precedenza, T1AM è un potente agonista in vitro del recettore TAAR1, un recettore delle ammine traccia accoppiato a proteine G, mentre manca di affinità per i recettori nucleari degli ormoni tiroidei (TH) sia di tipo TRalfa che TR beta. Gli effetti farmacologici più importanti delle TAs sono discussi di seguito.
Attività cardiaca
Ci sono prove che gli effetti rapidi di TH potrebbero avere un impiego terapeutico. In pazienti affetti da insufficienza cardiaca congestizia possiamo ottenere un rapido aumento della performance cardiaca a seguito di una consistente iniezione di T3. Recentemente è stato dimostrato che il T1AM provoca risposte rapide sia in vitro che in vivo.1
A seguito di un’iniezione intraperitoneale di 3-T1AM e T0AM (EC50 di 59 e 178 μmol/kg di peso corporeo) a topi wild-type (C57BL), è stata osservata una bradicardia reversibile. Dopo circa 1 ora dall’iniezione, la frequenza cardiaca passava da circa 600 a circa 350 battiti al minuto, per tornare poi alla normalità dopo circa 6-8 ore (dall’iniezione). Questo effetto cronotropo negativo è stato anche confermato da esperimenti condotti su preparati di cuore denervato di ratto adulto, evidenziando che l’attività sul cuore cessava solo una volta rimosso il 3-T1AM dal mezzo di perfusione, indicando anche un’attività dose dipendente.9
Oltre all’effetto cronotropo negativo, 3-T1AM ha mostrato effetti marcati anche sulla gittata cardiaca
Fig. 12
L’effetto inotropo negativo, inibito da 3-T1AM è risultato essere reversibile. Ad oggi sono state identificate solo poche molecole endogene che, come il T1AM, hanno sul cuore effetti inotropo e cronotropo negativo con potenze differenti; le principali sono l’adenosina, il TNFα e IL-6.2
Nei cardiomiociti e in cuori isolati di ratto H9c2 perfusi, è stato osservato un significativo assorbimento Na-dipendente di T1AM esogeno.10
L’effetto del 3-T1AM sul cuore non si limita a effetti cronotropo e ionotropo negativi dose-dipendenti, ma si estende anche all’inibizione di alcuni parametri cardiaci, come la pressione sistolica aortica e il flusso coronarico.
E’ stata infatti rilevata una maggiore durata del potenziale d'azione, che sembra essere dovuta all’inibizione di correnti ioniche anomale di ioni Ca++
e/o K+, e una riduzione dell'ampiezza e della durata della depolarizzazione indotta dal transito di calcio a concentrazioni farmacologiche di 3-T1AM.
Per comprendere la causa degli effetti del 3-T1AM sul cuore sono state condotte analisi sui cardiomiociti di ratto, e dai risultati è emerso che l’omeostasi del calcio e del potassio erano alterate, infatti c’era una diminuita concentrazione di calcio nelle vescicole del reticolo sarcoplasmatico.12
Tuttavia, non c’è nessuna prova che dimostri un effetto diretto di 3-T1AM sull’attività della proteina chinasi A.
E’ stata avvalorata l’ipotesi che a livello del cuore la T1AM entri nei cardiomiociti per la maggior parte grazie ad un sistema mediato dal Na e venga deaminata ossidativamente attraverso una via sensibile all’Iproniazide (una molecola ad attività antidepressiva che agisce inibendo irreversibilmente le monoaminossidasi MAOA e MAOB). Inoltre, nelle cellule H9c2 è stata osservata la produzione di T1AM a partire da T3 esogeno, ma questo risultato resta da confermare.
Da questi studi sugli effetti cardiaci delle tironamine è emerso che T0AM provoca effetti singolari. La perfusione con T0AM, ad una IC50 (concentrazione massima per inibire al 50% un processo) di 83 μM su cuori di ratto denervati, porta dopo pochi minuti alla diminuzione della gittata cardiaca; ma non influenza la frequenza cardiaca. Analogamente al T1AM, anche il T0AM è un agente inotropo negativo; ma differisce dal suo omologo superiore per l’attività cronotropa.
Ipotermia
Il controllo della temperatura è svolto dagli ormoni tiroidei; infatti la percezione del freddo stimola il SNC (sistema nervoso centrale). Questo porta alla stimolazione dell’ipotalamo, che induce la tiroide a liberare T4, e del sistema nervoso simpatico (SNS) a liberare NA (Noradrenalina), che, attraverso un’ampia cascata di effetti coordinati con la tiroide, porta all’aumento della temperatura.
A seguito dell’iniezione intraperitoneale di 3-T1AM e T0AM (EC50 di 59 e 178 μmol/kg di peso corporeo) in 6 topi wild-type (C57BL), è stata osservata una riduzione della temperatura corporea di circa 8°C entro 30 minuti.
Fig. 14
I topi diventato inattivi ma rimangono conservati i riflessi; ciò indica che non erano anestetizzati. Inoltre, la loro pelle, fresca al tatto, non mostrava alcuna reazione di compensazione (brividi o pilo erezione). L'effetto risultava essere completamente reversibile (6-8 ore) e, anche dopo trattamenti ripetuti per un periodo di 2 mesi, finora non si sono manifestati effetti collaterali.2
Anche se meno pronunciato, l’effetto ipotermico è stato osservato anche nei Djungarian, dopo l’iniezione di 50 mg di 3-T1AM per kg di peso corporeo. In entrambi i casi l’effetto ipotermico (30 minuti) è stato preceduto dalla riduzione del metabolismo (5 min). In generale l’ipotermia e il metabolismo sono due fenomeni molto correlati, quindi è probabile che l’ipotermia sia un effetto risultante dalla riduzione del metabolismo.13
Neuroprotezione
Studi scientifici hanno dimostrato che una lieve ipotermia conferisce neuroprotezione contro l’ischemia causata dagli ictus.14
E’ stato recentemente scoperto che i due derivati naturali della T4, la 3-iodotironamina (T1AM) e la tironamina (T0AM), una volta somministrati provocano una rapida ipotermia, che protegge dalle lesioni cerebrali causate da ictus.
Studio fondamentale per dimostrare l’attività neuroprotettiva è stato quello condotto nel 2007 da Scanlan e coll. sui topi. Lo studio consisteva nel confronto di gruppi di topi sottoposti a un modello murino d’ischemia. Ad alcuni gruppi era stato somministrato per via intraperitoneale T1AM e T0AM (50 mg/kg), con conseguente riduzione delle temperatura corporea da 37°C a 31°C. La riduzione della temperatura corporea è stata verificata in assenza di brividi o piloerezione. Dopo 24 ore dall’iniezione, la temperatura corporea dei topi trattati con T1AM o T0AM era ritornato al livello dei topi trattati solo con il veicolo. Il risultato dello studio è stato che i topi a cui erano state somministrate (prima o dopo l’induzione dell’ictus) le tironamine avevano subito infarti significativamente piccoli rispetto al gruppo di topi di controllo.
