Ab initio
ground potential energy surface
„
3A
⬙
…
for the O
„
3P
…¿
N
2O reaction
and kinetics study
Miguel Gonza´lez,a)Rosendo Valero, and R. Sayo´sa)
Departament de Quı´mica Fı´sica i Centre de Recerca en Quı´mica Teo`rica, Universitat de Barcelona, C/ Martı´ i Franque`s, 1. 08028 Barcelona, Spain
共Received 30 January 2001; accepted 2 May 2001兲
An ab initio CASPT2//CASSCF study of the 3A
⬙
ground potential energy surface for the O(3P) ⫹N2O(X1⌺⫹) reaction has been performed, investigating the two predominant reactive channels. Symmetry breaking is reported for some of the structures. Rate constants are calculated by means of the transition state theory yielding values in almost quantitative agreement with experiment for the 2 NO(X2⌸) channel, but at variance with experiment for the N2(X1⌺g⫹)⫹O2(X3⌺g⫺) one. Apreliminary study on the possible existence of surface crossings (3A
⬙
–1A⬘
,3A⬙
–1A⬙
, and3A⬙
–3A⬘
intersections兲 reveals that more efforts are warranted to fully explain the origin of this discrepancy. © 2001 American Institute of Physics. 关DOI: 10.1063/1.1381010兴
I. INTRODUCTION
The N2O molecule plays a relevant role in combustion processes as a generator of NOx species. Also, its thermal decomposition leads to oxygen atoms in their first singlet excited state, which participate in the stratospheric ozone cycle. In combustion processes, the reaction of the nitrous oxide molecule with ground state O(3P) atoms is of major
importance. The O(3P)⫹N2O reaction presents two product channels: O共3P兲⫹N2O共X1⌺⫹兲→NO共X2⌸兲⫹NO共X2⌸兲, ⌬H0 K 0 ⫽⫺36.0 kcal mol⫺1 共1兲 →N2共X1⌺g⫹兲⫹O2共X3⌺g⫺兲, ⌬H0 K 0 ⫽⫺79.6 kcal mol⫺1 . 共2兲 Under Cs symmetry one3A
⬘
and two 3A⬙
potentialen-ergy surfaces 共PESs兲 correlate with the products of channel
共1兲 while only one 3A
⬙
PES correlates with the products of channel共2兲. Thus, in Cssymmetry both reactions are feasible only through the ground PES (3A⬙
).Many experimental studies have been devoted to the de-termination of the rate constants for reactions 共1兲 and 共2兲.1 However, it was not until very recently that direct measure-ments of the total rate constant at an intermediate tempera-ture range were performed.2Later, a critical assessment of all the available information was carried out.1From the selected works, the expression k1⫽1.52⫻10⫺10exp(⫺13 930/T) 共1370–4080 K兲 cm3molecule⫺1s⫺1was derived for reaction 共1兲. Analogously, for reaction 共2兲 the expression k2⫽6.13 ⫻10⫺12exp(⫺8020/T) 共1075–3340 K兲 cm3molecule⫺1s⫺1 was finally obtained. Thus, activation energies of 27.7 and 15.9 kcal mol⫺1are deduced for reactions共1兲 and 共2兲, respec-tively.
On the other hand, to our knowledge no theoretical stud-ies of the system exist, apart from a quotation in Ref. 1 of an unpublished ab initio study yielding a barrier height for re-action共1兲 of 28 kcal mol⫺1, in good accordance with experi-ment, and a much higher barrier for reaction 共2兲 at variance with the results of Ref. 1. In the latter case, a crossing of the 3A
⬙
surface with the ground singlet surface (1A⬘
) of the O(1D)⫹N2O system is suggested as a lower-energy alterna-tive pathway to products, N2⫹O2.
II. METHODOLOGY
In the study of a PES, it is important to introduce all the configurations that have an important weight in one or an-other of its regions. In this way, a balanced description of, e.g., reactants and products, transition states, avoided cross-ings, etc., should in principle be achieved. In the CASSCF
共complete active space self-consistent-field兲 method3 all the configuration state functions 共CSFs兲 generated from a fixed set of molecular orbitals 共MOs兲, called the active space, are included. This method is intended to account for most of the nondynamical electronic correlation. As such, it is not ex-pected to yield reliable relative energies within the PES. Therefore, the dynamical correlation must also be accounted for. To this aim, we have chosen the CASPT2 approach4,5in which the CASSCF wave function is taken as the zeroth-order function to calculate the energy at the second zeroth-order of perturbation theory. This method has been employed with great success in the prediction of geometries and binding energies6as well as in the calculation of electronic spectra of small to medium-sized molecules.7 In this work we have adopted the CASPT2//CASSCF methodology, that is, opti-mal geometries are obtained at the CASSCF level and CASPT2 pointwise calculations are performed on the result-ing structures. The CASPT2//CASSCF method has an
esti-a兲Authors to whom correspondence should be addressed. Electronic mail:
[email protected] and [email protected]
2540
mated error in exoergicities of ⫾2 kcal mol⫺1, provided all the valence electrons are active and a large enough basis set is employed.
In practice, the most important aspect of the CASSCF method is the election of the active space, to which different approaches can be found in the literature.8,9In general, the selected MOs must be such that their corresponding natural orbital occupation numbers 共NOONs兲 are within the range 0.02–1.98, so as to account for the nondynamical correlation only. Also, it has been shown that to obtain the lowest energy for a given size of the active space the NOONs must be as different as possible from two or zero,8 and that the active space should be homogeneous throughout the PES so as to avoid discontinuities. However, the lowest energy and homo-geneity criteria are not easy to fulfil simultaneously for all nuclear configurations.10 The problem is more acute the smaller the active space is. These considerations are impor-tant for the present study, as it will be shown below.
In reactants关O(3P)⫹N2O兴, the dominant configuration presents two triplet-coupled electrons in two 2 p atomic oxy-gen orbitals, leaving the third 2 p orbital doubly occupied. A reasonable election is to take the MOs generated from all the 2 p atomic orbitals of the N2O2 system as the active space, i.e., a 14-electrons-in-12-orbitals or 共14,12兲 active space. However, it leads to an NOON of two for the atomic oxygen orbital carrying two paired electrons. Thus, it should not be taken as active according to the 0.02–1.98 criterion. Further, the energy drops considerably on substituting this orbital for one of the 2s-derived MOs in the N2O molecule, which pre-sents a NOON of about 1.98. This situation was found not only in reactants but also all along the entrance region of the PES. To the contrary, for the intermediate and exit regions of channel 共1兲 and for the exit region of channel 共2兲 all the 2 p-derived MOs are required, as all have NOONs essentially between 1.98 and 0.02. For channel共1兲 this fact generates an important energy gap between the entrance region and the other regions, so that the transition states 共TSs兲 in the en-trance region are lower in energy than the minima in the intermediate region. On the other hand, if the 2s-derived MO is kept in the intermediate region of channel共1兲 the CASSCF wave function is not well balanced, leading incorrectly to unsymmetrical stationary points. Following parallel work we are performing on the O(1D)⫹N
2O reaction, we added two more 2s-derived MOs to the active space to yield an 18-electrons-in-14-orbitals or 共18,14兲 active space. Again, one has two共18,14兲 active spaces, one in which three 2s-derived MOs are present 共reactants and entrance region, A-type space兲 and one with two 2s-derived MOs and all the 2 p-derived ones 共rest of channel 共1兲 and N2⫹O2, B-type space兲. In this manner, the TSs turn out to be placed above the minima to which they lead. In the reactants region, the
A-type active space is the only one that can be made to
converge; all attempts to obtain a B-type active space in this region were unsuccessful.
