UNIVERSITÀ DI PISA
Dipartimento di Farmacia
Corso di Laurea Specialistica in Chimica e
Tecnologia Farmaceutiche
Tesi di Laurea:
DESIGN, SYNTHESIS AND BIOLOGICAL
EVALUATION OF HETEROCYCLIC DERIVATIVES
AS POTENTIAL MAGL INHIBITORS
Relatori: Prof.ssa Clementina Manera
Candidato:
Alberico Droghetti
(matricola N° 304329)
Dott.ssa Chiara Arena
Settore Scientifico Disciplinare: CHIM-08
ANNO ACCADEMICO 2012 – 2013
INDEX
Introduction
……….…….21
Endocannabinoid System (ECS) ... 3
1.1 Cannabinoid Receptors ... 4
1.2 Endocannabinoids and Metabolizing Enzymes ... 6
1.2.1 Endocannabinoids Biosynthesis ... 6
1.2.2 Enzymes Involved in Endocannabinoid Degradation ... 9
2
MAGL like Pharmacological Target ... 13
2.1 The Role of MAGL in Pain and Inflammation ... 13
2.2 The Role of MAGL in Neurodegenerative Diseases ... 15
2.3 The Role of MAGL in Cancer and Cancer-Related Symptoms ... 18
3
Monoacylglycerol Lipase (MAGL) ... 20
3.1 MAGL: Structure and Most Important Residues ... 20
3.2 Binding of the Natural Substrate ... 23
3.3 MAGL Catalytic Mechanism... 25
4
MAGL Inhibitors ... 27
4.1 General Serine Hydrolase Inhibitors... 27
4.2 Inhibitors Inspired by the Endogenous Substrate ... 29
4.2.1 2-AG Analogues ... 29
4.2.2 1-AG Homologues ... 30
4.3 Organophosphoric (OP) Derivates as MAGL Inhibitors ... 32
4.4 Inhibitors Targeting the Essential Sulfhydril Group of MAGL ... 34
1
Equipments and Methods ... 51
2
Biological Assay ... 74
2.1 MAGL Inhibition HPLC Assay ... 74
2.2 Materials and methods ... 74
2.2.1 Reagents ... 74
2.2.2 Analytical Instruments ... 74
2.2.3 Instrumentation for the Preparation of Aqueous Solutions ... 74
2.2.5 Preparation of Solutions for the Performance of the Assay ... 75
2.2.6 Inhibitory Activity of Compounds Tested ... 75
3
Screening of Compounds Synthesized ... 77
3.1 Compound 2 ... 77 3.2 Compound 3 ... 77 3.3 Compound 4 ... 78 3.4 Compound 7 ... 78 3.5 Compound 8 ... 79 3.6 Compound 9 ... 79 3.7 Compound 10 ... 80 3.8 Compound 11 ... 80 3.9 Compound 12 ... 81 3.10 Compound 14 ... 81 3.11 Compound 15 ... 82 3.12 Compound 16 ... 82 3.13 Compound 19 ... 83 3.14 Compound 20 ... 83 3.15 Compound 21 ... 84 3.16 Compound 22 ... 84 3.17 Compound 23 ... 85 3.18 Compound 24 ... 85
Reference
………..………862
3
1 Endocannabinoid System (ECS)
The Endocannabinoid system (ECS) is a neuromodulatory network known to be targeted by Δ9–tetrahydrocannabinol (Δ9–THC), the main psychoactive component of Cannabis sativa.1 Since antiquity, cannabis extracts have been used not only for recreational purposes but also as medicinal agents. The endocannabinoid signaling system consists of two G-protein-coupled-cannabinoid receptors (CBRs), that can be activated by small lipid mediators, known as endocannabinoids (eCBs), and by Cannabis-derived drugs, together with the associated biochemical machinery (precursors, synthesis and degradative enzymes, and transporters).
Since their discovery, the endocannabinoids where found to be derivatives of arachidonic acid conjugated with a polar head group, ethanolamine or glycerol, by amide (such as anandamide or N-arachidonoylethanolamine (1), AEA), ester (2‐arachidonoylglycerol or 2‐AG (2) and virodhamine (3)) or ether moieties (2‐arachidylglycerol or noladin ether (4)). Within this class of lipid messengers, anandamide, and 2-arachidonoylglicerol acid are the best characterized, in particular concerning their pharmacology and metabolism.
Fig. 1 Structures of the cannabinoid receptor ligands anandamide (1), 2‐arachidonoylglycerol (2), virodhamine (3) and noladin ether (4).
4
1.1 Cannabinoid Receptors
Nowadays, two cannabinoid receptors, CB1 (CB1R) and CB2 (CB2R), have
been cloned, both of which belonging to the superfamily of G-protein-coupled receptors.
CB1 receptors are highly expressed in neuronal cells, basal ganglia, cerebellum,
hippocampus, and cortex2, 3, 4 in according with the effects of cannabinoids on cognition, memory, nociception and movement. Despite their principal localization in the central system, CB1Rs are also present in the periphery, though
in lower levels, including immune and reproductive systems, liver, intestine, vascular endothelium, and peripheral nerve synapses.
