UNIVERSITÀ DEGLI STUDI DI CATANIA
DOTTORATO DI RICERCA IN BIOLOGIA, GENETICA UMANA E
BIOINFORMATICA: BASI CELLULARI E MOLECOLARI DEL
FENOTIPO XXV CICLO
DIPARTIMENTO DI ANATOMIA, BIOLOGIA E GENETICA, MEDICINA
LEGALE, NEUROSCIENZE, PATOLOGIA DIAGNOSTICA, IGIENE E
SANITÀ PUBBLICA
G.F. INGRASSIA
UNITÀ DI BIOMEDICINA MOLECOLARE GENOMICA E DEI SISTEMI COMPLESSI, GENETICA, BIOLOGIA COMPUTAZIONALE
Dott. Marco Maugeri
__________
Analysis of the involvement of exosomal miRNAs and proteins in the
response of CRC cells to Cetuximab
Tesi di Dottorato
Coordinatore e Tutor:
Chiar.mo Prof. MICHELE PURRELLO
TABLE OF CONTENTS
1.ABSTRACT 1
2.INTRODUCTION 3
2.1 Colorectal cancer 3
2.1.1 Molecular basis of CRC 4
2.1.2 Prevention and screening of CRC 10
2.1.3 EGFR targeted therapy in CRC 11
2.1.4 Molecular markers for the prediction of anti EGFR
treatment efficacy in CRC 16
2.2 MicroRNA: an overview 18
2.2.1 Genomics of miRNAs 20
2.2.2 Molecular pathway of miRNA biogenesis 24
2.2.3 Mechanisms of control of miRNA biogenesis 29
2.2.4 MiRNAs and EGFR pathway 32
2.2.5 Post transcriptional repression by miRNAs 33
2.2.6 MiRNA targets 37
2.2.7 MiRNAs and implications for cancer and drug response 39
2.2.8 Circulating miRNAs for cancer diagnosis 43
2.2.9 MiRNAs involvement in CRC 45
2.2.10 MiRNAs in CRC therapy 50
2.3 Exosomes 51
2.3.1 Exosomal RNA delivery and miRNAs role 57
2.3.2 Exosomes and miRNAs: clinical relevance in cancer 58
3.MATERIALS AND METHODS 65
3.1 Cell Lines 65
3.2 Determination of chemosensitivity to Cetuximab 66
3.3 Cetuximab treatment 66
3.4 Exosome isolation and characterization 67
3.5 RNA isolation 68
3.6 Reverse transcription and miRNA profiling 68
3.6.1 MiRNA expression data analysis 70
3.7 MiRNA targets analysis 72
3.8 Transfection of exosomes and cell viability assay 73 3.9 Exosomal proteins profiling through antibody microarrays 74
3.9.1 Antibody microarrays printing 75
3.9.2 Exosomal proteins extraction 75
3.9.3 Proteins labeling 76
3.9.4 Sample incubation 76 3.9.5 Antibody microarrays data analysis 77
3.9.6 RNA binding proteins prediction analysis 78
4.RESULTS 79
4.1 Confirmation of different sensitivity of CRC
cells to Cetuximab treatment 79
4.2 CRC cell lines exosomes characterization 79
4.3 Exosomes and cellular miRNAs profiling in Caco 2 and
HCT 116 after Cetuximab treatment 81
4.3.1 Normalizators selection using different methods 81 4.3.2 MiRNA profiling after Cetuximab treatment
and asymmetric distribution of specific
miRNAs between exosomes and cells 82
4.4 Gene ontology analysis of exosomal and cellular miRNAs in CRC cells before and after
4.5 Exosomal proteins profiling of Caco 2 and HCT 116
cell lines after Cetuximab treatment 95
4.5.1 Antibody microarrays normalization 95
4.5.2 Exosomal proteins profiling 97
4.6 Exosomal proteins gene ontologies analysis 105
4.7 RNA binding proteins prediction analysis 110
4.8 Cell Viability assay after transfection of exosomes in CRC cells 111
5.DISCUSSION 116
6.CONCLUSIONS AND FUTURE PERSPECTIVES 126
1. ABSTRACT
It has been demonstrated that intercellular communication via cell released vesicles is very important both for normal and tumor cells, and specifically to determine tumor development and progression as well as invasion and angiogenesis. Cell communication could involve the exosomes, small vescicles of endocytic origin, which are released from different kinds of donor cells; they can transfer molecular signals as proteins and RNAs through the extracellular environment to specific recipient cells in a autocrine, paracrine or endocrine way. Exosomes can strongly influence the recipient cells phenotype by transferring oncogenes that could influence the response of cells to drugs or immune reactions. Recently, it has been demonstrated the presence of miRNAs inside the exosomes and their potential involvement in cancer development. Considering the important role of miRNAs in colorectal cancer, one of the most diffused and studied tumor, it was considered interesting to investigate the role of exosomal miRNAs and associated proteins in the response of CRC cells to Cetuximab (an anti EGFR therapeutic antibody). The EGFR signaling pathway is very importantly in relationship both to CRC and miRNA biogenesis and expression, and recently also to the exosomal communication system. Therefore, one of the major aims of this thesis was to analyze the possible involvement of exosomes in the response to Cetuximab of two CRC cell lines (wild type KRAS Caco 2 cells and KRAS mutated HCT 116 cells) through the transfer of specific miRNAs and proteins to recipient cells. To carry out this analysis, we performed cellular and exosomal miRNA profiling after Cetuximab treatment, for 745 miRNAs by using Real Time PCR. The results of the analysis showed that exosomal miRNA profiles globally reflect those of whole cells at steady state, but there exists an important quantitative asymmetrical distribution. After Cetuximab treatment, Caco 2 sensitive cells showed several exosomal differentially expressed (DE) miRNAs in comparison to HCT 116 cells. Many DE miRNAs
are involved in cancer and immunity. These data could be explained by considering that the EGFR pathway can regulate miRNA biogenesis via the MAPK/ERK cascade. Exosomal proteins analysis was performed for 741 cancer related proteins through a specific antibody microarrays platform. Also the profile of exosomal proteins from Caco 2 cells showed important alterations after Cetuximab treatment. Globally, several DE miRNAs and proteins from Caco 2 exosomes were related to cancer, stimulation of immunity and inflammation. Interestingly, exosomes transfection experiments between Caco 2 and HCT 116 cell lines (performed to investigate their effect on cell viability) showed that the transfection of steady state Caco 2 exosomes in the HCT 116 cell line determined a decrease of cell viability of recipient cells, while Cetuximab treated Caco 2 cells exosomes, transfected in HCT 116 cells, increased their viability. These data could be explained considering that exosomes from Cetuximab treated cells are enriched in oncogenic and immune stimulation related miRNAs. Finally, DE proteins were searched to find potential RNA binding proteins. Globally, the results of this thesis could be useful to: (1) verify the existence of horizontal transfer of genetic informations in eukaryotes; (2) search for potential miRNAs and proteins biomarkers of Cetuximab response in CRC in vivo. Eventually, it will be interesting to perform the characterization of exosomal miRNAs and proteins expression profiles of plasma from CRC patients after Cetuximab treatment. Moreover, the characterization of the asymmetrical distribution of miRNAs between cells and exosomes could be important to further investigate the potential and specific mechanism of miRNA sorting within exosomes.