In this work we have employed the共14,12兲 active space in the search and characterization of the stationary points on the 3A
⬙
PES by means of CASSCF analytical gradients. These stationary points were then reoptimized with the共18,14兲 active space. The CASSCF 共18,14兲 calculations
amount to about 900 000 CSFs in Cs symmetry. Eventually,
the active space has been reduced so as to carry out studies on surface crossing 共see below兲. Numerical characterization was needed for some of the structures 共see below兲. To this aim, the SURVIBTM program of rovibrational analysis was employed.11 Pointwise CASPT2 calculations with both ac-tive spaces were performed on all the structures. The stan-dard Cartesian Gaussian 6-31G(d) 共Ref. 12兲 and 6-311G(2d) 共Refs. 13 and 14兲 basis sets of Pople and co-workers were selected. This basis set of triple-zeta quality plus two sets of polarization basis functions allowed us to achieve a rather correct description of the N(2D) ⫹NO(X2⌸) reaction15 as well as of the highly demanding van der Waals (NO)2dimers16in previous works of our own, in which the CASPT2 and CASPT2//CASSCF methods were employed, respectively. All the stationary points were char-acterized as either minima共MIN兲 or transition states by nu-merical calculation of their CASSCF harmonic vibrational frequencies. The G2 variant of the CASPT2 method was selected.17 In hereafter, the acronym CASPT2 will stand for the CASPT2 G2 method. The calculations were performed by using theMOLCAS 4.1共Ref. 18兲 and theGAUSSIAN 98共Ref. 19兲 packages of electronic structure calculations.
III. RESULTS AND DISCUSSION
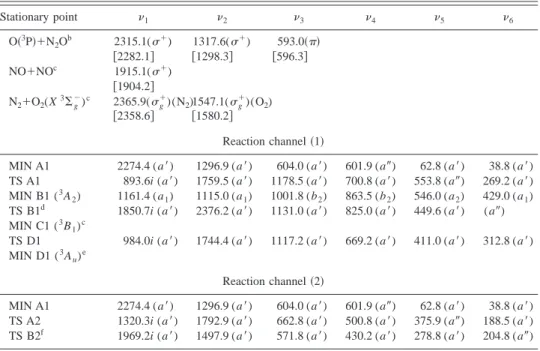
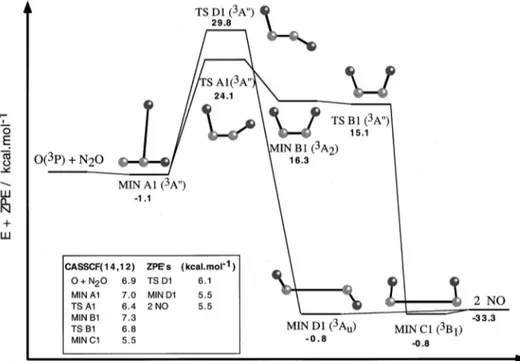
In Tables I, II, and III we show the geometries, harmonic vibrational frequencies and energies of the stationary points obtained, respectively. In Figs. 1 and 2 the energy profiles of channels 共1兲 and 共2兲, respectively, are presented.
As for the properties of reactants and products, the ge-ometries optimized at the CASSCF level are quite accurate
共Table I兲, although the bond distances are always slightly
overestimated 关0.6–1.1 % and average value of 0.6% for CASSCF共18,14兲兴 with respect to experiment. The frequen-cies show a maximum deviation of 33 cm⫺1共Table II兲 with the relative errors being in the interval 0.3–2.1 % for CASSCF共18,14兲 共average value of 1.1%兲. The exoergicities of reaction are also in good agreement with experiment
共Table III兲, although the error for channel 共1兲 关3.4 kcal mol⫺1 共9.3%兲 for CASPT2//CASSCF共18,14兲兴 is slightly larger than
the average estimated accuracy of the CASPT2 method. A qualitative description of the3A
⬙
PES can be done in terms of the dominant electronic configurations, which are detailed in Table IV. It is remarkable that O⫹N2O, MIN A1, TS A1, and 2 NO 关channel 共1兲兴, as well as N2⫹O2关channel 共2兲兴, present only one main CSF, with a CI coefficienthigher than 0.90. Note also that in MIN C1 and MIN D1
关channel 共1兲, NO-dimers兴 and in TS B2 关channel 共2兲兴 the two
dominant CSFs have almost an equal weight. As one can see in Fig. 1 关CASPT2//CASSCF共18,14兲 energy plus CASSCF共14,12兲 zero-point energy 共ZPE兲兴, in channel 共1兲 the oxygen atom can follow either a cis- or a trans-approximation to the N2O molecule passing through the shal-low van der Waals structure MIN A1. This structure is com-mon to both channels, as the oxygen atom is closer to the N central atom in the N2O molecule. A slight distortion either to the terminal N atom or to the oxygen atom leads to channels共1兲 or 共2兲, respectively. The barrier height including ZPE, though 5.7 kcal mol⫺1 smaller for the
cis-approx-imation, is in both cases relatively large 共24.1 and 29.8 kcal mol⫺1 for the cis- and trans-approaches, respectively兲. This fact can be justified if one considers that the N2O(X1⌺⫹) molecule is a closed-shell species in which an electronic promotion must take place prior to forming a bond with the open-shell oxygen atom. From TS A1 关cis-TS, Cs structure (3A
⬙
)] the system can evolve to the more symmet-ric (C2v) MIN B1 (3A2). However, TS D1 关trans-TS, Cs
structure (3A
⬙
)] can connect directly with MIN D1 关C 2h structure (3Au), NO-dimer trans-structure兴, and from this stationary point it can reach products. On elongation of the NN bond in MIN B1, an avoided crossing or a conical inter-section between the ground and the first excited 3A⬙
PES derived from O(3P)⫹N2O is patent, their CASSCF energies being very close to each other. As in the region near MIN B1 (C2v symmetry兲 the excited 3A⬙
state belongs to the 3B1 irreducible representation, it cannot interact with the ground state, which is classified as3A2. On distortion of the system to Cs symmetry, both states mix with each other becausethey belong to the same irreducible representation. Besides, a
nearby transition state关TS B1, Csstructure (3A
⬙