By contrast, CB2 receptors are primarily located in cells involved in immune
and inflammatory responses.5 Nevertheless initially thought to be exclusively
expressed in the periphery, CB2Rs were detected in the cerebellum and the
brainstem6, 7, 8 as well,and in microglial cells, where CB2R may play a central role
in modulation of their mobility and function both in vitro and in vivo.
At the molecular level, both CB1R and CB2R are seven-transmembrane domain
receptors (7TM), belonging to the superfamily of G-protein-coupled receptors (GPCRs), whose principal function is to transduce extracellular stimuli into intracellular signals. In particular, cannabinoid receptors bind to a Gi/o protein.
Therefore, their stimulation involves significant changes in cellular activity mainly due to the inhibition of adenylyl cyclase (AC) and the resulting decrease of the intracellular cAMP levels, thereby regulating cAMP-dependent protein kinase A (PKA) functions. Furthermore, interaction with activates other protein kinase, such as p38 mitogen-activated protein kinase (MAPK), focal adhesion kinase (FAK), c-jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK). In contrast to CB2 receptor activation, CB1 receptor activation modulates
calcium or potassium conductance, properties linked to the suppression of neuronal excitability and neurotransmitter release.
In the nervous system the mechanisms of endocannabinoid signaling differ considerably from the classic model of neurotransmission (e.g., cholinergic, aminoacidergic, and monoaminergic), where neurotransmitters, released after depolarization of the presynaptic neuron caused by an action potential, traverse
5 the synaptic cleft to bind and activate their associated receptors located on the postsynaptic neuron.
Differently, endocannabinoid signaling seems to take place via a retrograde mechanism, where stimulation of the postsynaptic neuron triggers the biosynthesis of endocannabinoids, which are released and transported by poorly understood mechanisms to activate cannabinoid receptors expressed primarily on the presynaptic terminal.
In this model, endocannabinoid signaling modulates transmission efficiency by facilitating communication from the postsynaptic to the presynaptic neuron. As CB1R activation acts to inhibit neurotransmission, the ultimate outcome of
endocannabinoid signaling depends on the nature of the participating cells. If CB1R is activated on glutamatergic neurons, for instance, endocannabinoid
signaling will be overall inhibitory, whereas if CB1 activation takes place on
GABAergic neurons, the net result will be “disinhibitory” (or excitatory) (Fig. 2).
MSN/PV CORTEX
Fig. 2 Activity-dependent generation of 2-arachidonoylglycerol (2-AG) for retrograde signalling.
Depolarization induced suppression of inhibition (DSI) is one of the best characterized forms of short-term synaptic plasticity where neuronal activity evokes post-synaptic depolarization via excitatory glutamatergic (Glu) action. (9: Acta Physiol 2012, 204, 267–276)
6 Besides CB1 and CB2 receptors, there are some evidences for additional
CB‐like receptors (such as GPR55, GPR18, TRPV channels…), but currently the evidence is not considered sufficiently strong for them to be formally included in the CB receptor family.10
1.2 Endocannabinoids and Metabolizing Enzymes
1.2.1 Endocannabinoids Biosynthesis
The endocannabinoids are a family of endogenous signaling lipids able to activate cannabinoid and non-CB1/CB2 receptors. They differ from the classic
neurotransmitters since they are not stored in synaptic vesicles, but are produced “on demand” by stimulus-dependent cleavage of membrane phospholipid precursors. To date, arachidonoyl ethanolamide (anandamide, AEA) and 2-arachidonoylglycerol (2-AG) are the best characterized.
Anandamide acts as a partial agonist at CB1 and CB2 receptors and mimics
most of the pharmacological effects produced by cannabinoid drugs in vitro and in vivo.
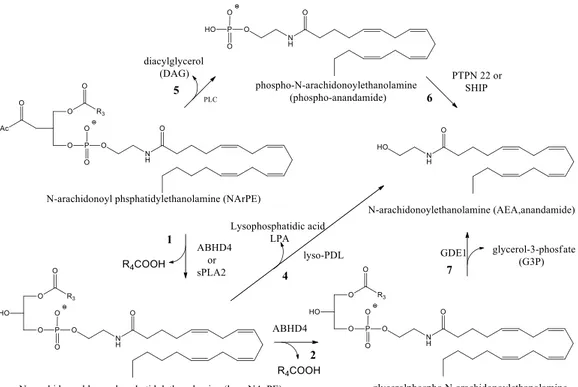
The mechanisms of anandamide biosynthesis in the nervous system are incompletely understood. It is generally accepted that anandamide is generated by calcium-dependent enzymatic transfer of arachidonic acid from the sn-1 position of membrane phospholipids to the primary amine of phosphatidylethanolamine (PE) to form N-arachidonoyl phosphatidylethanolamine (NArPE), followed by hydrolysis to give anandamide.11, 12 However, the calcium-dependent transacylase enzyme (CDTA) that forms NArPE has still not been molecularly identified, and the route by which NArPE is converted into anandamide remains unclear (Fig.3)