Da questo studio è emerso che T1AM e T0AM sono neuroprotettori potenti in fase acuta; e il T1AM può essere usato come trattamento di neuroprotezione preventivo per l’ischemia.
T1AM e T0AM offrono spunti di studio per possibili trattamenti farmacologici preventivi delle lesioni cerebrali da ictus.15
Iperglicemia
Somministrazioni intraperitoneali di T1AM (50 mg / Kg) in 6 topi adulti (wild-type C57BL) portavano, oltre agli effetti sopra descritti, un aumento dei livelli di glucosio nel sangue. Questo effetto iperglicemico si manifesta dopo pochi minuti dalla somministrazione fino a raggiungere, 2 ore dopo l’iniezione, livelli massimi di glicemia del 250 % al di sopra dei livelli fisiologici di controllo. La situazione risulta essere completamente invertita 8 ore dopo l'iniezione.
L’effetto iperglicemico è accompagnato da una diminuzione dei livelli plasmatici d’insulina e da un aumento dei livelli plasmatici di glucagone. I tessuti periferici mantenevano la sensibilità all’insulina; e questo è stato dimostrato somministrando insulina esogena 2 ore dopo l’iniezione di 3-T1AM, con l’effetto di normalizzazione dei livelli di glucosio. Coerentemente con questi risultati, uno studio in vitro su cellule pancreatiche umane e murine ha dimostrato che T1AM induce una inibizione della degradazione del glucosio stimolata dal rilascio di insulina.16
Studi più recenti (2009) condotti da Kieverik e coll. hanno dimostrato che l’iniezione i.c.v. (intracerebroventricolare) di T1AM (0.5 mg / Kg) induce nei ratti una rapida iperglicemia. Inoltre è stato dimostrato che la somministrazione ip (intraperitoneale) di 50 mg/kg di T0AM nei ratti aumenta le concentrazioni plasmatiche di glucosio e glucagone, anche se in misura minore rispetto T1AM.17
Fig. 17
Azione anoressizzante
Nel 2008 Braulke e coll. hanno condotto studi analizzando la risposta metabolica al T1AM (50 mg / kg) nel criceto Djungarian e in 6 topi BL. Da questo studio è emerso che la somministrazione di T1AM porta ad un blocco dell'utilizzazione dei carboidrati, e i percorsi metabolici vengono reindirizzati all’utilizzazione lipidica. Questa ipotesi è stata ulteriormente supportata dall'osservazione che il trattamento con T1AM causa chetonuria e una notevole perdita di grasso corporeo.13
L’azione anoressizzante, quindi è il risultato di molti effetti sull’organismo:
Riduzione metabolismo glucidico e aumento di quello lipidico a seguito della
somministrazione di 3-T1AM
Chetonuria e riduzione della massa grassa
L'ipotesi che il fabbisogno energetico venga coperto dal metabolismo lipidico in risposta al trattamento con 3-T1AM è stata confermata da due osservazioni distinte. Primo, i corpi chetonici sono stati rilevati nelle urine di criceti Djungarian 8-28 ore dopo l'iniezione. Secondo, è stata rilevata una significativa perdita di peso dovuta ad una riduzione della massa grassa in criceti mantenuti in condizioni climatiche controllate (caldo e freddo).13
Fig.18
Riduzione del peso corporeo
L’infusione per via intracerebroventricolare (icv) con dosi 100 volte inferiori a quelle impiegate per via i.p. (0,5 mg / kg di peso corporeo), ha prodotto effetti ancora più pronunciati sul metabolismo del glucosio in ratti Wistar maschi. Il mancato accumulo di sostanze infuse a livello periferico, supporta l’ipotesi di un’azione centrale di 3-T1AM e T0AM sulla produzione epatica di glucosio e quindi sul metabolismo. Gli autori hanno sottolineato un aumento del tono simpatico sulle isole pancreatiche con effetto della stimolazione centrale da parte delle TAMs. Inoltre, proiezioni dei sistemi simpatico e parasimpatico possono indurre a livello epatico la produzione di glucosio. Per spiegare l'aumento della concentrazione sierica di corticosterone è stato postulato che le TAMs (più forte per 3-T1AM che per T0AM) hanno un potente effetto attivante dell'asse ipotalamo-ipofisi-surrene.
Un altro studio indipendente ha dimostrato che l’iniezione intraperitoneale o icv di basse dosi di 3-T1AM ha un’azione diretta sul nucleo arcuato causando l’aumento dell'assunzione di cibo nei roditori senza influenzare il consumo di ossigeno e l’attività locomotoria. Questi studi suggeriscono che l'effetto anoressizzante di 3-T1AM sia regolato a livello ipotalamico.18
Fig.19..
Riduzione del quoziente respiratorio
Pochi minuti dopo l'iniezione ip (intraperitoneale) di 3-T1AM, il RQ (quoziente respiratorio) diminuisce da 0.9 a 0.7 sia nei topi C57BL wild-type che nei criceti Djungarian. Questo è indicativo di una rapida variazione di utilizzazione del combustibile metabolico prevalentemente da carboidrati (RQ = 0.9) prima del trattamento, ai lipidi (RQ = 0.7) dopo il trattamento. I valori minimi di RQ sono stati ottenuti da 3 a 4.5 ore dopo il trattamento. L'RQ è stato mantenuto per più di 24 ore, cioè anche dopo il recupero del tasso metabolico, ed è tornato alla normalità solo molto dopo la normalizzazione della funzione cardiaca. Questo indica che gli animali trattati hanno impiegato diverse ore per normalizzare il loro catabolismo metabolico.
Inibizione dei trasportatori delle monoammine
Nel tentativo di chiarire i meccanismi associati con effetti in vivo di TAM, sono state studiate le interazioni tra TAM e i trasportatori delle monoamine neuronali. Queste indagini sono state motivate dalla presenza di TAM nel cervello e dalla loro somiglianza strutturale con trasmettitori di monoamine e ammine traccia.19
Fig. 21
In sistemi di espressione eterologhi, tutte le TAMs, fatta eccezione di rT3AM, inibiscono l'attività della membrana plasmatica umana e del trasportatore della Dopamina del ratto (DAT). Allo stesso modo, tutte le TAMs (ad eccezione di T4AM) inibiscono il trasportatore vescicolare 2 delle monoamine di ratto (VMAT2), che non è localizzato sulla membrana plasmatica cellulare, ma sulla membrana vescicolare intracellulare.