)] is locatedon the lowest PES. Numerical characterization was needed to obtain the structure and frequencies of TS B1, since single-root CASSCF gives rise to a cusp behavior共i.e., the energy is not a smooth function of the nuclear coordinates兲. Another, previously reported example of this kind of behavior is the weakly avoided crossing between the neutral and the ionic states of the LiF system.24 In both cases, discontinuities in the wave function arise at points close to the crossing, where the wave function should switch smoothly from one domi-nant configuration to the other. This phenomenon can be ex-plained by the poor description of anyone configuration in terms of the MOs optimized in a situation where the other one is dominant. Furthermore, in the LiF system MRCI cal-culations on single-root CASSCF wave functions, although intended to remedy the deficiencies of the latter, lead to sharp discontinuities in the dipole moment function. The only way around this problem seems to be to introduce a state-average CASSCF wave function, as we have also done, with the inconvenient that no analytical gradients were available to us TABLE I. Geometries of the stationary points on the3A⬙PES at the CASSCF共18,14兲 level.a
Reactants and products RNN⬘ /Å RN⬘O⬘ /Å ROO⬘/Å ⬍NN⬘O⬘/° ⬍N⬘O⬘O/° Dihedral/°
O共3P兲⫹N2O c 1.1343 1.1931 180.0 共1.1323兲 共1.1901兲 共180.0兲 关1.1273兴 关1.1851兴 关180.0兴 NO⫹NOd 1.1605 共1.1586兲 关1.1508兴 N2⫹O2(X 3 ⌺g⫺) d 1.1045 1.2207 共1.1037兲 共1.2197兲 关1.0977兴 关1.2075兴
Reaction channel共1兲 RNN⬘ /Å RN⬘O⬘ /Å ⬍NN⬘O⬘/°,⬍ONN⬘/° Dihedral/°b
MIN A1 1.1339 1.1931,3.5925 179.9,65.2 180.0 共1.1320兲 共1.1900,3.5926兲 共179.9,65.1兲 共180.0兲 TS A1 1.2140 1.2011,1.6059 148.2,108.4 0.0 共1.2175兲 共1.1983,1.6085兲 共146.9,107.5兲 共0.0兲 MIN B1 (3A2) 1.2822 1.3043,13043 113.0,113.0 0.0 共1.3243兲 共1.3000,1.3000兲 共108.6,108.6兲 共0.0兲 TS B1e 共1.3578兲 共1.2960,1.2971兲 共108.7,110.9兲 共0.0兲 MIN C1 (3B1) 3.6767 1.1632,1.1632 84.3,84.3 0.0 共3.6794兲 共1.1628,1.1628兲 共84.0,84.0兲 共0.0兲 TS D1 1.2094 1.2040,1.6530 146.8,105.7 180.0 共1.2124兲 共1.2025,1.6555兲 共146.0,104.1兲 共180.0兲 MIN D1 (3A u) 3.4547 1.1634,1.1634 108.7,108.7 180.0 共3.4927兲 共1.1624,1.1624兲 共105.9,105.9兲 共180.0兲
Reaction channel共2兲 RNN⬘ /Å RN⬘O⬘ /Å ROO⬘ /Å ⬍NN⬘O/° ⬍OO⬘N⬘/° Dihedral/°b
MIN A1 1.1339 1.1931 3.3650 179.9 75.9 0.0
共1.1320兲 共1.1900兲 共3.3650兲 共179.9兲 共75.8兲 共0.0兲
TS A2 1.1483 1.3942 1.6620 138.9 115.6 0.0
共1.1527兲 共1.3715兲 共1.6518兲 共137.8兲 共114.6兲 共0.0兲
TS B2f 共1.1618兲 共1.4025兲 共1.8072兲 共130.5兲 共100.4兲 共180.0兲
aThe CASSCF共14,12兲 geometries are shown in parentheses. The available experimental data are given in brackets. b
The connectivity of the atoms is assumed to be O–N–N⬘– O⬘for channel共1兲 and N–N⬘– O⬘– O for channel共2兲, where O is the attacking O atom. Hence, in each case the dihedral angle is given according to this connectivity. Only the relevant bonding parameters for each structure are reported.
cExperimental data taken from Ref. 20. dExperimental data taken from Ref. 21. eDerived from an analytical fit of 60 C
2v/Cs50:50 average3A2/3B1(1/23A⬙) points located near an avoided crossing or a conical intersection of these states.
A second order polynomial was employed obtaining a RMSD of 4.5⫻10⫺5a.u. (3⫻10⫺2kcal mol⫺1).
f
Derived from an analytical fit of 72 Cs/C150:50 average 1/2 3
A⬙(1/23A) points located near an avoided crossing or a conical intersection of these states.
for this kind of calculation. The energies for TS B1 in Table III are reported at the state-average level with the two states equally weighted. An estimate of the error committed with respect to the single-root energy can be made by calculating
both energies at the closer stationary point, MIN B1. The obtained difference is slightly below 2 kcal mol⫺1at both the CASSCF and the CASPT2 levels. The relevant CSFs and their CI coefficients for the lowest 3A
⬙
state in TS B1 are TABLE II. Harmonic vibrational frequencies共in cm⫺1兲 of the 3A⬙stationary points at the CASSCF共14,12兲level.a Stationary point 1 2 3 4 5 6 O共3P兲⫹N 2Ob 2315.1(⫹) 关2282.1兴 1317.6(⫹) 关1298.3兴 593.0共兲 关596.3兴 NO⫹NOc 1915.1(⫹) 关1904.2兴 N2⫹O2(X3⌺g⫺) c 2365.9( g ⫹)(N 2) 关2358.6兴 1547.1(g ⫹)(O 2) 关1580.2兴 Reaction channel共1兲 MIN A1 2274.4 (a⬘) 1296.9 (a⬘) 604.0 (a⬘) 601.9 (a⬙) 62.8 (a⬘) 38.8 (a⬘) TS A1 893.6i (a⬘) 1759.5 (a⬘) 1178.5 (a⬘) 700.8 (a⬘) 553.8 (a⬙) 269.2 (a⬘) MIN B1 (3A 2) 1161.4 (a1) 1115.0 (a1) 1001.8 (b2) 863.5 (b2) 546.0 (a2) 429.0 (a1) TS B1d 1850.7i (a⬘) 2376.2 (a⬘) 1131.0 (a⬘) 825.0 (a⬘) 449.6 (a⬘) (a⬙) MIN C1 (3B 1)c TS D1 984.0i (a⬘) 1744.4 (a⬘) 1117.2 (a⬘) 669.2 (a⬘) 411.0 (a⬘) 312.8 (a⬘) MIN D1 (3A u)e Reaction channel共2兲 MIN A1 2274.4 (a⬘) 1296.9 (a⬘) 604.0 (a⬘) 601.9 (a⬙) 62.8 (a⬘) 38.8 (a⬘) TS A2 1320.3i (a⬘) 1792.9 (a⬘) 662.8 (a⬘) 500.8 (a⬘) 375.9 (a⬙) 188.5 (a⬘) TS B2f 1969.2i (a⬘) 1497.9 (a⬘) 571.8 (a⬘) 430.2 (a⬘) 278.8 (a⬘) 204.8 (a⬙) aThe symmetries of the normal vibrational modes are given in parentheses, and the available experimental data
are given in brackets. The modes are ordered from larger to smaller frequencies.
b
Experimental data taken from Ref. 20.
cExperimental data taken from Ref. 21.
dFrequencies from analytical fit共see Table I兲. The out-of-plane frequency could not be calculated because of
numerical problems共see text兲.
e
Frequencies of the dimers not calculated because of the symmetry-breaking problem.
fFrequencies from analytical fit共see Table I兲.