7
Fig. 3 Direct anandamide biosynthesis
Multiple mechanisms and putative anandamide biosynthetic enzymes have been suggested, including direct liberation of anandamide by NAPE-PLD, an N-acyl phosphatidylethanolamine (NAPE)-selective phospholipase D (PLD): 13 1) sequential O-deacylation of NArPE by the lyso(NAPE)-lipase a-b hydrolase 4
(ABHD4; Fig. 4, steps 1-2) and cleavage of the phosphodiester bond by the glycerophosphodiesterase GDE1 (Fig. 4, step 3);
2) O-deacylation of NArPE by phospholipase A2 (Fig. 4, step 1)14 and
hydrolysis of the phosphodiester bond by a lyso-PLD enzyme (fig. 4, step 4); 3) conversion of NArPE to phospho-anandamide by a phospholipase C (PLC)-like enzyme (Fig. 4, step 5) followed by dephosphorylation by the tyrosine phosphatase PTPN22 or the inositol 592phosphatase SHIP (Fig. 4, step 6).15
8
Fig. 4 Alternative anandamide biosynthesis (11. Jacqueline L. Blankman et al. Pharmacol Rev 65:849–871,
April 2013)
2‐arachidonoylglycerol is a full agonist at cannabinoid receptors, although less potent than AEA, stimulates GPR55 receptors in transfected cells, acts as a retrograde messenger on pre‐synaptic CB1R located on excitatory and inhibitory
synapses to inhibit neurotrasmitter release, and is an autocrine mediator of post‐synaptic slow selfinhibition (SSI) of neocortical interneurons. Unlike AEA, 2‐AG does not directly activate TRPV1 or PPAR receptors but can bind to non‐CB1/CB2 targets as suggested by the observation that the behavioral effects
induced by N‐arachidonyl maleimide (NAM), a selective MAGL inhibitor with in vivo efficacy, are only partially reversed by the CB1 antagonist Rimonabant.
2-AG is synthesized from arachidonoyl-containing diacylglycerol (DAG) species by sn-1-specific diacylglycerol lipase-a and-b (DAGLa and DAGLb) 16 (Fig. 5). Characterization of DAGL(–/–) mice confirmed a primary role for DAGLa in 2-AG formation in the brain and DAGLb in peripheral tissues, such as the liver.17 DAG precursors are themselves synthesized from membrane phospholipids with most evidence suggesting that the major 2-AG biosynthetic
9 pathway is the hydrolysis of sn-2 arachidonoyl phosphatidylinositol 4,5-bisphosphate (PIP2) species by PLCb18 (Fig. 5).
Fig. 5 2-Arachidonoylglycerol biosynthesis
1.2.2 Enzymes Involved in Endocannabinoid Degradation
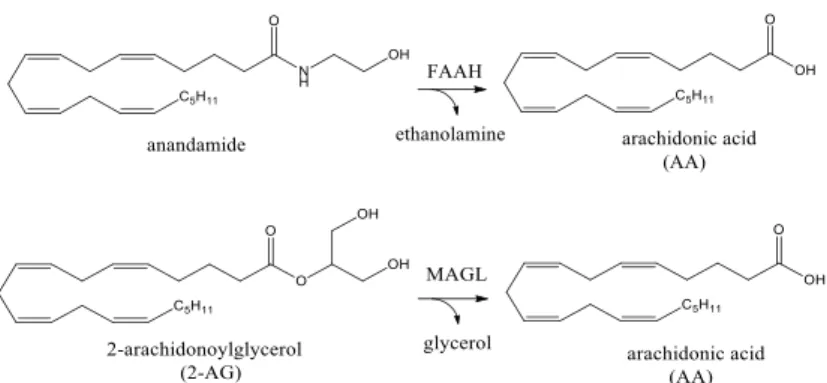
The biological actions of anandamide and 2-AG are terminated via a two‐step process consisting of facilitated diffusion into cells via a carrier-mediated transport, followed by enzymatic cleavage via two serine hydrolase enzymes, fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively (Fig. 6).
Fig. 6 Endocannabinoid hydrolysis. In the nervous system, anandamide and 2-AG are degraded primarily by
FAAH and MAGL, respectively
FAAH is a ≃60-kDa integral membrane protein that is highly expressed in the mammalian brain where is localized in intracellular membranes of postsynaptic soma and dendrites.19 FAAH is both a serine hydrolase, an enzyme class that utilizes a nucleophilic serine for catalysis, and a member of the amidase signature enzyme family. Unlike most serine hydrolases, which use a histidine residue as a catalytic base, FAAH engages a lysine for the same function, a distinction that enables FAAH to hydrolyze both amides and esters at equivalent rates. Structural
10 determination of the transmembrane-truncated rat enzyme bound to the nonselective inhibitor methyl arachidonoylfluorophosphonate confirmed the presence of a serine-serine-lysine (Ser241-Ser217-Lys142) catalytic triad, typical for amidase signature enzymes and different from the serine-histidine-aspartatic acid motif common to most serine hydrolases.20
Despite FAAH is the main responsible of its metabolism, anandamide signal may be disrupted via other mechanisms. Anandamide is, for instance, substrate of COX-2 enzymes. Indeed, administration of exogenous AEA to FAAH−/− mice leads to the production of detectable levels of prostamides, suggesting that the COX‐2 metabolic pathway becomes physiologically relevant under conditions affecting FAAH activity or promoting COX‐2 upregulation, as in the case of neurodegenerative diseases or tissue damage. In addition, lipoxygenases (12‐LOX and 15‐LOX) and P450, including its major brain isoform CYP2D6 [128], convert AEA into biologically active metabolites that exert their biological actions via cannabinoid, PPAR and TRPV1 receptors.21 However, the functional role of these substances in the CNS has not been determined.