Contrariamente, il trasportatore (umano e di ratto) della serotonina viene inibito solo da T2AM. Inoltre, il trasportatore umano della noradrenalina (NET) ha mostrato un
comportamento misto; infatti è attivato da molte TAMs e inibito da altre, tra cui 3-T1AM.
In generale possiamo dire che T1AM inibisce DAT, VMAT2 e NET a valori di IC50 nel range inferiore al micromolare.
Ad oggi il 3-T1AM è l’unica feniletilammina endogena identificata che inibisce VMAT2.
Nel loro insieme, la potenza delle TAMs nell’inibire i trasportatori di monoamine è paragonabile a quella delle ammine traccia biogene, per cui sono stati individuati valori di IC50 nella fascia più bassa del micromolare.19
La somministrazione di dosi elevate di T1AM in ratti provoca una significativa riduzione del consumo di ossigeno e causa iperglicemia. Questo effetto sul metabolismo glucidico potrebbe essere mediato dalla stimolazione del rilascio di glucagone e dall’inibizione della secrezione insulinica.
L’ipotalamo riveste un ruolo chiave nella regolazione dell’omeostasi energetica, integrando segnali provenienti sia dai tessuti periferici che da altre aree del sistema nervoso centrale. Numerosi nuclei ipotalamici, compreso il nucleo arcuato (ARC) e il nucleo paraventricolare (PVN), sembrano rivestire un ruolo importante nella regolazione dell’assunzione di cibo e del bilancio energetico. L’RNA messaggero dei recettori TAAR1 è espresso in alte concentrazioni nei nuclei ipotalamici. La presenza di T1AM endogena nei cervelli di ratto e la distribuzione dei recettori TAAR1 nei ratti e nell’ipotalamo dell’uomo suggeriscono che T1AM svolga una funzione importante nel controllo e nella regolazione dell’omeostasi energetica.
7. RECETTORI DELLE TIRONAMINE
E’ stato evidenziato che il T1AM non lega o attiva il TRα e il TRβ, mentre è attivo sul recettore TAAR1, recettore AMPc dipendente, sul recettore α2adrenergico e sul recettore 5HT2c serotoninergico.20
In vitro, il T1AM può attivare i recettori accoppiati a proteine G (GPCR), stimolando la produzione di cAMP (adenosina 3',5'-monofosfato ciclico) tramite il recettore associato
alle ammine traccia 1 (TAAR1) e la degradazione del cAMP attraverso il recettore adrenergico α2.9
Inoltre, T1AM ha un ruolo da neuromodulatore, inibendo il reuptake della dopamina (DAT), trasportando la norepinefrina (NET) e andando a inibire il trasporto e il confezionamento vescicolare delle monoamine 2 (VMAT2).
Ad oggi, T1AM è la molecola più potente endogena che può attivare il recettore TAAR1 sia di ratto che di topo. TAAR1 è un membro della famiglia dei recettori orfani associati alle ammine traccia GPCR; se ne riconoscono 19 sottotipi nel ratto, 16 nel topo e 9 nell’uomo.21
Fig. 22
Questi recettori sono espressi in tracce in diversi tessuti compresi cuore, rene, fegato, milza e pancreas; la funzione biologica di ogni sottotipo TAAR è ancora da definire. Il sottotipo TAAR1 umano e dei roditori è marcatamente attivato dalle ammine endogene come β-PEA (β-fenetilammina) nonché dalle psicostimolanti amfetamina e metamfetamina. Fino a poco tempo fa l'esplorazione delle funzioni biologiche di TAAR1 è stata ardua, a causa della mancanza di ligandi selettivi. Solo nel 2010-2011 Hoener e coll. hanno proposto l'identificazione dei primi ligandi selettivi per TAAR1, in particolare RO5166017, agonista selettivo TAAR1, che risulta essere molto più potente dell’ammina traccia β-PEA.22
Fig. 23
TAAR1 è espresso in numerose regioni cerebrali, comprese le zone contenenti nuclei monoaminergici e le regioni limbiche.
Evidenze scientifiche indicano che TAAR1 è coinvolto nella modulazione dei sistemi dopaminergici e serotoninergici, rendendo questo recettore un nuovo promettente bersaglio per la scoperta di molecole impiegabili in patologie monoaminergiche, come la schizofrenia, la depressione, deficit di attenzione disturbi iperattività (ADHD) e il morbo di Parkinson.22
Un secondo recettore proposto per mediare gli effetti di 3-T1AM è il recettore adrenergico α2A (o Adra2A), che è un recettore accoppiato a proteine G espresso in molti tipi cellulari incluse cellule pancreatiche.16
Studi in vitro hanno dimostrato che il recettore Adra2A umano e murino hanno una maggiore affinità per 3-T1AM rispetto all’adrenalina, un ligando endogeno di Adra2α. La somministrazione concomitante della yohimbina, antagonista Adra2A, con 3-T1AM in topi wild-type ha inibito gli effetti iperglicemici di 3-T1AM. Inoltre, 3-T1AM non ha mutato i livelli di glucosio e insulina nel sangue dei topi aventi uno specifico Gα1PCR knockout alle cellule pancreatiche, e non è riuscito a indurre iperglicemia nei topi che non presentavano il recettore Adra2A.16
8. ANALOGHI SINTETICI DELLA T1AM
Un primo studio su possibili analoghi sintetici delle TAMs ha permesso di evidenziare dei requisiti strutturali fondamentali sia per la loro attività farmacologica che per la loro eliminazione.23
L'iniezione ip (intraperitoneale) del derivato sintetico di T1AM privo del gruppo OH in posizione 4’ (composto 91) in 6 topi wild-type di tipo C57BL causa una rapida e pronunciata diminuzione della temperatura corporea (da 36,7° C a 23,6° C entro 120 min) ma ha anche effetti tossici, quali irrigidimento arti posteriori, che portano alla morte 24 h dopo l'iniezione.1
Fig.33
Analogamente, un derivato di 3-T1AM N-metilato contenente un atomo di fluoro al posto del gruppo OH in posizione 4’ causa un effetto ipotermico comparabile a quello indotto da 3-T1AM, ma con concomitante effetto tossico riscontrabile entro 1 settimana di utilizzo. Altri derivati sintetici di 3-T1AM sono risultati più potenti della 3-T1AM nell’attivare TAAR-1 eterologamente espresso, e presentano effetti ipotermici simili ma nessuna tossicità. Dall’analisi di questi dati si evidenzia che i composti privi di OH in posizione 4’ presentano una maggiore tossicità rispetto a T1AM. Il gruppo OH in posizione 4’ rappresenta un sito bersaglio di solfatazione e glucuronazione, ed è stato ipotizzato che la mancanza di 4’-OH può impedire la solfatazione e la glucuronidazione dei composti e quindi compromettere la loro eliminazione, che può essere un fattore di tossicità.