2. INTRODUCTION
2.1 Colorectal cancer
Colorectal cancer (CRC) is one of the most pervasive causes of cancer morbidity and mortality all over the world, in both sexes, specifically in the western society. In particular, about 150,000 U.S. residents are diagnosed annually with CRC, and approximately one third of CRC patients die from the disease (1). Moreover, the lifetime risk of CRC in the United States is 6%, and the average age at diagnosis is 66 years (1). In particular, CRC begins as a benign adenomatous polyp, which develops into an advanced adenoma with high grade dysplasia and then progresses to an invasive cancer. Invasive cancers that are confined within the wall of the colon (tumor–node–metastasis stages I and II) are curable, but if untreated, they spread to regional lymph nodes (stage III) and then metastasize to distant sites (stage IV). Stage I and II tumors are curable by surgical excision, and up to 73% of cases of stage III disease are curable by surgery combined with adjuvant chemotherapy. Recent advances in chemotherapy have improved survival, but stage IV disease is usually incurable (2). Moreover, CRC cases can be classified on the basis of the histological features; since CRC is a heterogeneous multifactorial disease and to better discriminate CRC cases, it is important to molecularly classify the tumors of the different patients on the basis of the specific and most common genetic alterations of CRC. This is important to predict the prognosis and the drug response of patients. In particular, CRCs can be classified on the basis of chromosomal instability (CIN), microsatellite instability (MSI), and CpG island methylator phenotype (CIMP). CIN, characterized by karyotypic variability resulting from gains and/or losses of whole/portions of chromosomes (3). MSI, due to inactivation of DNA mismatch repair genes was
found in 15% of sporadic CRCs, and is associated to TGFβRII, EGFR, and BAX genes mutations; many other genes, involved in cell proliferation, apoptosis and DNA repair are often altered in MSI (3). CIMP (CpG island methylator phenotype) is determined by alterations of methylation patterns, crucially involved in the transcriptional silencing of regulators of tumor suppression, cell cycle, DNA repair, and apoptosis: 35 40% of CRC (3). Based on these molecular features, CRCs can be further classified in 5 molecular subtypes:
Type 1 (CIMP high MSI H BRAF mutation);
Type 2 (CIMP high MSI L or MSS BRAF mutation) Type 3 (CIMP low MSS or MSI L KRAS mutation) Type 4 (CIMP neg MSS)
Type 5 or Lynch syndrome (CIMP neg MSI H)
2.1.1 Molecular basis of CRC
Although in last decades were conducted numerous studies about the pathogenetic mechanism that underlies both the CRC onset and progression, much remains to be clarified about the etiological factors of CRC, that is properly a complex and heterogeneous disease. However, it was shown that different mechanisms and factors could contribute as risk factors to CRC pathogenesis: environmental factors, dietary habits, lifestyle, inherited and somatic mutations. Concerning dietary habits and lifestyle, a diet rich of unsaturated fats and red meats, high total energy intake, frequent assumption of alcohol and limited physical activity are all factors that could promote the onset of the cancer (4 6). On the other hand, some drugs and substances could protect the organism against the onset of CRC, e.g. non steroidal anti inflammatory drugs, estrogen and calcium (7, 8). Also inflammation, hormones and gut flora could be involved in CRC progression (9). Anyway, it is clear that different and not well defined
environmental factors could interact with others factors (e.g. genetics, dietary) so influencing the carcinogenesis of CRC. Notwithstanding the modest progresses achieved to identify environmental and lifestyle risk factors in CRC development, in the last years different mutations affecting specific genes involved in CRC were identified. These genetic defects could underlie both the inherited predisposition to CRC (mutations in germ cells) and the onset of sporadic forms of the cancer, in which the patients acquire specific somatic mutations that arise during their lifetime. However, the most important genetic mechanism in CRC pathogenesis is represented by the appearance of genetic alterations that lead to novel or increased functions of oncogenes and alterations that lead to loss of function of tumor suppressor genes (TSGs) (1). Principally, the conversion of the cellular genes into oncogene alleles can result from particular point mutations and from alterations of the structure and the function of the genes such as chromosome alterations (e.g. rearrangements or amplifications that can lead to an altered gene expression regulation). To date, the detected oncogenes related mutations are principally somatic. The TSGs inactivation in CRC arises from localized mutations, complete loss of genes and epigenetic alterations and any other mechanisms that lead to their altered regulation. Prominently, the TSGs mutations are somatic. However, it is estimated that 15 30% of CRCs cases may have a major hereditary component given the occurrence of the pathology in first or second degree relatives (10, 11). Approximately, one quarter of these familial cases (i.e., <5% of all CRC cases) occurs in a setting with family history and/or clinical features that indicate a highly penetrant, Mendelian cancer syndrome that predisposes to CRC (1). However, most cases of inherited CRCs are represented by the hereditary nonpolyposis colorectal cancer (HNPCC) syndromes, and another significant subset is associated with familial adenomatous polyposis (FAP) and closely related variant syndromes. Moreover, CRC is related also with different types and less common inherited gastrointestinal (GI) tumor syndromes. In particular, in the Table 1.1 are summarized the common features of the most important forms of these syndromes with the involved germline mutations. For example, FAP is associated with APC
gene mutation. In HNPCC it is important the germline inactivation of one allele of either of the mismatch repair genes MSH2 or MLH1 in combination with somatic inactivation of the other alleles. Although the fraction of inherited CRC cases is small, the acquired knowledge about their development and progression have allowed to gain important new knowledge about the factors and the mechanisms that could be the basis of the sporadic forms of the cancer (the most frequent cases). However, the colorectal tissue could be affected by different benign GI tumors that are represented by lesions that are originated from the epithelial tissue and that project above the surrounding mucosa and are commonly termed polyps.
_______________________________________________________________________________
Table 1.1: Molecular genetics of the inherited forms of CRC. The most important gene alterations and the features of the
syndromes are shown in the table (From Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479 507).
In human CRC, most of these polyps have normally a size less then 5mm and are hyperplastic (1), and it was shown that they aren`t the principal cause of CRC tumorigenesis. On the other hand, adenomatous polyps (adenomas) (specific lesions derived from the glandular epithelium and characterized from hyperplastic morphology and altered differentiation of the epithelium cells), seem to be most strongly related to CRC onset: accordingly, it is thought that the adenomas are the precursor lesions of the CRC. The prevalence of adenomas in the United States is approximately 25% by age 50 and approximately 50% by age 70 (1). There is a
high risk of CRC in individuals whose adenomas are not removed, and polypectomy decreases the risk of CRC (1). Moreover, it`s important to consider that individuals affected by syndromes that strongly predispose to adenomas, such as FAP, invariably develop CRCs by the third to fifth decade of life if their colons are not removed (1). Nevertheless, only a fraction of adenomas progress to cancer, and progression probably occurs over years to decades. For instance, adenomas roughly 1 cm in size may have an approximately 10% to 15% chance of progressing to carcinoma over a 10 year period (1). So, because most colorectal carcinomas appear to arise from adenomas, in the last decades numerous studies analyzed the events that lead from adenomas to CRC. Concerning this, in 1990 Volgelstein hypothesized a general model to describe the linear histological and genetic events that lead to CRC, the adenoma to carcinoma sequence (ACS) (1,
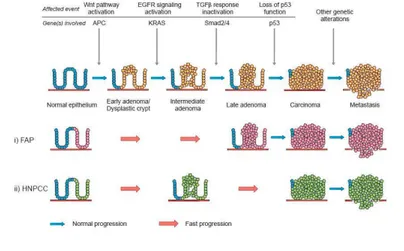
12). This model is likely to be an oversimplification, but it aligns observed clinicopathological changes with genetic abnormalities in the progression colorectal cancer (Figure 1.1).