After the discovery of 2-AG as a second endocannabinoid its inactivation in the nervous system was hypothesized to proceed through monoacylglycerol lipase (MAGL), a soluble serine hydrolase that peripherally associates with cell membranes.22 In the rat brain, MAGL expression is highest in the cerebellum, cortex, thalamus, and hippocampus where, like CB1, it is primarily localized to
presynaptic axon terminals. MAGL was originally isolated and cloned from adipose tissue, where it was characterized as an enzyme responsible for the last step of triglyceride catabolism. Multiple subsequent studies implicated MAGL as a key mediator of 2-AG degradation in the nervous system.
Although MAGL is the major responsible for its hydrolysis in intact neurons 22, 23, 2‐AG can be metabolized by FAAH as well, and, similarly to AEA, it is a
putative substrate of COX and LOX enzymes.
The COX‐2‐derived metabolite, PGE2 glycerol ester, has been shown to modulate miniature inhibitory currents and to affect long‐term potentiation (LTP) in mouse hippocampus in a CB1‐independent manner.
11 Confirmation of MAGL as the primary brain 2-AG hydrolase in nervous system and in several peripheral tissues was achieved through different findings: both the administration of a selective and in vivo active MAGL inhibitor, JZL184, and the genetic disruption of MAGL activity, reduced brain 2-AG hydrolase activity by ≃85%, 24 dramatically elevated 2-AG levels, and provided a select
subset of cannabinoid behaviors.
Thanks to a functional proteomics approach, the 2-AG hydrolysis activity of mouse brain membranes insensitive to MAGL inhibition (≃15% of total activity) was recently well-established, and attributed to the previously uncharacterized enzymes a/b-hydrolases 6 and 12 (ABHD6 and ABHD12).25
ABHD6 is a ≃30- kDa integral membrane serine hydrolase predicted to adopt an intracellular orientation.26 Murine ABHD6 expression is abundant in brain and multiple peripheral tissues and cell types.In the mouse brain, ABHD6 is highly expressed in cortical areas, where it preferentially localizes to postsynaptic dendrites that are often juxtaposed to presynaptic CB1 receptors.
ABHD12, instead, is a ≃45-kDa membrane glycoprotein predicted to contain a single-pass transmembrane domain and face the extracellular/luminal cellular space.26 High ABHD12 mRNA expression has been detected in mouse brain, bladder, prostate, white adipose, macrophages, and microglia.
Despite their limited contribution to bulk 2-AG degradation, it is possible that the different subcellular or cellular localizations occupied by ABHD6 and ABHD12 compared with MAGL might allow each enzyme access to distinct pools of 2-AG in the nervous system. Indeed, ABHD6 has recently been shown to regulate 2-AG degradation and signaling in murine primary neurons and cortical slices and control 2-AG accumulation in the Neuro2A cell line, which lacks MAGL.
Additionally, ABHD6 inhibition in vivo produced CB1- and CB2-mediated
anti-inflammatory and neuroprotective effects in a mouse model of traumatic brain injury.27 Human genetics has implicated ABHD12 as a critical regulator of neurologic function. For instance, loss-of-function mutations in ABHD12 cause the human neurodegenerative disease PHARC (polyneuropathy, hearing loss, ataxia, retinosis pigmentosa, and cataract), 28 although whether
ABHD12-12 disruption leads to neuronal death by dysregulation of 2-AG or an alternate mechanism remains to be determined.
Fig. 7 Schematic representation of 2‐AG inactivating pathways. 2‐Arachidonoylglycerol signaling is terminated by MAGL, ABHD6 or ABHD12‐ mediated hydrolysis or by COX2 oxidation into PGH2‐G. Note that arachidonic acid, PGH2‐EA and PGH2‐G can be further transformed into other bioactive lipids, such as prostaglandins and endocannabinoid‐derived prostaglandins, respectively.
In summary, the endocannabinoids are emerging as important signaling lipids in the CNS that modulate excitatory and inhibitory neurotransmission and synaptic plasticity.
These lipids do not bind metabotropic cannabinoid receptors exclusively, but display significant promiscuity as they activate ionotropic and nuclear targets (TRPV1 and PPAR receptors) as well as other non‐CB1/CB2 receptors.
The characterization of the endocannabinoid biosynthetic and metabolic pathways has led to the discovery of novel molecular entities, such as NAPE‐PLD, FAAH and MAGL, which can be targeted pharmacologically not only to manipulate endocannabinoid levels, but also to investigate the role of the endocannabinoid system in physiological and pathological conditions. In this regard, the development of more selective FAAH/MAGL inhibitors with significant in vivo activity is crucial to understand the specific biological actions of AEA, 2‐AG and their metabolites in living animals.
13
2 MAGL like Pharmacological Target
MAGL is the main enzyme involved in degrading 2-AG in vivo with consequential production of arachidonic acid (AA), precursor of pro- inflammatory prostaglandin synthesis. Furthermore, it is upregulated in aggressive human cancer cells, where it has a unique role of providing lipolytic sources of free fatty acids for synthesis of oncogenic signaling lipids that promote cancer aggressiveness.