La prima generazione di analoghi delle tironamine presenta modifiche sul gruppo amminico e sul sostituente in posizione 3.
Fig.34
Queste molecole sono state sperimentate testando l’accumulo di cAMP in cellule che esprimono stabilmente sia rTAAR1 (ratto) o mTAAR1 (topo).
Fig. 35
La sostituzione del gruppo amminico primario (NH2) con un gruppo idrossilico (OH) nel derivato 72 del T1AM ha portato alla totale perdita di affinità per entrambi i recettori (rTAAR1 e mTAAR1); mentre la sostituzione dello iodio in posizione 3 con un suo isostero, quale un gruppo etereo, ha portato all’ottenimento del derivato 77, che mostra un’attività simile a T1AM su entrambi i recettori (composto 77: mTAAR1 EC50 116; rTAAR1 EC50 33).
Fig.36
Mantenendo lo iodio in posizione 3, sono state fatte delle modifiche sul gruppo fenolico e monosostituzioni sull’ammina.
Fig.37
Il derivato N-metilato del T1AM (composto 96) manifesta una potenza sul recettore rTAAR1 leggermente maggiore rispetto al T1AM, ma una potenza leggermente minore sul mTAAR1.
La sostituzione dell’idrossile fenolico con un gruppo elettronattrattore è risultata deleteria (composto 93). Tuttavia, il composto 92, che contiene un atomo di fluoro, ha mostrato una maggiore potenza sia sul rTAAR1 che sul mTAAR1.
Un’altra parte dello studio SAR tratta gli effetti sull’attività di modifiche a livello del linker etereo e l’inserimento di un sostituente sull’anello aromatico distale rispetto al
Fig.38
Il O-feniltiramina derivato 32 mostra una maggiore attività per entrambi i recettori TAAR1 (sia di topo che di ratto) rispetto al T0AM (di circa 3 volte), ma come già anticipato mostra una marcata tossicità a causa della mancanza dell’OH fenolico.
L’aumento della distanza tra i due anelli arilici, attraverso l’inserimento di un linker carbonioso, è risultato deleterio per l’attività su entrambi i recettori. L’unica eccezione è rappresentata dai composti 40 e 41 in cui il linker è di 4 o 5 atomi di C.
L’inserimento di un carbonile nel linker (composto 42) ha prodotto un aumento della potenza sul mTAAR1, mentre sul rTAAR1 l’effetto osservato è stato opposto.
L'aggiunta di gruppi ingombranti e/o elettrondonatori sull’anello aromatico distale rispetto al gruppo amminico (composti 50-52) è tollerata sia dal rTAAR1 che dal mTAAR1. In buona parte anche l’inserimento di sostituenti ad elevata densità elettronica sul gruppo benzilico induce un significativo aumento della potenza su entrambi i recettori, suggerendo che tali sostituenti potrebbero rappresentare le interazioni π importanti per l’attivazione del recettore.
Analogamente ai composti di prima generazione, nei composti di seconda generazione è stata considerata la variazione dell’attività su mTAAR1 e su rTAAR1 in funzione di modifiche strutturali sull’ammina, nella posizione 3 e sull’anello aromatico distale rispetto all’ammina.
Fig.40
L’inserimento di sostituenti alchilici (composti 64-66) diminuisce l’attivazione di entrambi i recettori (sia rTAAR1 che mTAAR1). L'unica eccezione è rappresentata dal
composto 62 (rTAAR1 EC50 119; mTAAR1 EC50 54) rispetto al suo omologo inferiore (composto 38: rTAAR1 EC50 209; mTAAR1 EC50 168).
Fig.41
Fig.42
L’inserimento di un atomo di iodio in posizione 3 (composto 82) provoca un aumento dell’attivazione del rTAAR1 (rTAAR1 EC50 77), mentre non ha alcun effetto sul mTAAR1, come già anticipato nel composto 38 (rTAAR1 EC50 209; mTAAR1 EC50 168).
Fig. 43
Tuttavia, l’inclusione dello iodio e la metilazione del composto amminico, nel composto 84, ha portato ad un aumento della potenza di attivazione del TAAR1.
Curiosamente il composto 83, che conteneva un trifluorometile e lo iodio, non ha mostrato attività significativa sul rTAAR1, ma attiva debolmente solo mTAAR1. Tuttavia, il composto 85, contenente un trifluorometile (come il composto 83) e l'ammina metilata, è risultato un potente attivante sia del rTAAR1 che del mTAAR1 ed è stato calcolato il valore di EC50 (pari a 61 e 12 nM rispettivamente).
Fig.44 Sulla base di questi studi, si può concludere che:
La presenza del gruppo amminico è essenziale per l’attività
la monometilazione dell'ammina può essere utile, ma gruppi alchilici più grandi e la bis-alchilazione sono deleteri;
i recettori mTAAR1 e rTAAR1 differiscono per quanto riguarda la tolleranza alle variazioni del linker diarilico, sia in lunghezza che in funzionalità;
la presenza di un atomo di iodio o un sostituente metilico in posizione 3 nel nucleo delle tironamine è ottimale ai fini dell’attività.
9. CONCLUSIONI E PROSPETTIVE
Dall’analisi dei dati pubblicati fino ad oggi è evidente che oltre ai classici ormoni tiroidei (T4 e T3), alcuni metaboliti e analoghi strutturali esercitano effetti importanti sull’omeostasi metabolica. In particolare, gli effetti farmacologici indotti dalle tironamine a carico del metabolismo lipidico e dei processi bioenergetici risultano particolarmente interessanti e costituiscono il punto di partenza per la progettazione e sintesi di analoghi sintetici delle tironamine. Lo sviluppo di analoghi sintetici delle tironamine rappresenta una nuova strategia per lo sviluppo di nuovi agenti terapeutici utili per il trattamento di alcune importanti patologie quali ad esempio l’obesità e l’ipercolesterolemia.
Pro Contro
Metaboliti naturali
Calo peso corporeo e lipidi Nessuna selettività Increment spesa energetica Nessuno o pochi studi sui
primati Nessuno scompenso cardiaco
Agonisti selettivi TRβ
Calo peso corporeo, lipidi e
colesterolo Soppressione del TSH
Incremento tasso metabolico e
angiogenesi Uptake selettivo tissutale Nessuno scompenso cardiaco
INTRODUZIONE ALLA PARTE
SPERIMENTALE
Le due tironine T4 e T3, che sono gli ormoni prodotti e secreti dalla ghiandola tiroidea in risposta all’ormone ipofisario TSH, svolgono un ruolo importante nella regolazione dello sviluppo e crescita nei vertebrati. Gli ormoni tiroidei hanno anche importanti effetti nella regolazione del metabolismo energetico. Ad esempio, nell’uomo, l’ipotiroidismo è associato all’ipercolesterolemia, all’ipertensione e ad un rischio molto elevato di malattie cardiache, tutti eventi clinici a sostegno dell’importanza fisiologica di questa modulazione.