_______________________________________________________________________________
Figure 1.1: The adenoma to carcinoma sequence. (In the upper side) Sporadic forms; (B) FAP; (C) HNPCC. (Adapted from
Davies, R. J., et al. 2005. Colorectal cancer screening: prospects for molecular stool analysis, Nature Review Cancer 5:199 209).
On the basis of the ACS model, the progression from normal epithelium to colorectal cancer, starting from the adenoma, requires an accumulation of mutations in specific genes that affect the balance between proliferation and
apoptosis. Generally, the steps in development of sporadically occurring cancer in a normal colon epithelium require several mutations in specific genes, but for FAP and HNPCC few gene mutations can lead to a fast formation or progression of CRC in comparison to the sporadic forms of the cancer (Figure 1.1). At any rate, crucial molecular events of CRC sporadic forms include derangement of the Wnt pathway and defects in the transforming growth factor β (TGF β) signalling pathways, which exert a synergistic effect on the cell cycle. Finally, with loss of p53 function, several cell cycle checks and balances are disrupted, which paves the way to gross chromosomal aberrations and aneuploidy. The exact sequence of changes and the subsequent interactions between the products of the altered genes and pathways are very much important to elucidate and to have a clear and global picture about the whole mechanism. The intact or mutated key molecules of the Volgelstein model interact in many ways and at different points, forming an intricate network of molecular events, which unfolds during CRC tumorigenesis
(13). Anyway, these alterations in CRC formation and progression could be point
mutations affecting specific genes, chromosome mutations that cause genomic instability, and epigenetic alterations. Specifically, the loss of genomic stability can drive the development of colorectal cancer by facilitating the acquisition of multiple tumor associated mutations. The most common type of genomic instability in colorectal cancer is chromosomal instability, which causes numerous changes in chromosomal copy number and structure in important tumor suppressor gene, as APC, P53, and SMAD family member 4 (SMAD4), involved in CRC as a gene that can oppose to the malignant phenotype. The first molecular event in CRC formation is the inactivation of the APC gene, that is the leading cause of FAP and is present in 80% of sporadic forms of the tumor. This tumor suppressor gene is importantly involved in the regulation of differentiation, adhesion, polarity, migration, development, apoptosis, and chromosomal segregation (14). The product of the APC gene is an important regulator of β catenin protein within the Wnt pathway that is involved in the promotion of cell proliferation and differentiation, particularly in epithelial cells. Normal APC can
bind β catenin and determine its inactivation and subsequently its degradation by ubiquitin mediated degradation through the proteasome (15). Active β catenin can enter the cell nucleus to promote the transcription of several genes involved in cell proliferation, such as cyclin D1 and the oncogene MYC. Therefore, APC inactivation leads to aberrant proliferation of normal cells and to the formation of early adenomas. Normally, the following step in CRC pathogenesis is the acquisition of KRAS gene mutations that lead to EGFR signaling pathway deregulation, promoting cell proliferation and apoptosis escape. This happens in early or larger adenoma, where GTPase KRAS can be mutated in exon 2 (codons 12 and 13) or exon 3 (codon 61) in 40 50% of CRCs and contribute to the development of colorectal adenomas and hyperplastic polyps. KRAS mutations lead to the production of the activated form of the protein that can trigger the EGFR pathway proliferating signals in a continuous way; in turn, this event can increase the growth and invasion capacities of adenoma cells. KRAS mutation is followed by biallelic loss of chromosome 18q in up to 70% CRCs, associated to the loss of tumor suppressor genes as DCC (a cell surface receptor for neuronal protein netrin 1, important in cell adhesion and apoptosis), SMAD2 and SMAD4 (involved in TGF β signaling pathway), and TP53 mutations in the switch from late adenoma to early carcinoma. For MSI forms of CRC, the cancer development is characterized by Wnt pathway alterations and BRAF mutations. BRAF protein is involved in Mitogen Activated Protein Kinases/ Extracellular Signal Regulated Kinases (MAPK/ERK) signaling pathway to promote cell proliferation and migration. Mutations in microsatellite sequences can affect TP53 and BAX genes
(16). Also PTEN tumor suppressor gene can be altered in CRC, and also the AKT
pathway that is commonly hyperactivated. Inactivating mutations of PTEN, which are a late event in CRC carcinogenesis correlated to advanced metastatic tumors, occur in about one third of CRCs (17). Moreover, gain of function mutations of the PIK3CA gene upstream the pathway, occurring in 20% of CRCs, cause AKT signaling even in absence of growth factors.
2.1.2 Prevention and screening of CRC
There is considerable evidence that screening of asymptomatic persons, who are at average risk, can detect cancers at an early and curable stage, resulting in a reduction in mortality (18, 19). Furthermore, some screening tests may also detect cancer precursor lesions, which, if removed, may result in a reduced incidence of CRC (20). There are several different screening tests, each with advantages and limitations: differences among strategies are related to the sensitivity and specificity of the tests, their complexity, and the associated risk that complicate the process of informed decision making. For CRC patients showing clear clinical features and a family history consistent with a given familial syndrome, diagnostic tests can be easily performed to identify the germline mutation in patients and other members of the family to check if they have inherited the mutation. In case of sporadic CRC, however, screening and prevention are the main instruments for early diagnosis and design of the best therapeutic strategy. A well defined precursor lesion (adenoma) and a long preclinical course make CRC a candidate for screening. In practice, two main screening strategies are available: faecal occult blood testing (FOBT), and endoscopic screening (flexible sigmoidoscopy, FS, or FS and colonoscopy). FOBT is the most widely used screening test for CRC
(21) and the only screening test currently recommended by the European Union (22). In particular, CRCs bleed and this blood can be detected in the stool and
FOBTs are non invasive, cheap, easy to use, and may be carried out at home. As CRCs only bleed intermittently, FOBTs have to be repeated either each year or every other year to increase sensitivity for cancer. FS allows inspection of the mucosa as well as tissue biopsies and polyp removal in the distal part of the colon. Screening with FS reduces CRC mortality by 22%–31% and incidence by 18%– 23% (23 25). Colonoscopy allows direct inspection of the entire colon mucosa, tissue biopsies and polyp removal throughout the colorectum in one single session. These qualities suggest that colonoscopy is an ideal test for both early detection and prevention of CRC. In experienced hands, the sensitivity and specificity of
screening colonoscopy to detect advancedadenomas and cancer approaches 100%, and it is the final conclusive examination following any other positive screening test. Although colonoscopy is usually considered the gold standard to detect colon pathology, it is invasive, time consuming, expensive and associated with possible pain. Finally, genetic and epigenetic markers (in faeces or blood) for the detection of adenomas or early invasive CRC is a rapidly emerging field. Markers that have been associated with cancer or adenomas include the well known KRAS, APC and p53 genes, methylation markers (as vimentin and septin 9), and proteins as CEA or M2 PK (26).