TABLE III. Energies of the3A⬙PES stationary points calculated at different levels.a,b
Stationary point CASSCF共14,12兲 CASPT2//CASSCF共14,12兲 CASSCF共18,14兲 CASPT2//CASSCF共18,14兲 Experimentc
O共3P兲⫹N2O 0.0共0.0兲 0.0共0.0兲 0.0共0.0兲 0.0共0.0兲 0.0共0.0兲 NO⫹NO ⫺26.0 共⫺24.6兲 ⫺32.4 共⫺31.0兲 ⫺28.2 共⫺26.8兲 ⫺33.3 共⫺31.9兲 ⫺36.7 共⫺35.3兲 N2⫹O2(X3⌺g⫺) ⫺71.2 共⫺69.9兲 ⫺81.2 共⫺79.9兲 ⫺72.7 共⫺71.4兲 ⫺80.7 共⫺79.4兲 ⫺80.2 共⫺78.9兲 Reaction channel共1兲 MIN A1 ⫺0.5 共⫺0.6兲 ⫺1.1 共⫺1.2兲 ⫺0.5 共⫺0.6兲 ⫺1.1 共⫺1.2兲 TS A1 50.5共51.0兲 23.7共24.2兲 48.9共49.4兲 24.1共24.6兲 MIN B1 (3A 2) 57.7共57.3兲 19.4共19.0兲 48.4共48.0兲 16.3共15.9兲 TS B1d 58.9共59.0兲 15.9共16.0兲 45.8共45.9兲 15.1共15.2兲 MIN C1 (3B 1)e ⫺0.2 共⫺0.2兲 ⫺0.8 共⫺0.8兲 ⫺0.2 共⫺0.2兲 ⫺0.8 共⫺0.8兲 TS D1 57.1共57.9兲 29.4共30.2兲 54.3共55.1兲 29.8共30.6兲 MIN D1 (3A u) e ⫺0.3 共⫺0.3兲 ⫺0.8 共⫺0.8兲 ⫺0.3 共⫺0.3兲 ⫺0.8 共⫺0.8兲 Reaction channel共2兲 MIN A1 ⫺0.5 共⫺0.6兲 ⫺1.1 共⫺1.2兲 ⫺0.5 共⫺0.6兲 ⫺1.1 共⫺1.2兲 TS A2 61.3共63.1兲 38.4共40.3兲 59.2共61.0兲 38.5共40.3兲 TS B2d 77.6共80.2兲 55.8共58.4兲 75.6共78.2兲 56.3共58.9兲
aEnergies are given in kcal mol⫺1with respect to reactants. For MIN C1 and MIN D1, it is more convenient to give them relative to products. The absolute
energies of reactants at each one of the levels considered are as follows: CASSCF共14,12兲, ⫺258.744 475 a.u.; CASPT2//CASSCF共14,12兲, ⫺259.337 241 a.u.; CASSCF共18,14兲, ⫺258.752 660 a.u.; and CASPT2//CASSCF共18,14兲, ⫺259.339 496 a.u.
bEnergies with共without兲 ZPE without 共in兲 parentheses. c
Data derived from Refs. 20–23.
dPointwise state-average calculations on the geometry obtained by means of an analytical fit共see Table I兲. eZPEs derive from frequencies assumed to be equal to those of 2 NO molecules共see Table II兲.
given in Table IV. These coefficients are interchanged in the second3A
⬙
state. Proceeding further along the reaction path, the wave function acquires an increasing3B1 character, lead-ing finally to MIN C1, NO-dimer cis-structure, and products共2 NO兲.
In channel 共2兲 关see Fig. 2 共CASPT2//CASSCF共18,14兲 energy plus CASSCF共14,12兲 ZPE兴, similar structures are found in the entrance region, i.e., apart from the weakly bound MIN A1关Csstructure (3A
⬙
)], two TSs of cis-共TS A2兲 and trans-共TS B2兲 geometry 关Cs structures (3A⬙
)] are char-acterized. The corresponding barrier heights are considerably higher than those in channel共1兲. Besides, TS B2 is about 18 kcal mol⫺1 above TS A2, while in channel 共1兲 the alreadyreported energy gap is only 5.7 kcal mol⫺1. The larger insta-bility of these structures could be justified by the weaker OO bond formed, which would compensate for a smaller portion of the required N2O promotion energy. An avoided crossing or a conical intersection was also evident near TS B2, for which the same comments as for TS B1 regarding the need of using a state-average wave function can be applied. Note that the second 3A
⬙
state correlates with products in an ex-cited state, while in TS B1 both the ground and first exex-cited states correlate with products in their ground state. As one can notice from Table IV, the two main CSFs have almost equal CI coefficients in TS B2, which would indicate the proximity to a crossing point. By analogy with the structures FIG. 1. Schematic representation of the structures of the stationary points for channel 共1兲 obtained at the CASSCF共18,14兲 level including ener-gies 关CASPT2//CASSCF共18,14兲 plus CASSCF共14,12兲 ZPE兴 relative to reac-tants关O(3P)⫹N2O兴 except for MIN
C1 and MIN D1, which are given rela-tive to products共2 NO兲. For TS B1, the MIN B1 out-of-plane frequency was assumed to calculate the ZPE共see Table II兲.
FIG. 2. Schematic representation of the structures of the stationary points for channel 共2兲 obtained at the CASSCF共18,14兲 level including ener-gies 关CASPT2//CASSCF共18,14兲 plus CASSCF共14,12兲 ZPE兴 relative to reac-tants关O(3P)⫹N
reported in channel 共1兲, two van der Waals minima of cis-and trans-geometries in the exit valley are expected, al-though they are not shown in Fig. 2. However, as no such minima have been reported experimentally, we did not try to locate these presumably very shallow structures.
In Table I we note the important quantitative differences that exist between the CASSCF 共14,12兲 and 共18,14兲 geom-etries of MIN B1, while they are very similar for the other stationary points. These discrepancies are due to the impor-tant 2s-derived active MOs lacking in the 共14,12兲 space, whose NOONs are 1.99 and 1.98, indicating that they intro-duce a strong nondynamical correlation. For TS B1, the NOONs for these MOs are about the same as for MIN B1, so a geometry shift should also be expected. Unfortunately, in this case a pointwise characterization at the CASSCF共18,14兲 level was deemed too expensive to be carried out. We should mention that a certain difference in the NN distance and the NNO angle between the CASSCF共14,12兲 and 共18,14兲 levels can also be noted in MIN D1. However, in this case the discrepancies can be attributed to the flatness of the PES rather than to an intrinsic better description at the CASSCF共18,14兲 level.