2.1 The Role of MAGL in Pain and Inflammation
The antinociceptive effects of Cannabis sativa have been known for millennia; however, the use of cannabinoids for therapeutic purposes is limited because of psychomimetic side effects that include sensorimotor, affective and cognitive injury. An interesting alternative to the use of cannabinoid receptor agonists may be represented by the inhibition of the catabolic enzymes, fatty acid amide hydrolase FAAH and monoacylglycerol lipase MAGL, involved in degradation of the endogenous cannabinoids, anandamide and 2-arachidonoylglycerol respectively. Inhibitors of endocannabinoid metabolic enzymes elevate brain endocannabinoid levels, allowing prolonged activation of their receptor targets.
To date, it is well established that FAAH inhibitors produce antinociceptive effects in multiple pre-clinical nociceptive assays, including neuropathic, inflammatory, and acute thermal nociception. Moreover, FAAH-disrupted mice showed antinociceptive phenotypes, without any evidence of CB1 receptor
functional tolerance, although the antinociceptive effects of the FAAH inhibitor in the rat carrageenan model were lost following repeated administration. While the impact of selective FAAH inhibition has been well-established, selective inhibitors of MAGL have only recently been developed to examine systematically the in vivo consequences of elevating 2-AG levels.
Recently studies have demonstrated that selective MAGL inhibitors are able to produce beneficial antinociceptive effects in various models of inflammatory and neuropathic pain, as well as gastro-protective actions, in the absence of cannabimimetic side effects. These studies were performed using the two most potent MAGL inhibitors available today, JZL184 and KML29. In particular,
14 KML29 is known to be the most selective MAGL inhibitor, and the first that increases 2-AG levels, but does not possess cross-activity with FAAH, compared to JZL184, or other serine hydrolases. It was demonstrated that this compound produced significant anti-allodynic and anti-edematous effects in the carrageenan model of inflammatory pain and partially reversed mechanical and cold allodynia in the chronic constriction injury (CCI) model of neuropathic pain. All these effects seem to be mediated through distinct cannabinoid receptor mechanisms of action. For instance, both CB1 and CB2 receptors play fundamental roles in the
anti-allodynic effects of MAGL inhibitors in the carrageenan model of inflammatory pain, but only CB1 receptors are necessary for the anti-allodynic
effects in the CCI neuropathic pain model. CB2 receptor component in the
anti-allodynic effects of MAGL inhibition in the various inflammatory pain models could be associated to infiltration of immune cells, which present a high CB2
expression. Differently, the CB1 receptor component may be mediated by
activation of these receptors located supraspinally, spinally or in peripheral nociceptors.29
It is well known that the antinoceptive effect of endocannabinoids involves both supraspinal structures (e.g. periaqueductal gray, thalamus, rostral ventromedial medulla and amygdala), and the dorsal horn of the spinal cord. In the CNS, MAGL is localized mainly presynaptically and is also expressed in microglia. Endocannabinoids are produced in dorsal root ganglion cells, a primary afferent input to spinal cord. However, anatomical localization of MAGL in the periphery is not known. Thus, the antinociceptive effects of MAGL inhibitors in pain models involving a strong inflammatory component, may involve both neuronal and non- neuronal sites of action, within the CNS and periphery.
Nomura et al. (2001) demonstrated that hydrolysis of 2-AG by MAGL represents an important pathway of formation of arachidonic acid in the brain. Thus, MAGL inhibition causes a reduction of brain levels of AA, and a subsequent decrease of prostaglandins and pro-inflammatory cytokines following inflammatory insults. Therefore, it is necessary to evaluate the possibility that decreased prostaglandin synthesis, as a consequence of decreased availability of
15 substrate for their production (i.e. AA), may contribute to the antinociceptive effects of MAGL inhibitors.
In addition, KML29 produced a dose-dependent CB1 receptor mediated
gastroprotective effect in the NSAID-induced gastric hemorrhage model. Thus, MAGL inhibition may be an effective strategy for the prevention of NSAID- induced gastric ulcers.29
Finally, these studies showed that the inhibitor KML29 not provide cannabimimetic side effects, such as catalepsy, hypothermia, or hypomotility, neither at high doses. Moreover, in the drug discrimination assay, KML29 did not substitute for THC, which suggested that 10-fold increases of 2-AG in whole brain are not sufficient to produce THC-like discriminative stimulus effects. This last discovery can be explained by the hypothesis that 2-AG acts as retrograde messengers in the brain and is synthesized “on demand” from phospholipid precursors. Therefore, blockade of its degradation may not result in sufficient levels of 2-AG to stimulate CB1 receptors in circuits mediating the discriminative
stimulus effects of THC.