2.1.3 EGFR targeted therapy in CRC
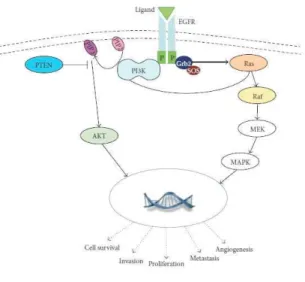
The ErbB family of receptor tyrosine kinases comprises ErbB1, 2, 3 and 4. ErbB1, also known as EGFR, is a typical member of the ErbB family having a tyrosine kinase activity stimulated upon ligand binding (27, 28). EGFR is a 170 kDa transmembrane receptor glycoprotein, composed of an intracellular tyrosine kinase (TK) domain, a transmembrane lipophilic segment, and an extracellular ligand binding domain that is important to bind its autocrine ligands: the most important of them are epidermal growth factor (EGF), transforming growth factor alpha (TGF α) (29), and neuregulin family proteins: these latter areinvolved in the activation of DNA synthesis and cell growth in numerous tumor types, including gastrointestinal diseases (GI) and especially CRC (29). EGFR catalyzes the transfer of phosphate molecules from ATP to an active site endowed with tyrosine kinase activity to mediate signals, triggering a cascade of well identified molecular events that will protect cells from apoptosis, facilitate invasion, and promote angiogenesis (29). Several intracellular signaling pathways are involved in the control of these functions and are functionally regulated by EGFR activation; mainly, they comprises MAPK/ERK, p38 Map Kinase, PI3K/AKT and JAK/STAT pathways (Figure 1.2).
_______________________________________________________________________________
Figure 1.2: EGFR signaling pathway general overview. The main downstream signaling pathways controlled by EGFR
receptor are reported as well as the biological results following their activation.
Normally, EGFR receptor acts as homodimer: in particular, after the binding of its ligand, it homodimerizes or heterodimerizes with other ErbB members, which promotes the autophosphorylation of several tyrosines within the intracellular domain; this in turn determines the serial activation of specific mediator proteins that finally lead to the activation of several and specific protein kinase cascades. In particular, the effects of EGFR activation comprise the activation of RAS and STAT proteins, SRC family kinases, AKT protein and MAP kinases, inducing the transcription of genes involved in cellular processes such as cell division and survival (29). In particular, AKT (a well established antiapoptotic protein) enables cell survival by indirectly regulating NF kB that in turn can lead to the blockage of the apoptosis. (29). AKT may also promote telomerase activity by phosphorylation of the human telomerase reverse transcriptase (30); by activating the matrix metalloproteinase protein it can induce tumor invasion and metastases (29). Activation of the MAPK kinase pathway signaling also increases the expression of major proteins involved in cell cycle regulation and in the negative regulation of the apoptosis: therefore, it is evident that EGFR pathway alterations are strongly involved in the genesis of several cancers (31). Enhanced activity or overexpression of EGFR has been found to be associated with tumor progression
and poor survival in various malignancies, such as head and neck, lung, breast, gastrointestinal tract and bladder cancers (29). Also EGFR ligands alterations are common, as for TGF α, heparin binding EGF like growth factor (HB EGF), and amphiregulin (32). In particular, EGFR, that is overexpressed in 25 77% of CRC, is associated with advanced stages of the disease (33), with a poor prognosis; it may predict a potential metastatic risk (34). In CRC, also KRAS, BRAF, PI3K3CA and the tumor suppressor PTEN can be altered. Therefore, EGFR was suggested as a potential target for antitumor agents, both for its position on the top of several cancer related pathways and because it is easily reachable by drugs
(Figure 1.3). EGFR signaling pathway can be targeted by either monoclonal
antibodies (C225) or tyrosine kinase inhibitors [ZD 1839 (gefitinib), OSI774 (erlotinib), CI1033, PKI166, GW572016], or even by antisense approaches (antisense molecules to EGFR or targeting key regulatory regions of the EGFR)
(29).
_____________________________________________________________________________
Figure 1.3: The figure shows the actual pharmacological approaches for EGFR targeted therapy in cancer, the drugs and the
relative molecular targets are reported. As showed, also several molecular component of the EGFR controlled downstream pathways can be used as therapeutic targets.
Therefore, each of these approaches has distinct mechanisms of action (Figure
binding of the specific ligands; TK inhibitors (TKIs) target the intracellular TK domain. About their clinical applications on the patients, several studies have substantiated and conferred significant benefits of anti EGFR agents in several types of solid tumors, including colorectal, head and neck cancer, non small cell lung cancer (NSCLC), and pancreatic cancer in terms of overall survival, progression free survival and overall response rate (35): therefore, some of these drugs have been approved by Food and Drug administration (FDA) for clinical applications for the patients (e.g., the monoclonal antibodies Cetuximab and Panitumumab). Anti EGFR monoclonal antibodies are specifically designed to bind the extracellular region of EGFR acting as competitive inhibitors, thus preventing receptor dimerization, autophosphorylation and downstream signaling
(36); in addition, they can induce receptor internalization, ubiquitinization,
degradation and prolonged downregulation. Cetuximab (IMC C225 / Erbitux) is an FDA approved human–murine chimeric anti EGFR monoclonal antibody (37) that binds to the second (L2) domain of EGFR, thereby blocking its downstream signaling by prompting receptor internalization and ligand receptor interactions. Cetuximab can be used as monotherapy or in combination with different chemotherapeutics. Importantly, it was approved by FDA in 2004; in 2008 the Committee for Medicinal Products for Human Use (CHMP) approved Cetuximab for patients with advanced colorectal cancer, who had 75% EGFR positive expression and wild type KRAS in their tissues and had failed oxaliplatin or irinotecan based chemotherapy. This is important because KRAS mutational status, specially for CRC, is a strong Cetuximab response predictor (biomarker); recently, it has been shown that using combined Cetuximab with oxaliplatin and capecitabine did not show any benefit even in patients with wild type KRAS tumors (35). Moreover, Cetuximab is currently used for the treatment of patients with squamous cell carcinoma of the head and neck with metastatic disease and in combination with radiation therapy for locally advanced cancer. Notwithstanding Cetuximab efficiency, the precise mechanisms through which it expresses its antitumor activity after downregulating EGFR are numerous and not completely
cleared yet. As predictable, EGFR impairment by Cetuximab negatively affects cell cycle progression (S phase controlled by growth factors) and promotes apoptosis (38). Moreover, angiogenesis and metastases processes can be involved in Cetuximab effects on tumor cells; about this, it was demonstrated that Cetuximab decreases tumor cell production of angiogenic growth factors, as vascular endothelial growth factor (VEGF), bFGF and interleukin 8 (IL 8); this in turn correlates with a significant decrease in microvessel density and an increase in apoptotic endothelial cells (38). In addition, Cetuximab antitumor effect can be driven by triggering antibody dependent cellular cytotoxicity (ADCC), through activation of cytotoxic host effector cells (39). Panitumumab (formerly ABX EGF) is a second, fully humanized IgG2 monoclonal antibody, FDA approved, used for the treatment of EGFR expressing metastatic colorectal cancer; similar to Cetuximab, the activating mutations in the KRAS gene resulting in EGFR indipendent activation of MAPK signaling in CRC, are important reasons to discriminate the patients that can undergo to Panitumumab treatment: clearly, the administration of this drug is suggested for patients harbouring wild type KRAS. EGFR targeted TKIs are small molecules that act as adenosine triphosphate (ATP) analogues and inhibit EGFR signaling by competing and binding at ATP binding pockets on the intracellular catalytic kinase domain of receptor Tyrosine Kinases, thereby preventing autophosphorylation and activation of several downstream signaling pathways. In particular, Gefitinib, Erlotinib and Lapatinib are important examples of TKIs (Figure 1.3): Erlotinib can inhibit cell proliferation, blocking the cell cycle in the G1 phase. Erlotinib is currently approved in patients with relapsed NSCLC (35). Finally, it is important to pinpoint that kinases inhibitors can be used to target molecular key components of the signaling pathways, which are under the control of EGFR. For example, MAPK inhibitors can be used to block MAPK cascade impairing the function of specific enzymes as MEK 1/2 and BRAF (Figure 1.3).