In Table II the harmonic vibrational frequencies are pre-sented. They have been calculated only at the CASSCF共14,12兲 level because the CASSCF共18,14兲 calcula-tions are computationally too expensive. For TS B1, the out-of-plane frequency could not be calculated because this co-ordinate is too flat to be numerically fitted. Also, problems with the CASSCF wave function were found for the (NO)2 dimers共see below兲, so that for them the frequencies of prod-ucts are assumed to evaluate the ZPE.
The energies of the stationary points are shown in Table III共see also Figs. 1 and 2兲. On moving from the CASSCF to
the CASPT2 method there is a general stabilization of the stationary points, by about 30 kcal mol⫺1in channel共1兲 and 20 kcal mol⫺1 in channel 共2兲 with respect to reactants. In particular, a marked decrease in the barrier heights is appar-ent. It is worth mentioning that partial optimization of the triplet (NO)2 dimers, MIN C1 共cis-兲 and MIN D1 共trans-兲 was performed in our previous work16 at the CASPT2// CASSCF 共18,14兲 level. Thus, the NO distance and NNO angle of the corresponding singlet dimers were assumed and energies for different values of the NN distance were fitted to a suitable polynomial expression. The resulting geometries and dissociation energies with and without the basis-set su-perposition error 共BSSE兲 included were RNN ⫽2.9885 (3.2920) Å, RNO⫽1.1594 Å, ⬍NNO⫽94.5°, De
⫽1.08(0.28) kcal mol⫺1 for MIN C1; and R
NN ⫽2.9764(3.1636) Å, RNO⫽1.1594 Å, ⬍NNO⫽109.1°, De ⫽1.02(0.35) kcal mol⫺1for MIN D1. The parameters
includ-ing the BSSE are put in parentheses. It is patent the influence of the dynamical correlation in the shortening of the NN distances, as can be seen on comparing with Table I. Also, the BSSE has a great influence both on the NN distance and on the dissociation energy (De) in these van der Waals dimers.
Problems with the symmetry of the CASSCF wave func-tion were present in MIN B1, in the region near TS B1 and in the 共NO兲2 dimers. We reported on the same problem for the latter species in the work referred to above.16 Briefly, when the molecular framework symmetry is not imposed on the CASSCF wave function, a lower-energy broken-symmetry solution is obtained. This phenomenon has been the subject of several theoretical studies. For a recent paper with many references see Ref. 25. Symmetry breaking is an example of the more general problem of instabilities in self-consistent-TABLE IV. Electronic configurations and active spaces of the3A⬙stationary points at the CASSCF共18,14兲 level.
Reaction channel共1兲 Inactive Active Electronic configurationsa
O⫹N2O (Cs) 6a⬘ 10a⬘⫹4a⬙ 关12a⬘
22a⬙2兴1a⬘11a⬙1共0.94兲
MIN A1 (Cs) 6a⬘ 10a⬘⫹4a⬙ 关12a⬘
2
2a⬙2兴1a⬘11a⬙1共0.94兲
TS A1 (Cs) 6a⬘ 10a⬘⫹4a⬙ 关12a⬘
2
2a⬙2兴1a⬘11a⬙1共0.91兲 MIN B1 (3A2) (C2v) 3a1⫹3b2(6a⬘) 5a1⫹5b2⫹2a2⫹2b1(10a⬘⫹4a⬙) 关7a1
2 5b2 2 1a2 2 1b1 2兴1b 2 1 1b1 1 /关6a1 2 6b2 2 2b1 2兴1a 1 1 1a2 1 共0.86/0.25兲
TS B1 (Cs) 6a⬘ 10a⬘⫹4a⬙ 11a⬘21a⬘11a⬘22a⬙21a⬙1/11a⬘21a⬘21a⬘12a⬙21a⬙1 共0.84/0.10兲b
MIN C1 (3B1) (C2v) 3a1⫹3b2(6a⬘) 5a1⫹5b2⫹2a2⫹2b1(10a⬘⫹4a⬙) 关6a1 2 6b2 2 1a2 2 1b1 2兴1a 1 1 1b1 1 /关6a1 2 6b2 2 1a2 2 1b1 2兴1b 2 1 1a2 1 共0.67/0.64兲
TS D1 (Cs) 6a⬘ 10a⬘⫹4a⬙ 关12a⬘22a⬙2兴1a⬘11a⬙1共0.90兲
MIN D1 (3A
u) (C2h) 3ag⫹3bu(6a⬘) 5ag⫹2bg⫹2au⫹5bu(10a⬘⫹4a⬙) 关6ag
2 1bg 2 1au 2 6bu 2兴1a g 1 1au 1 /关6ag 2 1bg 2 1au 2 6bu 2兴1b g 1 1bu 1 共0.68/⫺0.64兲
2 NO (Cs) 6a⬘ 10a⬘⫹4a⬙ 关12a⬘22a⬙2兴1a⬘11a⬙1共0.94兲
Reaction channel共2兲
O⫹N2O (Cs) 6a⬘ 10a⬘⫹4a⬙ 关12a⬘
22a⬙2兴1a⬘11a⬙1共0.94兲
MIN A1 (Cs) 6a⬘ 10a⬘⫹4a⬙ 关12a⬘
2
2a⬙2兴1a⬘11a⬙1共0.94兲
TS A2 (Cs) 6a⬘ 10a⬘⫹4a⬙ 关12a⬘
2
2a⬙2兴1a⬘11a⬙1共0.89兲
TS B2 (Cs) 6a⬘ 10a⬘⫹4a⬙ 12a⬘
2
1a⬘11a⬘02a⬙21a⬙1/12a⬘21a⬘01a⬘12a⬙21a⬙1
共0.59/0.58兲b
N2⫹O2(X3⌺g⫺) (Cs) 6a⬘ 10a⬘⫹4a⬙ 关12a⬘
2
2a⬙2兴1a⬘11a⬙1共0.93兲
aLeading configurations and CI coefficients for each stationary point are given. For the configurations, the number of orbitals belonging to each irreducible
representation is indicated. The doubly occupied orbitals are enclosed in brackets while the singly occupied ones are given in the last place.