Il ruolo cruciale svolto dagli ormoni tiroidei nel metabolismo ha suggerito lo sviluppo di tireomimetici selettivi per una delle due isoforme dei recettori tiroidei TR (TRα e TRβ), in particolare per l’isoforma TRβ, ed il loro utilizzo in protocolli sperimentali.
Nel 1998 nei laboratori diretti dal Prof. Thomas S. Scanlan presso l’Universita’ della California di San Francisco fu progettato e sintetizzato in ottime rese un analogo della T3, denominato originariamente GC-1, poi designato come QRX-431 e successivamente come Sobetirome, che differiva strutturalmente dall’ormone tiroideo in 3 aspetti fondamentali:
1) gli atomi di iodio nelle posizioni 3 e 5 erano sostituiti da gruppi metilici (CH3), mentre l’atomo di iodio in posizione 3’ era sostituito da un gruppo isopropilico [CH(CH3)2]. 2) l’atomo di Ossigeno a ponte tra I due gruppi arilici era sostituito da un gruppo
metilenico (CH2).
3) la catena aminoacidica in posizione 1, era sostituita dal gruppo ossiacetico (OCH2COOH). C H3 CH3 CH3 C H3 O H O OH O Figura 1: GC-1
Questo nuovo analogo di sintesi della T3 è risultato essere un agonista selettivo per l’isoforma beta del recettore tiroideo (TRβ), e somministrato ad animali di laboratorio, ha condotto ad una riduzione dei livelli plasmatici di colesterolo confrontabile a quella prodotta dal T3 a concentrazioni che non modificano significativamente la funzione cardiaca. In tempi recenti Sobetirome, ha completato la Fase 1 di clinical trials per lo sviluppo come farmaco per il trattamento dell’ipercolesterolemia.
Un aspetto di particolare importanza è che le modifiche strutturali apportate alla molecola di T3 nella creazione di GC-1 hanno prodotto uno scaffold molecolare che, a differenza della struttura
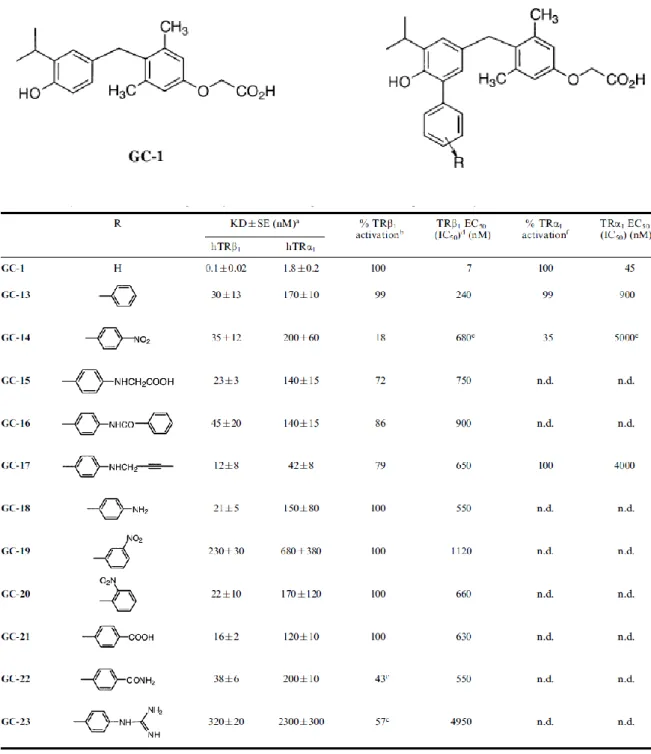
dell’ormone tiroideo, offre ampie possibilità di derivatizzazione chimica. Di conseguenza nel corso di questi ultimi anni sono stati prodotti numerosi analoghi di sintesi di GC-1. Tra questi alcuni hanno consentito di raggiungere una selettività per il TRβ superiore a quella propria di GC-1 (es. GC-23), mentre altri si sono dimostrati efficaci antagonisti TRβ selettivi (es. GC-14 e GC-18), fornendo utili strumenti per lo studio dell’azione dell’ormone tiroideo sia in vitro che in vivo.
Una nuova prospettiva sulla risposta agli ormoni tiroidei è scaturita dalla scoperta di derivati endogeni dell’ormone tiroideo, quali la tironamina (T0AM) e la 3-iodotironamina (T1AM), che è stato proposto avere origine dalla tiroxina per azione metabolica di deiodinasi e di decarbossilasi. O I I I I O H COOH N H2 O I I O H I NH2 O I O H NH2 I I I O I I O H COOH N H2 I T 4 rT 3 Dio3 Dio1, Dio2 ? ? CO2 O I I O H I NH2 O I I O H NH2 Dio1 O I O H NH2 CO2 Dio3
x
T4AM rT3AM T3AM 3,3'T2AM T1AMTali metaboliti non risultano essere ligandi dei recettori nucleari tiroidei né dei recettori classici per le amine biogene, ma sono invece potenti agonisti dei recettori associati alle ammine traccia di tipo 1 (TAAR1), che sono dei recettori orfani accoppiati alle proteine G di recente scoperta, considerati un promettente target per lo sviluppo di farmaci destinati al trattamento di patologie di natura psichiatrica e neurodegenerativa24-25-26.
Lo studio in modelli in vivo degli effetti indotti dalle tironamine endogene (T0AM e T1AM), ha permesso di evidenziare che alcuni di questi effetti si manifestano in tempi molto brevi (pochi minuti) e risultano essere opposti a quelli normalmente svolti dall'ormone tiroideo. Tra questi, in particolare per T1AM che è la più potente delle due tironamine, sono stati evidenziati: riduzione dell'attività locomotoria, profonda ipotermia, e riduzione dell'attività cardiaca. Nel criceto la somministrazione acuta di T1AM ha determinato una riduzione del grasso corporeo, associata a chetonuria e ad una riduzione del quoziente respiratorio. Questi risultati suggeriscono che T1AM favorisca il catabolismo lipidico a spese del catabolismo glucidico. È tuttavia importante sottolineare che le tironamine possono agire da inibitori dei trasportatori delle monoamine biogene, influenzando l’immagazzinamento ed i livelli extracellulari di queste ultime, e di conseguenza questo effetto può complicare l’interpretazione degli effetti fisiologici osservati in seguito alla somministrazione di tironamine 19.