2.1.4 Molecular markers for the prediction of anti EGFR
treatment efficacy in CRC
As described, anti EGFR Monoclonal Antibody therapy is one of the most promising interventions to contrast CRC, especially for metastatic CRC (mCRC). Although clinical applications of Cetuximab and Panitumumab extend the median survival time for metastatic mCRC patients beyond 2 years (40, 41), several studies have shown that the efficacy of these drugs for mCRC patients who had been previously treated with chemotherapy is only around 10%. Accordingly, the current challenge is the personalization of anti EGFR therapy for mCRC. Mutations in the EGFR gene or its increased gene copy number detection were positively associated with efficacy of therapies, although they are poor predictors for anti EGFR therapy clinical efficacy. In addition, the incidence of somatic mutations of EGFR in advanced colorectal cancer is less than 1%. These mutations are independent of KRAS mutation (42) and not associated with Cetuximab and Panitumumab treatment responses (43, 44). In contrast, genetic variations in the two EGFR dependent signaling pathways (RAS RAF MAPK and PI3K PTEN AKT) may reveal more informations for predicting the clinical efficacy of anti EGFR therapies (45). In particular, RAS activates several key molecules of the RAS RAF MAPK signal cascade, as RAF, MEK1/2, and ERK to control cell growth, differentiation, and apoptosis. Previous studies have demonstrated that the RAS RAF MAPK signaling pathway is a primary contributor to the anti EGFR treatment response in mCRC patients, specifically for Cetuximab / Panitumumab resistance and for EGFR TKIs (e.g., Erlotinib) (45). KRAS activating mutations are considered to be an independent predictor of poor anti EGFR treatment efficacy and approximately 35% 45% of mCRC patients exhibit KRAS mutations. The efficacy of Panitumumab monotherapy for chemotherapy resistant mCRC patients, exhibiting the wild type KRAS gene, is 10% 17% compared with 0% for patients with KRAS mutations (45). The efficacy of Cetuximab is 12.8% for patients with wild type KRAS and 1.2% for patients with KRAS
mutations (46). Normally, KRAS is a GTP binding protein, involved in the activated EGFR transduction signaling to downstream pathways. Activated KRAS is involved in the hydrolysis of GTP to GDP, after which it is turned off. Activating mutations of the KRAS gene lead to an increase of cell proliferation signals that cannot be significantly inhibited by Cetuximab, since this mAb acts upstream the KRAS protein (Figure 1.4). KRAS mutational status is normally used to discriminate the patients that can use or not Cetuximab or Panitumumab.
___________________________________________________________________________________________________
Figure 1.4: The effects of anti EGFR treatment on wild type and mutated KRAS target cells.
Also BRAF mutational status could be a good predictor of anti EGFR treatment response. Approximately 6% 10% of mCRC patients exhibit BRAF gene mutations; in several studies, these mutations are associated with Cetuximab resistance. In particular, the most common mutation of BRAF is V600E that induces structural changes in RAF protein, which is the first effector identified downstream of RAS, increasing its kinase activity (47). This finding suggests that testing for the BRAF V600E mutation complements KRAS mutation analysis and may be as important as KRAS testing for treatment decisions, although the relationship between BRAF mutation and Cetuximab resistance is not as clear as for KRAS (48). However, it is important to stress that KRAS and BRAF mutations are mutually exclusive. Also PTEN deregulation (e.g., deletion), which plays a key role in CRC pathogenesis, is detected in CRC patients; PTEN inactivation is not only a predictor of non response to anti EGFR therapy (45), but also is a marker for poor prognosis specially for mCRC. Also PIK3CA and AKT mutations are
detected in many CRC cases: they could be useful as predictors of anti EGFR therapy response. Finally, it seems that the best prediction approach for anti EGFR response is the shared mutational status analysis of all of the involved genes, to identify the best approach to treat the patients.
2.2 MicroRNA: an overview
MiRNAs are small RNAs, 18 26 nucleotides in length, that serve a central biologic function in regulating gene expression (49, 50). In particular, instead of being translated into proteins, the mature forms of miRNA can bind to one or more specific 3’ untranslated region (3’UTR) of messenger RNA (mRNA), in a sequence specific manner, to interfere with its translation or to cause its degradation: therefore, miRNAs represent an important post transcriptional gene silencing (PTGS) mechanism. Globally, this silencing mechanism that involves other small noncoding RNAs, is defined RNA interference (RNAi) (51). The
discovery of miRNAs and their target mRNAs has uncovered novel mechanisms regulating gene expression beyond the central dogma. To date, only a single possible instance of positive gene regulation miRNA mediated has been described
(52). It is estimated that only 1% of the genomic transcripts in mammalian cells
encode miRNAs, whereas nearly one third of the genes may be regulated by miRNAs (50, 53). This it is very important also from the point of view of the
evolution, because the central role of the miRNAs in the cellular gene expression network emphasizes the emerging importance of the major fraction of the human genome, which doesn't code proteins: prior to the complete characterization of the human genome sequence, this had been previously considered as useless DNA
(54, 55). The ENCODE project, a large and high throughput study to identify and
characterize novel functional sequences of the human genome, has further shown the important regulatory role of the human noncoding sequence. Besides miRNAs,
the very large molecular group of non coding RNA comprises siRNAs (small interfering RNAs), piRNAs (Piwi interacting RNAs) and lncRNAs (long non coding RNAs). Also siRNAs and piRNAs are involved in the RNA interference mechanism (56). About the discovery of the miRNAs, the founding members of the this class were originally identified in C. elegans as genes that were required for the timed regulation of developmental events. Since then, hundreds of miRNAs have been identified in all metazoan genomes, including worms, flies, plants and mammals. Functionally, miRNAs have diverse expression patterns and it has been shown that they can regulate various physiological processes, such as development, cell differentiation, cell proliferation, cell death, chromosome structure, virus resistance, signaling transduction; most important, they are involved in pathological processes, especially for tumors (Table 1.2) (57 59).
___________________________________________________________________________________________________
Table 1.2: Mammalian miRNAs expression patterns in specific tissues and abnormal miRNAs expression in tumors.