In conclusion, the relation between MAGL and antinociceptive action of his inhibition is explained with increase of the endocannabinoid activities on CB1 and
CB2 receptors at different levels. MAGL inhibition produces beneficial
antinociceptive and gastroprotective effects, without unwanted cannabimimetic side effects similar to those produced by Cannabis or THC. The findings that both KML29 and JZL184 produce antinociceptive effects in a wide variety of animal models of pain, suggest that inhibition of MAGL represents a promising strategy to treat inflammatory and neuropathic pain, with no or minimal cannabimimetic side effects.29
2.2 The Role of MAGL in Neurodegenerative Diseases
In the last years, inflammation was recognized to be the hallmark of multiple neurological disorders, including chronic pain, traumatic brain injury, stroke, and neurodegenerative diseases, such as Parkinson’s and Alzheimer’s diseases and multiple sclerosis.30
16 The inflammation response in nervous system is triggered by prostaglandins, which are produced from arachidonic acid by cyclooxygenase enzymes (COX1 and COX2) that are localized in neurons and glial cells. Indeed, both pharmacological treatment with COX inhibitors or genetic depletion of COX enzymes provide protection in models of neurodegenerative disorders that have an inflammatory component, such as Parkinson’s (PD) and Alzheimer’s disease (AD).
Up until a few years ago, phospholipase A2 (PLA2) enzymes and cytosolic
PLA2 (cPLA2) in particular were considered the primary source of arachidonic
acid for cyclooxygenase (COX)–mediated biosynthesis of prostaglandins. It has been recently demonstrated the presence, in brain, of a distinct pathway, governed by monoacylglycerol lipase (MAGL), that is able to generate arachidonic acid (AA) for neuroinflammatory prostaglandins by hydrolyzing the endocannabinoid 2-arachidonoylglycerol.31
The existence of this alternative mechanism was confirmed by the observation that cPLA2-deficient mice have unaltered AA levels in brain, collected with the finding that genetic or pharmacological inactivation of MAGL in mice causes significant reductions in brain AA.
Furthermore, it has already proved that cannabinoid agonists exert protective or palliative benefits against neurodegenerative diseases as well. Indeed, in Parkinson’s disease model CB agonists provided decrease of dopamine depletion in the substantia nigra, promoted survival of dopaminergic neurons, and reduced oxidative stress and pro-inflammatory cytokine release from microglia, thus improving motor function; whereas in Alzheimer’s disease models they attenuated microglia activity, COX2 expression and amyloid β plaque levels.
In accord with the beneficial effects of cannabinoid agonists and COX inhibitors in neuroinflammation and neurodegenerative disease models, both genetic and pharmacological blockades of MAGL also provide anti-inflammatory effects in the brain and neuroprotective effects in PD and AD models. It is interesting the finding that the impairment in brain eicosanoid production in MAGL-deficient animals was not reversed by CB1 or CB2 receptor antagonists or
17 is a direct consequence of reductions in AA rather than an indirect consequence of enhanced endocannabinoid signaling.
Furthermore, the toxicities caused by COX inhibitors, like gastrointestinal bleeding, was not observed after treatment with the MAGL inhibitors JZL184; on the contrary inhibition of MAGL by JZL184 exerts a CB1 receptor–dependent
protective effect on COX inhibitor–induced gastric bleeding.
These findings identify MAGL as a distinct metabolic node that couples endocannabinoid to prostaglandin signaling networks in the nervous system and suggest that inhibition of this enzyme may be an innovative way to suppress the pro-inflammatory cascades that underlie neurodegenerative disorders.
Fig. 8 Connecting cannabinoid and prostaglandin pathways: inhibition of MAGL (a) increased the levels of
2-arachidonoylglycerol (2-AG) in the brain, which added through the cannabinoid receptors to decrease pain (b) and lowers the production of arachidonic acid to decrease levels of inflammatory prostaglandins and blunt inflammation in the brain (c).
18
2.3 The Role of MAGL in Cancer and Cancer-Related Symptoms
It was well-established that cannabinoid receptor agonists exhibit anti-cancer proprieties through inducing apoptosis in vitro and angiogenesis and metastasis in vivo. Beyond their direct effects against cancer, cannabinoid have been clinically used to relieve chemotherapy side effects, such as nausea, pain, and lack of appetite.32
Consistent with these clinical benefits of direct cannabinoid agonists, MAGL inhibition exerts anti-hyperalgesic effect via a CB2-depent manner, and shows
anti-emetic and anti-nausea effects.
Instead, it is more recent the finding that the conversion of cells from a normal to cancerous state is accompanied by reprogramming of metabolic pathways, including those that regulate glycolysis, glutamine-dependent anaplerosis, and the production of lipids.
Lately, increased de novo lipid biosynthesis, with the resulting development of a “lipogenic” phenotype, has been individuated, among all dysregulated metabolic pathways, as the physiological process playing the major role in cancer. For instance, elevated levels of fatty acid synthase (FAS), the enzyme responsible for fatty acid biosynthesis from acetate and malonyl CoA, seem to be correlated with poor prognosis in breast cancer patients, and inhibition of FAS may result in decreased cell proliferation, loss of cell viability, and decreased tumor growth in vivo. From these evidences, it was presumed that FAS may support cancer growth, at least in part, by providing metabolic substrates for energy production. Furthermore, other features of lipid biochemistry have been found to be critical for supporting the malignancy of cancer cells:
generation of building blocks for newly synthesized membranes to allow high rates of proliferation;
composition and regulation of membrane structures that coordinate signal transduction and motility;
biosynthesis of a pool of protumorigenic lipid signaling molecules.