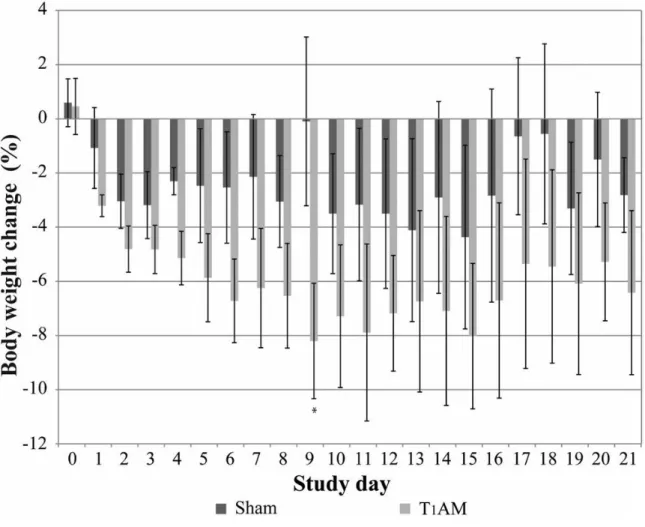
Studi preliminari, somministrando T1AM in maniera cronica a topi naturalmente obesi, sembrano confermare una riduzione del peso corporeo conseguente a stimolo del catabolismo lipidico. In aggiunta, dopo sospensione del trattamento è stato osservato solo un modesto recupero del peso corporeo 27.
Figura 4: calo di peso delle cavie nei giorni della somministrazione del T1AM 27
Nel complesso i risultati sperimentali suggeriscono che T1AM abbia le potenzialità per essere sviluppato come farmaco antiobesità e/o per il mantenimento del peso corporeo, pertanto l’identificazione del target molecolare responsabile dell’azione di T1AM appare di fondamentale importanza.
Un utile contributo al chiarimento della natura degli effettori di T1AM, responsabili in ultimo degli effetti fisiologici conseguenti alla somministrazione di tironamine, potrebbe derivare dallo sviluppo di agonisti ed antagonisti TAAR1 selettivi.
A tale scopo, essendo T1AM un potente agonista TAAR1 selettivo con attività nanomolare, l’inserimento di specifiche modifiche strutturali nello scaffold molecolare “tironaminico” di T1AM può costituire una valida strategia per lo sviluppo di nuovi ligandi dei recettori TAAR1, sia ad attività agonista che antagonista. In effetti, recenti studi hanno mostrato che anche alcuni metaboliti dell’amiodarone, che è un farmaco largamente usato per il trattamento di aritmie cardiache che presenta una certa analogia strutturale con le tironamine, si sono rivelati specifici agonisti dei TAAR1 28.
O I I N CH3 CH3 O CH3 O Figura 5: Amiodarone
Inoltre, il gruppo del Prof. Thomas S. Scanlan (OHSU, Portland, OR, USA) ha sintetizzato degli analoghi della 3-iodotironamina che sono stati valutati come agonisti del recettore TAAR1.
In particolare le principali modifiche strutturali apportate allo scaffold molecolare di T1AM includono sostituzioni dell’ammina primaria con raggruppamenti N-alchilamminici o N,N- diN-alchilamminici, variazioni strutturali a carico del linker che unisce i due anelli aromatici (A e B) e sostituzioni aromatiche.
O H O NH2 I T1AM X O N R4 R3 R2 R1 4' A B R1: OMe, CF3, F X: CH2, CH2CH2 R2: Me, H R3 = R4 = Me, Et
Figura 6: Modifiche strutturali apportate alla molecola di T1AM 1
L’ esame dei rapporti struttura-attività (SAR) di tali derivati, ha evidenziato che:
a) la presenza di un gruppo amminico primario sulla catena etilamminica è essenziale al fine dell’attività biologica mediata dal recettore TAAR1;
b) la sostituzione dell’atomo di iodio in posizione 3 di T1AM con un bioisostero alchilico non influenza l’attività farmacologica (EC50 14 e 33 nM, rispettivamente);
c) l’allungamento del linker tra i due anelli aromatici, A e B, è deleterio per l’attività;
d) il sostituente OH in posizione 4’ non sembra essere essenziale per l’attività sul recettore;
e) l’allungamento della catena etilamminica in posizione 1 provoca una riduzione dell’attività.
Sulla base di questi risultati e con lo scopo di individuare potenziali nuovi analoghi delle tironamine, nel laboratorio presso il quale ho svolto questa tesi di laurea sono state progettate nuove molecole usando come scaffold molecolare la 3-iodotironamina. In particolare, nel corso dello svolgimento di questa tesi di laurea, sono state sintetizzate le molecole 1-4, che presentano uno scheletro tironaminico al quale sono state apportate, in maniera singola o combinata, le seguenti modifiche strutturali:
1) Sostituzione dell’Ossigeno a ponte tra I due gruppi arilici con un gruppo metilenico. 2) Sostituzione dello Iodio in posizione 3 con un atomo di idrogeno (H)
3) Sostituzione del gruppo idrossilico (OH) in posizione 4’ con un gruppo amminico (-NH2, -NHR o -NR1R2) o con un nitrogruppo (-NO2).
4) Sostituzioni della catena laterale etilamminica con il raggruppamento ossietilamminico o ossiacetico. O2N COOH N H2 COOH N H2 NH2 O H O NH2 1 2 3 4
4-[4-(2-aminoetil)benzil]anilina acido [4-(4-nitrobenzil)fenil]acetico
SCHEMA 1 O H B(OH)2 O H BF3K O2N CH2OH O2N CH2Cl O2N CH2CN O2N CH2COOH N H2 CH2COOH H2N NH2 a b c d e f g 5 6 7 8 1 2 3
Reagenti e condizioni: a: KHF2, MeOH/H2O, t.a, 30'; b: 4-Nitrobenzil bromuro, PdCl2 dppf, Cs2CO3, H2O/Diossano, 95°C, 24 h; c: SOCl2, CHCl3, t.a, 2h; d: NaCN, H2O/CH3CN, 100° C, 150 W, 8 bar, 4 cicli di 20 min.; e: LiAlH4, AlCl3, THF, riflusso, 12 h; f: H2SO4 50%, riflusso, 30'; g: Idrazina idrata, Carbone, FeCl3, MeOH, riflusso, 12 h.