field methods, like the single determinantal SCF method26 and the CASSCF method.27–28 We note that not even the state-average CASSCF method is completely free from this problem.29The broken symmetry orbitals are valence-bond-like, i.e., they are located in one atom or fragment rather than being spread over the whole molecule. In our case, localiza-tion of one electron in each one of the NO moieties in the (NO)2 C2v-symmetric structures is encountered. This effect
becomes more acute on increasing the NN distance; thus, it does not take place in the entrance TSs. Whether symmetry breaking takes place or not depends on the balance between the so-called orbital size effect and resonance interactions.30 Hence, the orbital sizes are optimal for localized electrons, while the delocalized solution can be regarded as a resonance between the localized solutions in which the orbitals are op-timal only in an average way. For the above-mentioned (NO)2species, the resonance is not strong enough to prevent the electrons from localizing. The occurrence of this effect could be justified by the complicated electronic structure of MIN B1 and TS B1, in which two radical electrons are placed in different in- and out-of-plane MOs共see electronic configurations in Table IV兲, since it is known that open-shell species are particularly prone to symmetry breaking. For in-stance, several cases of broken symmetry are reported on open-shell C2v-symmetric systems somewhat similar to the
current triplet N2O2, like the doublet formyloxyl 共HCOO•兲 radical,30 the doublet and quartet states of the NO2 molecule31 or different triplet states of the CO2molecule.32
The spurious symmetry breaking must be distinguished from the real, physically motivated one, taking place in or near a conical intersection or an avoided crossing and caused by the nonadiabatic couplings, like that in TS B1.33To dis-tinguish both types of effects, two calculations can be done at a symmetric structure, i.e., one in which the wave function keeps the molecular symmetry and another one in which it does not. If these energies differ from each other, as it hap-pens for the above-mentioned structures, one faces a spuri-ous symmetry breaking.
A different kind of broken symmetry was present in the (NO)2 dimer region. In the 2 NO(X2⌸) asymptote, if C2v
or C2h symmetry is imposed on the CASSCF wave function, each of the NO fragments looked at separately keeps C⬁v symmetry with the radical electron distributed between the two degenerate * orbitals. However, if the symmetry is relaxed to Csthe electrons locate in only one orbital per NO
molecule, thereby breaking the cylindrical symmetry. These two situations lead to an energy difference of several kcal mol⫺1. Thus, even though the product calculation was performed in Cs symmetry 共see Table IV兲, the dissociation
energy of the dimers was calculated with respect to their same-symmetry C2v or C2h asymptotes. In this way, we are assuming that the Cs dissociation curve would be almost parallel to the C2v or C2h ones. In our work on the NO dimers we found this assumption to be a good compromise for this kind of symmetry breaking.16
To solve the firstly reported symmetry-breaking prob-lem, we have employed the CAS SI共CAS–state interaction兲 method.34,35 Application examples are available in the literature.36,37In this method, a nonorthogonal CI is applied
to two broken-symmetry solutions, i.e., the one obtained in the actual calculation and that generated from it by means of application of the lacking molecular symmetry elements. Thus, for a symmetric structure 共e.g., of C2v symmetry兲
these solutions are mirror images of each other. The resulting CAS–SI MOs belong to the molecular symmetry point group, and can be given as input CASSCF orbitals. For Cs
structures, that do not keep the molecular (NO)2 mirror plane, the two solutions are no longer equivalent. Even in this case, CAS–SI is applied in the same way to yield delo-calized solutions that join smoothly with the C2v one.25We have found that by applying this method, the CASSCF wave function recovers the correct symmetry in the affected re-gions of the PES.
Finally, we have performed conventional transition state theory共TST兲38rate constant calculations based on TS A1 and TS D1关channel 共1兲兴 and TS A2 关channel 共2兲兴, as obtained at the CASPT2//CASSCF共18,14兲 level. We discarded TS B2, which would have a minor contribution to the rate constant. The level splitting owned to the spin–orbit interaction in O(3P) has been taken account since it is expected to have an
important effect on the electronic partition function for all but very high temperatures. Therefore, the rate constant for reaction 共1兲 can be expressed as
k1,TST⫽
3
5⫹3e⫺E共3P1兲/kT⫹e⫺E共3P0兲/kT
⫻共k共3A
⬙
兲⫹k共3A⬘
兲兲, 共3兲where E(3P1) and E(3P0) are, respectively, the energies of the3P1and3P0levels of O(3P), 158.29 and 226.99 cm⫺1,23 with respect to the lowest energy level (3P2). In this expres-sion, k(3A
⬙
) and k(3A⬘
) derive from the classical expression of the TST rate constant excluding the electronic partition functions. In all transition state calculations, the rotational symmetry numbers are omitted. At this point we introduce the approximation that the contribution of the3A⬙
and of the 3A⬘
PESs are equal, since our preliminary results on the3A⬘
PES indicate strong similarity of their corresponding TS structures and energies. With this approximation, the follow-ing expression results:k1,TST⫽
6
5⫹3e⫺E共3P1兲/kT⫹e⫺E共3P0兲/kT
⫻关k共3A
⬙
,TS A1兲⫹k共3A⬙
,TS D1兲兴. 共4兲 As for channel 共2兲, since only the 3A⬙
PES correlates with reactants and products, one arrives at the expression:k2,TST⫽
3
5⫹3e⫺E共3P1兲/kT⫹e⫺E共3P0兲/kT关k共
3A
⬙
,TS A2兲兴. 共5兲Two levels of calculation have been considered: 共a兲 simple TST level; 共b兲 TST plus complete tunneling Wigner correction.39In the latter case the rate constant from a given TS takes the form
where⌫ is the Wigner correction factor of the corresponding TS that is expressed as ⌫⫽1⫺241
冉
hs kT冊
2冉
1⫹kT E0冊
. 共7兲In this expression, s is the imaginary frequency and E0 is the barrier height 共including the ZPE兲 for each one of the transition states considered.