Globally, the discovery of miRNAs adds a new dimension to our understanding of complex gene regulatory networks: certainly, this kind of regulation (strictly linked to transcription factors) represents a novel level of control of gene
regulation; more recently, it has become apparent that miRNAs themselves are subject to complicated control, at the levels of both miRNA metabolism and function, particularly during rapid developmental transitions or changes in cellular environment. It is important to stress that the numbers of individual miRNAs expressed in different organisms are comparable to those of transcription factors or RNA binding proteins (RBPs); the nature of miRNAs interactions with their mRNA targets, which involve short sequence signatures, makes them well suited for combinatorial effects with other miRNAs or RBPs that associate on the same mRNA. Potentially, miRNAs could target dozens or even hundreds of different mRNAs, therefore individual miRNAs can coordinate or finely tune the expression of a plethora of cellular proteins (60). For these reasons, the considerable number of studies and publications about miRNAs biogenesis, genomics, targeting mechanism and functional role are increasing the knowledge about them, although much still remains to clarify about miRNAs function and their activity regulation within cells.
2.2.1 Genomics of miRNAs
Since the discovery of RNAi, efforts to identify endogenous small RNAs have led to the identification of hundreds of miRNAs in nematodes, fruit flies and humans. In the last decade, more than 500 different miRNAs have been identified in animals and plants, where the number of miRNA genes is expected to increase to 500–1000 per species, which would comprise 2–3% of protein coding genes
(61). In humans, it`s estimated that the human genome may encode over 1000
miRNAs (for more informations: http://www.mirbase.org/). Moreover, from the evolutionary point of view, nearly all miRNAs are conserved in closely related species and many have homologous genes in distant species (61). These data are important because they stress the evolutionary importance of miRNAs within RNAi. In order to identify novel miRNA genes, three different approaches have
been used. The first approach is through forward Genetics: normally it identifies the miRNA genes mutations associated with specific and generally altered phenotypes, such as in the case of lsy 6 in C.elegans. This was the first miRNA with a role in neuronal patterning, and provided new insights into left / right axis formation (62). A second approach has used the directional cloning to construct a cDNA library for endogenous small RNAs (63, 64). This methodology has allowed a large scale miRNA identification: hundreds of miRNAs have been cloned from various cell lines, diverse tissues of mouse, fly and zebrafish, and a wide range of developmental stages in mouse, fly, worm, frog and zebrafish (65
68). However, this approach shows limitations, as the difficulty to find miRNAs
expressed at low level or only in specific conditions (e.g. the cell type). To bypass these limitations, the bioinformatic approach and the deep analysis of genomic sequences are very precious. Computational identification of miRNA genes is based largely on phylogenetic conservation and the structural characteristics of miRNA precursors. Simple homology searches by specific algorithms can reveal homologous copies of known miRNAs; also the identification of conserved genomic segments in the intergenic area, which potentially fold into a stem loop structure, represents another important parameter to find miRNA genes. More recently, some algorithms to identify novel miRNA sequences have exploited the conservation of the seed region of the miRNA, a sequence of nucleotides in the 5` region of mature miRNAs. In particular, nucleotides at 2–7 positions (relative to the 5` end of miRNAs) are responsible for recognizing the 3`UTR of target mRNAs and forming RNA duplexes. By retrieving all the RNAs able to form these duplexes with mRNAs, it´s possible to find candidate miRNA genes. Moreover, an intensive integrative approach was taken by Bentwich et al., who combined bioinformatics predictions with microarray analysis and sequence directed cloning and found 89 novel human miRNAs (69). Bioinformatically predicted miRNAs are validated for their expression, generally by PCR and microarray (70). Recently, the development and use of high throughput sequencing techniques (71) and the continuing refinement of computational prediction
algorithms (71) have contributed to increase the number of miRNAs identified. miRNA genes are scattered in all chromosomes in humans, except for the Y chromosome. Approximately 50% of known miRNAs are found in clusters (61,
71 73) and are transcribed as polycistronic primary transcripts. miRNAs in a given
cluster are often related to each other, suggesting that the gene cluster is a result of gene duplication. A miRNA gene cluster often contains unrelated miRNAs. A plausible but yet to be validated possibility is that the clustered miRNAs are functionally related by virtue of targeting the same gene or different genes in the same pathway. Although an important fraction (it is estimated about 70%) of miRNA genes is located in defined transcription units (TUs) (74), these loci can be differentially organized in the genome (71):
• Intergenic miRNAs,
• Intronic miRNAs, both protein coding and non coding genes,
• Exonic miRNAs, both protein coding and noncoding genes
Intergenic miRNAs are located between known TUs (Figure 1.5). These miRNAs can be monocistronic with their own promoters or polycistronic, where several miRNAs are transcribed as cluster of primary transcripts with a common promoter. Intronic miRNAs are found in the introns of annotated genes, both protein coding and noncoding. These miRNAs can be present as a single miRNA or as a cluster of several miRNAs. Intronic miRNAs are thought to be transcribed from the same promoter as their host genes and processed from the introns of host gene transcripts. In the particular case of mirtrons, the intron is the exact sequence of the pre miRNA with splice sites on either side (75, 76). Exonic miRNAs are far more rare than either of the types above and often overlap an exon and an intron of a noncoding gene. These miRNAs are also thought to be
transcribed by their host gene promoter and their maturation often excludes host gene function (77).
__________________________________________________________________ Figure 1.5: Genomic organization of the miRNA genes. These genes can be associated with both coding and noncoding
proteins genes.
Concerning the complete definition of the structure of the miRNA genes, this type of research is ongoing and much remains to be done on this topic. Generally, the most important aim is to identify the putative promoters of miRNA genes. About this, recently the identification and biochemical confirmation of 59 putative promoters for 79 miRNAs in human cells have been reported (78). Additionally, polymerase II chromatin immunoprecipitation of regions surrounding known miRNA genes has revealed promoters for miRNAs that are located several kb upstream (79). Analysis of transcription start sites, expressed sequence tag matches, CpG island predictions, 5’ and 3’ end identifiers, transcription factor binding sites, and polyadenylation signals were combined to analyze the structure of intergenic human miRNAs (80). Based on these analyses, it was discovered that the majority of intergenic human miRNAs have a primary transcript between 3 and 4 kb long, with a clearly delineated transcription start site and poly(A) signal serving as the boundaries of the RNA transcript.
2.2.2 Molecular pathway of miRNA biogenesis
Animals miRNAs are processed through a complex multi step process
(Figure 1.6) from long precursor molecules (pri miRNA), which are transcribed in
the nucleus by the RNA polymerase II, either from independent genes or as part of host gene (representing introns or exons) (Figure 1.6): therefore, it seems that miRNA genes might be transcriptionally regulated through their host gene promoters. However, in a first nuclear step of the canonical biogenesis pathway, the transcribed pri miRNA is cleaved into ~70 nucleotide precursor (pre miRNA) that is transported to the cytoplasm and in the second event that follows, this precursor is cleaved to generate ~21–25 nucleotide mature miRNA; then, through its association with a specific effector complex this is able to repress the expression of specific mRNA targets. The miRNA biogenesis is a highly regulated process, whose steps are finely controlled (81). Although the canonical miRNA biogenesis pathway is the most conserved and widely used, it exists a non canonical processing mechanism, called mirtron pathway, which allows the processing of “mirtrons”, intronic miRNA that are located within mRNA encoding host genes (82). Normally, the introns that enter the mirtron pathway are transcribed together with the host mRNA, and then spliced and debranched by
Lariat Debranching Enzyme (Ldbr), after which they fold into pre miRNA
hairpins. Even though mirtrons are independent from Drosha cleavage, they are known to function in gene expression regulation similar to canonical miRNAs.