Some of these lipid messengers that contribute to cancer development have been characterized: phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3], which activates protein kinase B/Akt to promote cell proliferation and survival;
19 lysophosphatidic acid (LPA), which stimulates cancer aggressiveness; and prostaglandins formed by cyclooxygenases, which support migration and tumor-host interactions.
Since newly synthesized fatty acids are rapidly incorporated into neutral- and phosphor-lipid stores, cancer cells have to possess complementary “lipolytic” pathway to liberate stored fatty acids for metabolic and signaling purposes.
In this context, MAGL, that was found to be upregulated in aggressive human cancer, plays a unique role of providing lypolitic sources of free fatty acids (FFAs), through hydrolysis of monoacylglycerols (MAGs). Therefore, the resulting MAGL-FFA pathway feeds into a diverse lipid network enriched in pro-tumorigenic signaling molecules and promotes migration, survival, and in vivo tumor growth. In this way aggressive cancer cells may couple lipogenesis with lipolytic activity to generate a pool of protumorigenic signals that support their malignant behavior.33
Consistent with this well-established function, it was showed that MAGL blockade in aggressive breast, ovarian, and melanoma cancer cells impairs cell migration, invasiveness, and tumorigenicity through lowering FFAs and protumorigenic signaling lipids, rather than enhancing endocannabinoid signaling.34 Differently, it was demonstrated that in prostate cancer, MAGL inhibitors decrease the cancer pathogenicity through simultaneously enhancing of anti-tumorogenic cannabinoid mechanism and reducing FFA-derived protumorigenic lipid signals.
To sum up, MAGL inhibitors could exert a double role in an anticancer therapy: reducing the malignancy of aggressive human cancer cells and alleviating cancer-associated symptoms such as pain and nausea.33
20
3 Monoacylglycerol Lipase (MAGL)
3.1 MAGL: Structure and Most Important Residues
As reported before MAGL is the enzyme that catalyzed the hydrolysis of 2‐AG.
In 2005, the first homology model of MAGL was presented based on 3D structure of chloroperoxidase from Streptomyces lividans as the template. As a result of poor sequence homology between MAGL and chloroperoxidase, the homology model offered limited insights into the overall structure and organization of MAGL protein.
However, as the central core of the α/β hydrolase superfamily members is highly conserved, the model offered first insights into the MAGL active site with the catalytic triad: serine 122 (S122), aspartic acid 239 (D239) and histidine 269 (H269), previously identified based on mutagenesis studies (Fig. 9). The model also suggested that two cysteine residues (C208 and C242) were located within a close distance from the active site and it was suggested that one or both of these cysteines were potential targets of the maleimide‐based inhibitor NAM (Fig. 10). Site‐directed mutagenesis studies have provided experimental support for these predictions.16
The crystal structure of MAGL was recently solved and this was achieved independently by two laboratories. Labar et al. solved the structure of human MAGL at 2.2 Å resolution and interestingly, MAGL crystallized in this study as a dimer.
21
Fig. 9 Overall structure of hMAGL. A) MAGL asymmetric unit. α4 helix is colored magenta. Membrane
representation is a palmitoyloleoylphosphatidylethanolamine bilayer minimized using molecular dynamics simulation. B) Left: Side view of a MAGL monomer, with catalytic triad represented as sticks, and cap domain colored magenta. Right: Top view (908 rotation) of the same MAGL subunit (35: Chembiochem. 2010 Jan 25;11(2):218-27.)
Fig. 10 Docking of NAM in the active site of MAGL, bound to Cys201. The main conformations found by using Gold software are represented with different colours. Catalytic triad is coloured in orange, and Cys201, Cys208 and Cys242 are represented (35: Chembiochem. 2010 Jan 25;11(2):218-27)
22 Bertrand et al. resolved the crystal structure of human MAGL both in its apoenzyme form and in complex with the potent covalent inhibitor SAR629 (fig.12). As expected, protein folds conserved in the α/β hydrolase superfamily were present also in the MAGL structure. These include an eight‐stranded β‐sheet, composed of seven parallel strands and one antiparallel strand, surrounded by α helices. Moreover, a flexible cap domain covers the structurally conserved β‐sheet and a lid domain guards the entrance of a relatively large, occluded hydrophobic tunnel (approx. 25 Å in length and approx. 8 Å in width), and the active site is buried at the bottom of the tunnel (Fig. 9 B). 36
Fig. 11 Docking of 2-AG in the active site of MAGL. The natural substrate is bound in the tetrahedral
intermediate state to Ser122. The four first conformations found by using Gold software are represented using different colours. A) Acyl-binding and B) alcohol-binding sites are highlighted, as well as C) the glycerol exit channel. Side chains of residues interacting with the 2-AG acyl moiety or lining the hydrophilic cavity are represented as sticks. The interactions in the oxyanion hole are represented with dashed lines. The α4 helix is also indicated by arrows (35: Chembiochem. 2010 Jan 25;11(2):218-27)
23
3.2 Binding of the Natural Substrate
Explanation of the MAGL structure provides the first structural basis for rational drug design. To illustrate this and to highlight some key structural features of the active site, 2-AG has been docked in MAGL (Fig. 11, Fig 12). This cavity becomes wider as one moves away from the catalytic triad (Ser122, Asp239 and His269), deeply buried in the protein, to the surface of the protein. Several hydrophobic residues cover the channel leading from the surface to the nucleophilic serine. Indeed, Leu148, Ala164, Leu176, Ile179, Leu205, Val207, Ile211, Leu213, Leu214, VaL217 and Leu241 side chains are properly located to interact with the arachidonoyl moiety of 2-AG, and mediate the MAGL substrate specificity for lipid substrates.