I composti 1-3 sono stati sintetizzati mediante la procedura descritta nello schema 1. Per reazione di salificazione dell’acido 4-idrossimetilboronico commerciale con KHF2 si è ottenuto il derivato 5, che è stato sottoposto a reazione di cross-coupling con il 4-nitrobenzil bromuro per dare il composto 6. La successiva reazione di clorurazione in presenza di SOCl2 ha fornito il corrispondente cloroderivato 7 che per sostituzione
nucleofila con NaCN ha condotto al cianoderivato 8. La riduzione del composto 8 in presenza di LiAlH4 e AlCl3 ha portato al composto amminico 1, mentre la sua idrolisi con H2SO4 ha permesso di ottenere l’acido 2. La successiva reazione del derivato 2 con idrazina idrata in presenza di FeCl3 e carbone ha fornito il corrispondente acido 4-(p.amminobenzil) fenil acetico 3.
SCHEMA 2 Br OH CHO OH BrCH2CN
+
OH MemO OCH2CN a b c O H OCH2CH2NH2 Br OMem CHO OCH2CN+
4 11 9 10Reagenti e condizioni: a: DIPEA, DBE, riflusso, 12 h; b: DMF, Cs2CO3, 30', t.a.; c: n-BuLi, THF, -78°C, 2 h.
Per la sintesi del composto 4 è stata effettuata una procedura riportata in letteratura (Chiellini et al) e riportata nello schema 2. Il 4-bromofenolo commerciale è stato protetto con il MEM per reazione dell’1-(clorometossi)-2-metossietano con MEMCl in BDE. La successiva reazione di 9 con il derivato aldeidico 10 (ottenuto per reazione di 4-idrossibenzaldeide con Br-acetonitrile) in presenza di n-BuLi non ha però portato al composto 11 desiderato. La procedura sintetica alternativa che ha portato al prodotto 4 è riportata nello schema 3
SCHEMA 3 MeO B(OH)2 O2N OH O2N OMe N H2 OCH2CN a b c O2N OCH2CN O H OCH2CN HO OCH2CH2NH2 d e f 12 13 14 15 16 4
Reagenti e condizioni: a: 4-Nitrobenzil bromuro, K2CO3, PdCl2, Acetone/H2O, t.a., 72 h; b: BBr3, CH2Cl2, 0°C, 1 h; c: BrCH2CN, DMF, Cs2CO3, t.a., 30'; d: Pd/C, AcOH, EtOH, t.a., 48 h; e: NaNO2, H2SO4, H2O, 100°C, 1 h; f: LiAlH4, AlCl3, THF, riflusso, 12 h.
Una reazione di Cross-Coupling tra l’acido 4-metossifenilboronico e p-nitrobenzil bromuro utilizzando PdCl2 come catalizzatore e K2CO3 come base ha fornito il composto 12. La successiva reazione di demetilazione in presenza di BBr3 a 0°C ha condotto al corrispondente idrossiderivato 13, che è stato alchilato con bromoacetonitrile per dare il cianoderivato 14. La successiva idrogenazione catalitica del composto 14 utilizzando Pd/C in CH3COOH concentrato ha fornito il derivato anilinico 15, che a seguito della reazione con NaNO2 in H2SO4 ha fornito il fenolo 16 attraverso la formazione del sale di diazonio quale intermedio. Il composto 16 è stato infine ridotto con LiAlH4/AlCl3 a fornire il prodotto finale 4.
I nuovi analoghi di sintesi di T1AM, saranno testati in vitro per valutarne la capacità di attivare i recettori TAAR1. Essendo quest’ultimi accoppiati a proteine Gs, tale valuzione potrà essere facilmente eseguita conducendo dei saggi di misurazione dell’accumulo di c-AMP (c-Amp agonist activity assay). Successivamente i composti che mostreranno attività in vitro saranno testati in vivo per analizzarne gli effetti sulla contrazione cardiaca e sul metabolismo energetico. I risultati di questi studi dovrebbero fornire utili informazioni sui rapporti struttura attività in molecole di natura tironaminica, evidenziando i requisiti strutturali dello scheletro tironaminico atti all’attivazione o al blocco dei recettori TAAR1, e stabilendo una specifica correlazione con gli effetti funzionali osservati. Tali informazioni amplieranno anche le attuali conoscenze sulle potenzialità terapeutiche dei recettori TAAR1.
MATERIALI E METODI
La struttura di tutti i composti è stata controllata per mezzo della spettrometria 1H NMR. Degli spettri 1H NMR sono stati riportati i particolari più significativi. Tutti i composti sintetizzati presentano dati spettrali in accordo con le strutture assegnate. Gli spettri di risonanza magnetica nucleare sono stati eseguiti con uno spettrofotometro Bruker operante a 400 MHz in CDCl3 o CD3OD. I chemical shift sigma sono espressi in ppm.
Le analisi elementari sono state eseguite nel nostro laboratorio di Chimica Analitica; la differenza tra i valori teorici e quelli calcolati risulta essere compresa in un intervallo di ± 0.4%.
Le evaporazioni sono state eseguite sottovuoto in evaporatore rotante e le disidratazioni delle fasi organiche sono state eseguite usando Na2SO4.
Le TLC analitiche sono state effettuate usando lastre MERK di gel di silice (G60) contenenti un indicatore di fluorescenza 20 x 20.2 mm. Le varie macchie sono state evidenziate per mezzo di lampada UV (256 nm).
SCHEMA 1 Sintesi del derivato 5
O H
BF3K
trifluoro[4-(4-(idrossimetil) fenil]-borato di potassio
Ad una soluzione di acido 4-idrossimetil-fenil boronico (1.3 g; 8.55 mmoli) in MeOH è stato aggiunto KHF2 (2.7 g; 34.2 mmoli) e poche gocce di H2O. La miscela è stata lasciata in agitazione a temperatura ambiente per 30 minuti. Trascorso questo periodo il solvente è stato evaporato e il solido ottenuto è stato purificato mediante cristallizzazione da iPrOH, fornendo il derivato 5 desiderato.
Resa: 63 % 1 H NMR (CD3OD) δ: 4.53 (s, 2H, CH2); 7.18 (d, 2H, J = 7.2 Hz, AA’XX’); 7.48 (d, 2H, J = 7.2 Hz, AA’XX’) ppm. ANALISI ELEMENTARE C7H7BF3KO C H Calc. % 39.28 3.30 Trov. % 39.55 3.43
SCHEMA 1 Sintesi del derivato 6
O2N CH2OH
[4-(4-nitrobenzil)fenil]metanolo
Ad una soluzione del sale 5 (509 mg; 2.38 mmoli) in diossano/H2O in rapporto 9:1 (17 ml) sotto azoto è stato aggiunto p-nitrobenzilbromuro (513 mg; 2.38 mmoli), carbonato di cesio (2.30 g; 7.1 mmoli) e PdCl2dppf (34.8 mg; 0.05 mmoli). La miscela così ottenuta è stata posta in agitazione a 95°C per 24 ore. Trascorso tale periodo il solvente organico è stato evaporato e la fase acquosa estratta con CH2Cl2. La fase organica è stata essiccata filtrata ed evaporata fornendo un grezzo che è stato purificato tramite cromatografia su colonna utilizzando come eluente una miscela n- Esano/AcOEt in rapporto 7:3
Resa: 32 %
1
H NMR (CDCl3) δ: 4.08 (s, 2H, CH2); 4.68 (s, 2H, CH2OH); 7.17 (d, 2H, J = 8.0 Hz AA’XX’); 7.34-7.32 (m, 4H, Ar); 8.14 (d, 2H, J = 8.4 Hz, AA’XX’) ppm.