__________________________________________________________________ Figure 1.6: Molecular pathway of miRNA biogenesis (From Winter J et al. S. Many roads to maturity: microRNA
biogenesis pathways and their regulation. Nat Cell Biol. 2009 Mar;11(3):228 34).
In the canonical pathway of biogenesis, the long pri miRNA (several kb) (79) folds into a hairpin, which is characterized by the presence of a bulge at a specific position and acts as substrate for two members of the RNAse III family of enzymes, Drosha and Dicer (Figure 1.7). These enzymes are crucial for miRNA biogenesis and are characterized by highly conserved RNAse III domains in animals and are dsRNA specific endonucleases, which generate 2 nucleotide long 3′ overhangs at the cleavage site (49). In particular, Drosha is a large protein of ~160 kD predominantly localized in the nucleus, which contains two tandem RNAse III domains (RIIIDs) and a dsRNA binding domain (dsRBD), which are essential for the catalysis, and an amino terminal segment of unknown function
(83). Regardless of the diverse primary sequences and structures of pri miRNAs,
during the catalysis process (Cropping) Drosha cleaves these into ~70 bp pre miRNA that consist of an imperfect stem loop structure (figure 1.6). It has been
shown that Drosha acts as a complex together its cofactor, the Di George syndrome critical region gene 8 (DGCR8) protein, in humans. This complex is known as the Microprocessor complex and it is able to recognize specific pri miRNAs.
__________________________________________________________________ Figure 1.7: The RNAe III domain (RIIID) is the catalytic domain that is responsible for the endonucleolytic reaction of
RNAe III enzymes such as Dicer and Drosha. The RIIIDs (shown as RIIIDa and RIIIDb) are well conserved motifs found in RNAe III type proteins of eubacterial, archaeal and eukaryotic origin.
After its nuclear generation, the pre miRNA is exported by Exportin 5 (Exp5), a Ran GTP dependent nucleo/cytoplasmic cargo transporter, to the cytoplasm where these hairpin precursors are cleaved by Dicer into a small (~20 25bp) imperfect dsRNA duplex (miRNA: miRNA*) that contains both the mature miRNA strand (guide strand) and its complementary strand miRNA* (passenger strand). This process is called “dicing”. In particular, Dicer is a highly conserved protein that is found in almost all eukaryotic organisms, as S. pombe, plants and animals. Some of these organisms contain more isoforms of this protein, with different roles, such as in flies; in particular, D. melanogaster has both Dicer1 (required for pre miRNA cleavage) and Dicer2 (needed for siRNA generation) (83). Mammals contain a single copy of Dicer. Dicer homologues are multidomain proteins of ~200 kDa, which contain two RIIIDs and a dsRBD, a long N terminal segment that contains a Dead box RNA helicase domain, as well as a DUF283 domain and
a PIWI Argonaute Zwille (PAZ) domain (Figure 1.7). The PAZ domain is important since it is responsible for the binding to the 3′ protruding end of small RNA (83). Normally, Dicer functions in association with many other proteins, such as transactivation responsive (TAR) RNA binding protein (TRBP) and the Argonaute family proteins, composed by a large number of components in the different species. Importantly, these proteins could have other important roles in miRNA stability and effector complex formation and action (83). However, during its catalysis Dicer`s efficient dsRNA cleavage requires dimerized RNAe III domains: on the basis of known RNAe III structures, functional catalytic sites can only be formed at the interface of the RNAe III dimer, and generate mature miRNA of 21 25 nucleotides: such differences in size possibly result from the presence of bulges and mismatches in the pre miRNA stem (49). Following processing, the guide strand of the miRNA/miRNA* duplex is preferentially incorporated into an effector complex, named miRNA induced silencing complex (miRISC), whereas the other strand (passenger or miRNA*) is released and degraded. In particular, the miRISC shares core components with that of siRNA, so that both are collectively referred to as the RNA induced silencing complex (RISC). The RISC is mainly composed of the Argonaute protein AGO2, TRBP, PACT (protein activator of PKR) and Dicer. The functions of Dicer might be broader than previously suspected, involving both the initiation and the effector steps of miRNA/siRNA mediated gene silencing. However, the most relevant protein in the mammalian RISC is AGO2 that acts as catalytic center of the complex (84). About AGO family proteins, which have a crucial role within RISC, most species express multiple AGO homologues: AGO1–AGO4 in mammals; dAGO1 and dAGO2 in flies; ALG 1 (argonaute like gene) and ALG 2 in C.
elegans. In particular, these proteins are involved in miRNA or both miRNA and
siRNA pathways; in mammals, each of the four AGO proteins (AGO1 AGO4) functions in the miRNA repression mechanism, but only AGO 2 is involved in the RNAi pathway through siRNA. The AGO 2 (Figure 1.8) protein is the main RISC component responsible for the mRNA target direct degradation, given that its
RNaseH like P element induces wimpy testis (PIWI) and PAZ domains. The RNAseH like P element induced wimpy testis (PIWI) domain is competent in endonucleolytic cleaving the mRNA.
__________________________________________________________________ Figure 1.8: Structure of AGO 2 and GW182 proteins.
Within the human RISC, the AGO proteins interact directly with miRNAs, but other accessory proteins can exert other important functions, as the glycine tryptophan protein of 182 kDa (GW182), which acts as downstream effectors in the repression (50, 60). The fragile X mental retardation protein (FMRP), a component of the RISC, is a RNA binding protein involved in the modulation of translation, specifically in the neurons. The target mRNA specificity, and probably also the functional efficiency of a miRNA, require that the mature miRNA strand from the miRNA:miRNA* duplex be selectively incorporated into the RISC for target recognition. About the mechanism of mature miRNA selection much still remains to be clarified, although it seems that the retained strand is the one that has the less stably base paired 5’ end in the miRNA/miRNA* duplex (49, 85). However, as Dicer generate the miRNA: miRNA* duplex, the stability of the 5′ ends of the two arms of the miRNA:miRNA* duplex is usually different depending on the intrinsic characteristics of the hairpin stem strands sequence (pre miRNA). However, miRNA* strands are not always by products of miRNA biogenesis and in specific cases can also be loaded into the RISC to function as miRNA. Differently from the guide strand, normally the miRNA* strand is degraded rapidly upon its exclusion from the RISC: in fact, the recovery rate of miRNA* from endogenous tissues is ~100 fold lower than that of miRNA (49). In
conclusion, these findings indicate that the relative instability at the 5′ end of the mature miRNA might facilitate its preferential incorporation into the RISC. The thermodynamic properties of the miRNA precursor determine the asymmetrical RISC assembly, and therefore the target specificity for post transcriptional inhibition.