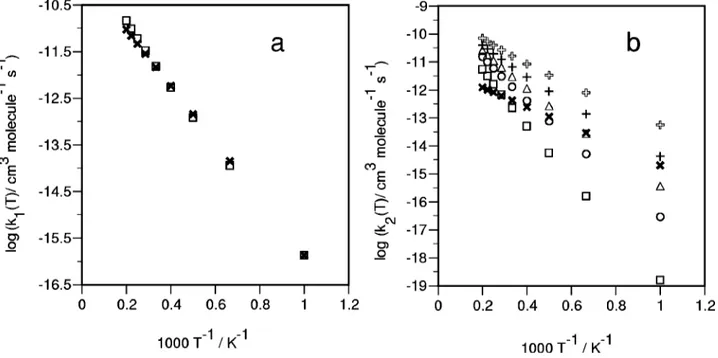
The TST results for the temperature interval 1000–5000 K, which brackets the experimental range, are collected in Table V. Note the almost perfect agreement between the pre-dicted rate constant values and the experiment for channel
共1兲, as the Arrhenius plots in Fig. 3 make evident. This
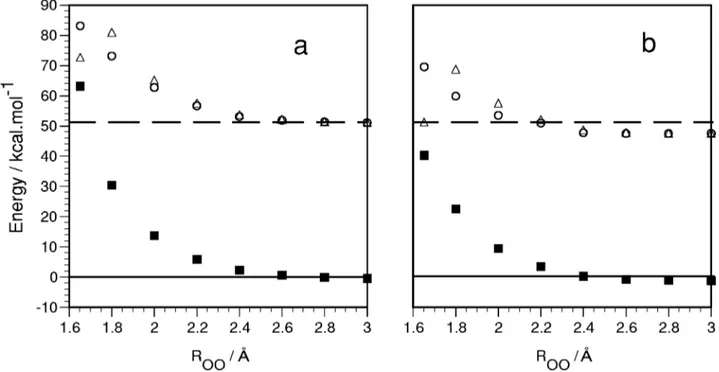
con-trasts with the results for channel共2兲, in which different val-ues for the classical barrier were considered, none of them giving even a reasonable agreement with experiment. To take deeper insight into this discrepancy, we have analyzed the possibility of a lower energy reaction path through an inter-system crossing 共ISC兲. To do this, we have constructed a minimum energy path共MEP兲 between reactants and the low-est TS A1 by freezing the OO distance and optimizing the rest of geometric parameters at the CASSCF共14,12兲 level. The energies of the corresponding structures were calculated for the 1A
⬘
, 1A⬙
, and 3A⬙
states. The results are shown in Fig. 4. In all points, a considerable energy gap between the triplet ground state and the singlet states is kept. Previously, an ISC for the 3A⬙
/1A⬘
PESs40 was located. However, itsFIG. 3. Arrhenius plot of the rate constants for reactions共1兲 and 共2兲 in the temperature interval 1000–5000 K. 共a兲 Reaction 共1兲: TST 共‘‘---䊐---’’兲; experimental
共‘‘---Ã---’’兲 共Ref. 1兲. 共b兲 Reaction 共2兲 for which several values of the classical barrier height have been considered in the TST calculation: E ⫽40.3 kcal mol⫺1 共‘‘---䊐---’’兲, E⫽30.0 kcal mol⫺1 共‘‘---䊊---’’兲, E⫽25.0 kcal mol⫺1 共‘‘---䉭---’’兲, E⫽20.0 kcal mol⫺1 共‘‘---⫹---’’兲, ⌬E⫽⫽15.0 kcal mol⫺1 共‘‘---open plus sign---’’兲; experimental 共‘‘---Ã---’’兲 共Ref. 1兲.
TABLE V. Rate constants for the O(3P)⫹N
2O reaction.a T/K Channel共1兲 Channel共2兲 TST TST共W兲b Experimentc TST TST共W兲b Experimentd 1000 1.26⫻10⫺16 1.36⫻10⫺16 1.36⫻10⫺16 1.38⫻10⫺19 1.60⫻10⫺19 2.02⫻10⫺15 1500 1.11⫻10⫺14 1.15⫻10⫺14 1.41⫻10⫺14 1.49⫻10⫺16 1.59⫻10⫺16 2.92⫻10⫺14 2000 1.19⫻10⫺13 1.22⫻10⫺13 1.44⫻10⫺13 5.41⫻10⫺15 5.63⫻10⫺15 1.11⫻10⫺13 2500 5.33⫻10⫺13 5.41⫻10⫺13 5.78⫻10⫺13 4.96⫻10⫺14 5.09⫻10⫺14 2.48⫻10⫺13 3000 1.51⫻10⫺12 1.53⫻10⫺12 1.46⫻10⫺12 2.25⫻10⫺13 2.29⫻10⫺13 4.23⫻10⫺13 3500 3.27⫻10⫺12 3.30⫻10⫺12 2.84⫻10⫺12 6.81⫻10⫺13 6.90⫻10⫺13 6.20⫻10⫺13 4000 5.98⫻10⫺12 6.02⫻10⫺12 4.67⫻10⫺12 1.59⫻10⫺12 1.61⫻10⫺12 8.25⫻10⫺13 4500 9.72⫻10⫺12 9.76⫻10⫺12 6.88⫻10⫺12 3.12⫻10⫺12 3.15⫻10⫺12 1.03⫻10⫺12 5000 1.45⫻10⫺11 1.46⫻10⫺11 9.37⫻10⫺12 5.42⫻10⫺12 5.46⫻10⫺12 1.23⫻10⫺12
aRate constants are given in cm3molecule⫺1s⫺1.
b
In this case the complete Wigner tunneling correction has been applied to the TST calculations. See the text.
cDetermined according to the expression: k
1⫽1.52⫻10⫺10exp(⫺13 930/T) cm
3molecule⫺1s⫺1共Ref. 1兲.
dDetermined according to the expression: k
geometry is rather different to those found in the MEP of the 3A
⬙
PES. Therefore, it seems that a triplet–singlet ISC does not exist at geometries around the MEP.This negative result led us to explore the alternative that the 3A
⬙
PES crossed with the3A⬘
PES, that does not corre-late with ground electronic state products 关N2(X1⌺g⫹) ⫹O2(X3⌺g⫺)兴 but instead with N2(X1⌺g⫹)⫹O2(C3⌬u). To this aim, we carried out a search of intersection points at the state-average CASSCF level employing the minimum energy crossing point algorithm41 as implemented in the GAUSSIAN 98 package.19 For practical reasons the active space had to be reduced to a minimum, i.e., four electrons in three orbitals, which corresponds to the oxygen atom 2 p-derived ones. In these calculations, the Cartesian Pople 6-31G共d兲 basis set was chosen. With this procedure we ob-tained the following structure: RNN⫽1.08 Å, RNO⫽1.29 Å,RON⫽1.96 Å, ⬍NNO⫽158°, ⬍OON⫽148°, ⬍OONN
⫽180°. We performed CASPT2 and CASSCF 共14,12兲
point-wise calculations on this structure, obtaining 3A
⬙
–3A⬘
en-ergy gaps of 1.1 and 1.3 kcal mol⫺1, respectively. Therefore, it can be inferred that the minimal共4,3兲 active space contains the relevant interactions to account for the surface crossing, so that a good prediction is achieved. The energy of this point relative to reactants for the 3A⬙
state is 32.4 共25.9兲 kcal mol⫺1 at the CASSCF 共CASPT2兲 共14,12兲 level. If this point is to act as the effective barrier to reaction, the latter will be lowered by about 14 kcal mol⫺1 with respect to TS A1 共see Table III兲.IV. SUMMARY
An ab initio study of the 3A
⬙
ground PES of the O(3P)⫹N2O(X1⌺⫹) reaction has been performed at the CASPT2//CASSCF level. The stationary points on the tworeactive channels have been characterized. Two paths are found in channel 共1兲, bearing predominant cis- and trans-arrangements. Channel 共2兲 presents similar structures in the entrance region. Most remarkable is the decrease in energy observed when introducing the dynamical correlation. A TST rate constant calculation was performed, reaching almost quantitative agreement with experiment for channel 共1兲. However, for channel 共2兲 there is a great discrepancy with experiment. The location of an ISC, that would reduce the energy barrier in this channel, was not feasible. Nevertheless, a 3A
⬙
–3A⬘
intersection point placed 25.9 kcal mol⫺1 above reactants was located. We suggest this to be a possible lower-energy pathway to products, that could lead to a better ac-cordance with the experiment in what respects to the N2⫹O2 formation.ACKNOWLEDGMENTS
This work has been supported by the Spanish Ministry of Education and Culture共MEC兲 through the project DGES PB98-1209-C02-01. Financial support from the ‘‘Generalitat de Calalunya’’共Autonomous Government of Catalonia兲, refs. 1998SGR 00008 and 2000SGR 00016, is also acknowl-edged. R.V. thanks also the ‘‘Generalitat de Calalunya’’ for a ‘‘Beca de Formacio´ d’Investigadors’’ predoctoral re-search grant. The authors are also grateful to the ‘‘Center de Supercomputacio´ i Comunicacions de Catalunya (C4-CESCA/CEPBA)’’ for computer time made available.