ANALISI ELEMENTARE
C14H13NO3 C H N
Calc. % 69.12 5.39 5.76
SCHEMA 1 Sintesi del derivato 7
O2N CH2Cl
1-(clorometil)-4-(4-nitrobenzil)benzene
Ad una soluzione d‘alcol 6 (86.4 mg; 0.35 mmoli) in CHCl3 posta a 0°C è stato aggiunto SOCl2 (0.009 ml). La miscela ottenuta è stata lasciata in agitazione a t.a. per 2 ore. Trascorso tale periodo il solvente è stato evaporato e il solido ottenuto ripreso con H2O e alcalinizzato con NaOH 1N; la fase acquosa è stata estratta con CH2Cl2. La fase organica è stata essiccata ed evaporata fornendo il grezzo desiderato 7 che è stato utilizzato nella successiva reazione senza ulteriore purificazione.
Resa: 83%
1
H NMR (CDCl3) δ: 4.08 (s, 2H, CH2); 4.57 (s, 2H, CH2Cl); 7.17 (d, 2H, J= 8.0 Hz, AA’XX’); 7.35-7.32 (m, 4H, Ar); 8.15 (d, 2H, J = 8.8 Hz AA’XX’) ppm.
ANALISI ELEMENTARE
C14H12ClNO2 C H N
Calc. % 64.25 4.62 5.35
SCHEMA 1 Sintesi del derivato 8
O2N CH2CN
[4-(4-nitrobenzil)fenil]acetonitrile
Ad una soluzione di 7 (152 mg; 0.58 mmoli) in CH3CN (0.78ml) è stato aggiunto NaCN (57.0 mg; 1.16 mmoli) in H2O (0.26 ml). La miscela così ottenuta è stata posta al microonde impostando i seguenti parametri: temperatura 100°C; power 150 W; pressione 8 bar; tempo 20 minuti. Dopo raffreddamento la miscela di reazione è stata estratta con CH2Cl2 e la fase organica è stata essiccata ed evaporata a pressione ridotta ottenendo il grezzo desiderato 8.
Resa: 86%
1
H NMR (CDCl3) δ: 3.73 (s, 2H, CH2CN), 4.08 (s, 2H, CH2); 7.19 (d, 2H, J = 8.0 Hz; AA’XX’); 7.33-7.28 (m, 4H, Ar); 8.15 (d, 2H, J = 8.8 Hz; AA’XX’) ppm.
ANALISI ELEMENTARE
C15H12N2O2 C H N
Calc. % 71.42 4.79 11.10
SCHEMA 1 Sintesi del derivato 1
N
H2 NH2
4-[4-(2-aminoetil)benzil]anilina
Ad soluzione di LiAlH4 (3.04 mmoli) e AlCl3 (405 mg; 3.04 mmoli) in THF, posta sotto azoto, lasciata in agitazione a t.a. per 5 minuti, è stata aggiunta una soluzione del cianoderivato 8 (85.0 mg; 0.34 mmoli) in THF. La reazione è stata posta in agitazione a reflusso a 66°C per 12 ore. Trascorso tale periodo la miscela è stata portata a 0°C, addizionata di H2O e acidificata con HCl 10%. La soluzione è stata quindi lavata con Et2O, la fase acquosa è stata alcalinizzata con NaOH 2 N ed addizionata con CHCl3. L’emulsione risultante è stata filtrata su setto a celite e la fase organica è stata infine lavata con NaCl saturo, anidrificata, filtrata ed evaporata a pressione ridotta fornendo un grezzo 1 che è stato purificato tramite formazione del cloridrato.
P.f.: 200-202 °C Resa: 70% 1 H NMR (CD3OD) δ: 2.93 (t, 2H, J = 7.8 Hz; CH2); 3.15 (t, 2H,J = 7.8 Hz; CH2NH2); 4.02 (s, 2H, CH2); 7.25-7.20 (m, 4H, Ar); 7.33 (d, 2H,J = 8.4 Hz, AA’XX’) 7.38 (d, 2H, J = 8.4 Hz A’XX’) ppm. 13 C NMR (CD3OD) δ: 142.83, 139.53, 134.58, 130.20, 129.12, 128.67, 128.50, 122.71, 40.54, 40.30, 32.75 ppm. ANALISI ELEMENTARE C15H18N2 C H N Calc. % 79.61 8.02 12.38 Trov. % 79.45 8.18 12.77
SCHEMA 1 Sintesi del derivato 2
O2N CH2COOH
acido [4-(4-nitrobenzil)fenil]acetico
Una soluzione del cianoderivato 8 (51.6 mg; 0.20 mmoli) in H2SO4 50% (0.2 ml).è stata posta in agitazione a reflusso per 30 minuti. Trascorso tale periodo la miscela di reazione viene stata posta a 0°C ed addizionata di H2O; il solido precipitato viene raccolto per filtrazione e lavato con H2O fornendo il prodotto desiderato 2.
P.f.: 119-121 °C Resa: 79%
1
H NMR (CDCl3) δ: 3.58 (s, 2H, CH2COOH); 4.09 (s, 2H, CH2); 7.19 (d, 2H, J = 7.6 Hz, AA’XX’); 7.37-7.27 (m, 4H, Ar); 8.17 (d, 2H, J = 7.6 Hz, AA’XX’) ppm.
13 C NMR (CDCl3) δ: 173.69, 148.41, 146.52, 138.46, 133.08, 129.77, 129.59, 129.51, 129.08, 123.75, 123.69, 42.65, 41.25 ppm. ANALISI ELEMENTARE C15H13NO4 C H N Calc. % 66.41 4.83 5.16 Trov. % 66.44 4.55 5.28
![Fig. 9: distribuzione di [ 125 I]-T1AM nei tessuti e nei 16 organi che sono stati considerati in cinque momenti diversi](https://thumb-eu.123doks.com/thumbv2/123dokorg/7990199.121018/16.892.169.817.104.587/distribuzione-tessuti-organi-stati-considerati-cinque-momenti-diversi.webp)