2.2.3 Mechanisms of control of miRNA biogenesis
Different mechanisms to regulate miRNAs biogenesis have been detected; in particular, recent studies have shown that various factors or growth factor signaling pathways control every step of the miRNAs biogenesis pathway (86). It is important to stress that the alterations of these mechanisms are strongly linked to cancer. The transcription of miRNAs is one of the major regulatory steps in their biosynthesis. Many studies suggest that several characteristics of miRNA gene promoters (CpG islands, TATA box, TFIIB recognition, initiator elements and histone modifications) are similar (and essentially shared with) to the regulatory sequences of the protein coding gene promoters. As previously reported, miRNA genes are transcribed by the RNA polymerase II. From this point of view, the transcription factors (TFs) that regulate the miRNA genes are shared with protein coding genes, including c myc and p53; this also applies to cell type specific transcription factors, as MEF2, PU.1 and REST (86). Moreover, miRNA gene transcription is dynamically regulated by the most important cellular signaling pathways through specific growth factors, as Platelet Derived Growth Factor (PDGF) and Transforming Growth Factor b (TGF b). It has been shown that c Myc binds to E boxes and activates transcription of the miR 17 92 cluster
(87). Often, c MYC hyperactivation (common in some tumors) is consistent with
high level of the miR 17 92 cluster, which has been shown to negatively control the E2F1 gene; this encodes a cell cycle regulator. Recenty, it has been shown also
that miRNAs can regulate the TFs level generating specific and important regulatory loops, negative or positive, that determine the fine regulation of miRNAs levels. Many of the mechanisms of epigenetic control known to regulate protein coding genes, as DNA methylation and modifications of histones, seem to apply to miRNA genes. For example, in bladder cancer the expression of miR 127 is decreased through promoter hypermethylation (88); miRNA gene promoters are also regulated by histone modifications during development and pathogenesis. MiRNAs biogenesis regulation can be importantly exerted at the level of pri miRNA processing by the Drosha complex. This kind of regulation can be reached with different mechanisms. One of them is the tune control of the total levels of Drosha and DGCR8 proteins in the cells: different experimental evidences show that there are mechanisms that increase the pri miRNA processing, but they are still unknown; actually, the most believable hypothesis considers possible that post transcriptional modifications or association with accessory factors could alter the activity of Drosha or DGCR8. For example, the DEAD box RNA helicases p68 (DDX5) and p72 (DDX17) are involved in this regulation by binding the Drosha/DGCR8 pre miRNA complexes together with multiple p68 interacting proteins, including the Smads, p53 and estrogen receptor a (ER α). These proteins can transduce cellular signals, as DNA damage (p53), estrogen signals (ER α), and TGF β stimulation. The interaction between p68/p72 proteins and Smads proteins and p53 can promote Drosha activity, increasing the level of different miRNAs. At any rate, this regulation appears to be very specific (86) (Figure 1.9).
__________________________________________________________________ Figure 1.9: Pri miRNA processing regulation by Drosha control through p68/p72. In the figure are shown the signals that
can influence miRNA biogenesis.
Moreover, there exist other accessory proteins that regulate Drosha activity modifying the pri miRNA structure: for example, the protein hnRNP A1 is required for the processing of a member of the miR 17 92 cluster, miR 18a; this protein can bind the mentioned miRNA to increase Drosha binding. Conversely, the NF90/NF40 proteins complex can bind the pri miRNA preventing Drosha binding with the result of decreased pre miRNA cellular levels. Proteins as KSRP can bind the loop region of pri miRNA and pre miRNA to promote the binding and the activity of Dicer and Drosha proteins. Moreover, the Lin28 protein can bind the pre miRNA to decrease the Dicer action and the effect of the TUT4 protein could determine the pre miRNA let 7 family degradation (86). The Dicer proteins level are also finely regulated, and the association with its cofactors as TRBP or PACT is important for Dicer stability and activity: in fact, the depletion of these proteins in cells decreases the steady state levels of Dicer protein. The function of these cofactors, and in turn Dicer activity, can be regulated by different signaling pathway and specifically by the MAPK/ERK pathway. The nuclear transport of pre miRNAs could be potentially regulated under specific physiological conditions, but actually the evidences about this aren’t so strong. Moreover, total levels of the Ago proteins within the cell also contribute to global miRNA regulation and biogenesis. Ectopic expression of Ago proteins results in a
dramatic increase in mature miRNA. The dramatic increase in mature miRNA mediated by increased Ago expression could indicate that Ago proteins are limiting in the cell and serve to stabilize miRNA (86). Finally, the level of pri miRNA and pre miRNA could be regulated by RNA editing through the proteins ADAR (adenosine deaminase) that can convert adenosine to inosine, altering the secondary structure and the stability of the RNA: such alterations of the seed region could affect target mRNA binding (60). The mature miRNA decay could permit the degradation of specific miRNAs, regulating in a global way the biological effects of these miRNAs (89).
2.2.4 MiRNA and EGFR pathway
Since miRNAs biogenesis and functions are strongly related to the main cellular pathways, it is very important to characterize the regulatory interplay between these pathways and miRNAs. About this, recently it was shown that the EGFR pathway can regulate the miRNAs biogenesis through the MAPK/ERK cascade. The EGFR signaling pathway is important to transduce growth factors
stimuli (inducing growth, survival, proliferation, regulation of apoptosis) from the
extracellular environment to the nucleus; alterations of EGFR pathway are implicated in cancer (31). Specifically, the MAPK/ERK cascade mediated miRNA biogenesis regulation is exerted through the ERK kinases (ERK1/2), which is activated by serum or the tumor promoter phorbol 12 myristate 13 acetate (PMA). It can promote the phosphorylation of TRBP, which enhances Dicer stability and miRNA production. In particular, TRBP protein is phosphorylated on Ser142, Ser152, Ser283 and Ser286. Interestingly, increased abundance and activity of Dicer is correlated with the general increase in miRNA production, mostly for growth promoting miRNAs and with reduced expression of let 7 tumor suppressor miRNA (90). Conversely, pharmacological inhibition of MAPK/ERK, for example using the MKK1 inhibitor U0126, resulted in the decrease of TRBP
phosphorylation and increase in an anti growth miRNAs profile. To reinforce this findings, in a recent paper, the authors showed that the p38 MAPK MK2 signaling pathway promotes miRNAs biogenesis by facilitating the nuclear localization of p68 (a key regulator of Drosha activity). In particular, p68 is phosphorylated at its Ser 197 by the kinase MK2 (91). Conversely, in 2013 Hong et al. (92) showed that EGFR pathway can lead to the suppression of the maturation of specific tumour suppressor like miRNAs in response to hypoxic stress through phosphorylation of AGO2 at Tyr 393; this in turn reduces the binding of Dicer to AGO2 and inhibits miRNAs processing from precursors to mature miRNAs. This evidences could depend from the hypoxia conditions, so it is possible to hypothesize that the miRNA generating complex regulation through EGFR pathway is strongly dependent on the specific conditions of growth. The connections between the EFGR pathway and miRNAs is very complex, considering the different downstream kinases cascades.
2.2.5 Post transcriptional repression by miRNAs
As components of RISC, the primary action of mature miRNAs is to target the complementary mRNA, a process governed by base pairing, to induce their translational repression or deadenylation and degradation. Although in plants most miRNAs pair to target mRNA in a nearly perfect match, leading to mRNA cleavage and subsequent degradation, in animal cells miRNAs typically make imperfect pairings (mismatches and bulges) with their mRNA targets (50). The base pairing between miRNAs and target mRNAs depends on specific sequences both in the miRNA and in the mRNA. In particular, mature miRNAs comprise a region called seed region that is characterized by a sequence located at nucleotides 2 8 of the 5’ region, which is responsible for the identification and binding of mRNA 3’UTR. The binding of the 3’ half of the mature miRNA to the 3’UTR of the mRNA is essential to stabilize the duplex miRNA mRNA, so that the RISC