1N. E. Meagher and W. R. Anderson, J. Phys. Chem. A 104, 6013共2000兲,
and references therein.
2A. Fontjin, A. Goumri, A. Ferna´ndez, W. R. Anderson, and N. E. Meagher,
J. Phys. Chem. A 104, 6003共2000兲.
3B. O. Roos, The complete active space self-consistent field method and its
applications in electronic structure calculations, in Advances in Chemical
FIG. 4. Energy relative to reactants关O(3P)⫹N
2O兴 for the3A⬙,1A⬙, and1A⬘PESs, in front of the OO distance关channel 共2兲兴, taken as reaction coordinate,
with the rest of parameters optimized at the CASSCF共14,12兲 level for the3A⬙ state:共a兲 CASSCF calculations: 3A⬙ 共‘‘---䊏---’’兲,1A⬘ 共‘‘---䊊---’’兲, 1A⬙
Physics; Ab Initio Methods in Quantum Chemistry II, edited by K. P.
Lawley共Wiley, Chichester, UK, 1987兲, p. 399.
4K. Andersson, P.-A. Malmqvist, B. O. Roos, A. J. Sadlej, and K. Wolinski,
J. Phys. Chem. 94, 5483共1990兲.
5K. Andersson, P.-A. Malmqvist, and B. O. Roos, J. Chem. Phys. 96, 1218
共1992兲.
6K. Andersson and B. O. Roos, Int. J. Quantum Chem. 45, 591共1993兲. 7B. O. Roos, K. Andersson, M. P. Fu¨lscher, P.-A. Malmqvist, L.
Serrano-Andre´s, K. Pierloot, and M. Mercha´n, Multiconfigurational perturbation theory—applications in electronic spectroscopy, in Advances in Chemical
Physics: New Methods in Computational Quantum Mechanics, edited by
S. A. Rice共Wiley, New York, 1996兲, p. 219.
8
P. Pulay and T. P. Hamilton, J. Chem. Phys. 88, 4926共1988兲.
9
J. M. Anglada and J. M. Bofill, Chem. Phys. Lett. 243, 151共1995兲.
10K. Wolinski and P. Pulay, J. Chem. Phys. 90, 3647共1989兲.
11SURVIBTM, W. C. Ermler, H. C. Hsieh, and L. B. Harding, Comput.
Phys. Commun. 51, 257共1988兲.
12
P. C. Hariharan and J. A. Pople, Theor. Chim. Acta 28, 213共1973兲.
13R. Krishnan, J. S. Binkley, R. Seeger, and J. A. Pople, J. Chem. Phys. 72,
650共1980兲.
14M. J. Frisch, J. A. Pople, and J. S. Binkley, J. Chem. Phys. 80, 3265
共1984兲.
15
M. Gonza´lez, R. Valero, and R. Sayo´s, J. Chem. Phys. 113, 10983共2000兲.
16R. Sayo´s, R. Valero, J. M. Anglada, and M. Gonza´lez, J. Chem. Phys. 112,
6608共2000兲.
17K. Andersson, Theor. Chim. Acta 91, 31共1995兲. 18
K. Andersson, M. R. A. Blomberg, M. P. Fu¨lscher et al.,MOLCAS, Version 4.1, Lund University, Sweden, 1998.
19M. J. Frisch, G. W. Trucks, H. B. Schlegel et al.,
GAUSSIAN 98, Revision A.7, Gaussian, Inc., Pittsburgh, PA, 1998.
20J. L. Teffo and A. Che´din, J. Mol. Spectrosc. 135, 389共1989兲. 21
K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure,
Vol. IV. Constants of Diatomic Molecules共Van Nostrand Reinhold, New
York, 1979兲.
22H. Okabe, Photochemistry of Small Molecules共Wiley, New York, 1978兲.
23S. Bashkin and J. O. Stoner, Jr., Atomic Energy Levels and Grotrian Dia-grams共North-Holland, Amsterdam, 1975兲, Vol. I.
24C. W. Bauschlicher, Jr. and S. R. Langhoff, J. Chem. Phys. 89, 4246
共1988兲.
25
R. Broer and W. C. Nieuwpoort, J. Mol. Struct.: THEOCHEM 458, 19
共1999兲, and references therein.
26R. Seeger and J. A. Pople, J. Chem. Phys. 66, 3045共1977兲. 27
A. Sanchez de Meras, M.-B. Lepetit, and J.-P. Malrieu, Chem. Phys. Lett.
172, 163共1990兲.
28N. Ghihery, J.-P. Malrieu, D. Maynau, and K. Handrick, Int. J. Quantum
Chem. 61, 45共1997兲.
29A. Zaitsevskii and J.-P. Malrieu, Chem. Phys. Lett. 228, 458共1994兲. 30A. D. McLean, B. H. Lengsfield III, J. Pacansky, and Y. Ellinger, J. Chem.
Phys. 83, 3567共1985兲.
31C. P. Blahous III, B. F. Yates, Y. Xie, and H. F. Schaeffer III, J. Chem.
Phys. 93, 8105共1990兲.
32
L. Engelbrecht and B. Liu, J. Chem. Phys. 78, 3097共1983兲.
33E. R. Davidson and W. T. Borden, J. Phys. Chem. 87, 4783共1983兲. 34P.-A. Malmqvist, Int. J. Quantum Chem. 30, 479共1986兲.
35
P.-A. Malmqvist and B. O. Roos, Chem. Phys. Lett. 155, 189共1989兲.
36M. Mercha´n, R. Pou-Ame´rigo, and B. O. Roos, Chem. Phys. Lett. 252,
405共1996兲.
37
L. Rodrı´guez-Monge and S. Larsson, Int. J. Quantum Chem. 61, 847
共1997兲.
38D. G. Truhlar, A. D. Isaacson, and B. C. Garrett, in Theory of Chemical Reaction Dynamics, edited by M. Baer共CRC, Boca Raton, FL, 1985兲, Vol.
4, p. 65.
39J. I. Steinfeld, J. S. Francisco, and W. L. Hase, Chemical Kinetics and Dynamics共Prentice-Hall, Englewood Cliffs, NJ, 1993兲, p. 318. 40D. R. Yarkony, Electronic structure aspects of nonadiabatic processes in
polyatomic systems, in Modern Electronic Structure Theory, Part I, edited by D. R. Yarkony共World Scientific, Singapore, 1995兲.
41
M. A. Bearpark, M. A. Robb, and H. B. Schlegel, Chem. Phys. Lett. 223, 269共1994兲.