The near environment of the catalytic triad presents a more hydrophilic character than the channel pointing towards the enzyme surface. Besides the backbone NH from Met123 and Ala51, which form the “oxyanion hole,” the Tyr58 hydroxyl group, the NH from the His121 and His272 side chains, the guanidinium from Arg57, the carboxylate from Glu53, and the backbone carbonyl from Ala51 delimit a polar cavity that accommodates the polar glycerol head group of 2-AG. A glycerol molecule found in this alcohol-binding pocket is in support of the proposed binding mode of 2-AG. A positive electron density feature was apparent in the vicinity of the oxyanion hole at the end of the structure-refinement process. This was located at the entry of this polar cavity and precisely at the same place as the glycerol moiety in 2-AG docking results. The glycerol alcohol moieties are in interaction with the Ala51 carbonyl group, Tyr194 alcohol and Glu53 carboxylate. The His121 lateral chain and a conserved water molecule inside the cavity are located close to the alcohol and therefore might also interact with the glycerol function and participate to the MAGL selectivity for monoacylglycerols (Fig. 11).37
24
Fig. 12 Crystal structure of the human MAGL with 2-AG docked in the active site or in complex with SAR629.
Left: surface representation. Right: the Ser122/His269/Asp239 catalytic triad and the main residues lining the sites that allow interaction with both ligands are represented as sticks. Two pockets filled with SAR629 fluorophenyl groups or with 2-AG acyl moiety (P1 and P2), as well as the alcohol binding pocket (P3), are highlighted. The hydrophobic �4-helix residues are also represented (38: Current Medicinal Chemistry, 2010, 17, 2588-2607).
Fig. 13 Electron density in MAGL active site. A glycerol molecule is bound in the alcohol-binding pocket (35:
25
3.3 MAGL Catalytic Mechanism
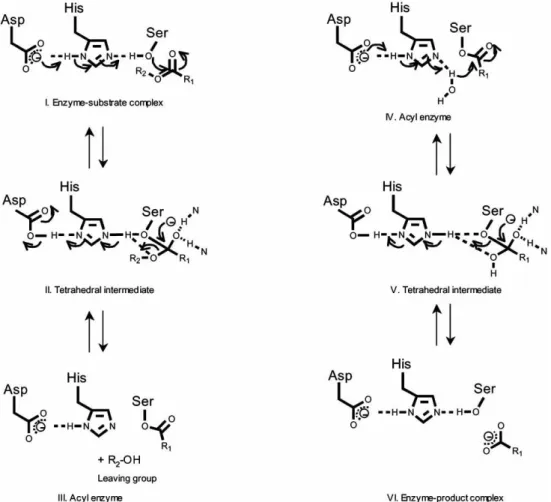
The reaction mechanism of lipolytic enzymes is a two-step kinetic process. The binding step is represented by the interaction of the enzyme with the water-lipid interface followed by the hydrolysis of the substrate as the second step, during which the active site accommodates a single substrate molecule.
When the substrate is positioned in the active site, hydrolysis of the ester bond occurs as a result of the nucleophilic attack of the catalytic serine’s activated hydroxyl group on the substrate’s activated carbonyl carbon by a universal acid-base mechanism.
The initial step of the mechanism is protonation of the His269 in the active site from the serine residue.
Histidine protonation is stabilized by the negative charge associated with Asp239, the third residue of the MAGL catalytic triad, which lies in a hydrophobic environment. In this manner, the catalytic Ser122 becomes a strong nucleophile and attacks the carbonyl carbon of the substrate with consequent formation of a tetrahedral intermediate that is stabilized by the oxyanion hole.
The role of the oxyanion hole is important in activating the carbonyl group of the substrate.
Generally, the oxyanion hole is not preformed, but is formed during lid opening. Once the tetrahedral intermediate is formed, the reaction proceeds by elimination of the leaving group and acylation of the serine.
Finally, deacylation of this serine occurs upon attack of a water molecule in the active site, formation of a charge-relay system, and consequent regeneration of the active MAGL enzyme (Fig. 14). 37
26
27
4 MAGL Inhibitors
Agents that modulate endocannabinoid system transmission are actively being sought as therapeutics to treat important behavioral, metabolic, and neurological diseases. For therapeutic upregulation of endocananbinoid signaling, pharmacological inhibition of endocannabinoid deactivating enzymes may offer higher selectivity and lower risk of unwanted psychotropic side effects as compared to cannabinoid-receptor agonists. Specifically, targeted inhibition of 2-AG deactivation is considered an attractive therapeutic approach against pain, inflammation, and neurodegenerative and immune disorders.
4.1 General Serine Hydrolase Inhibitors
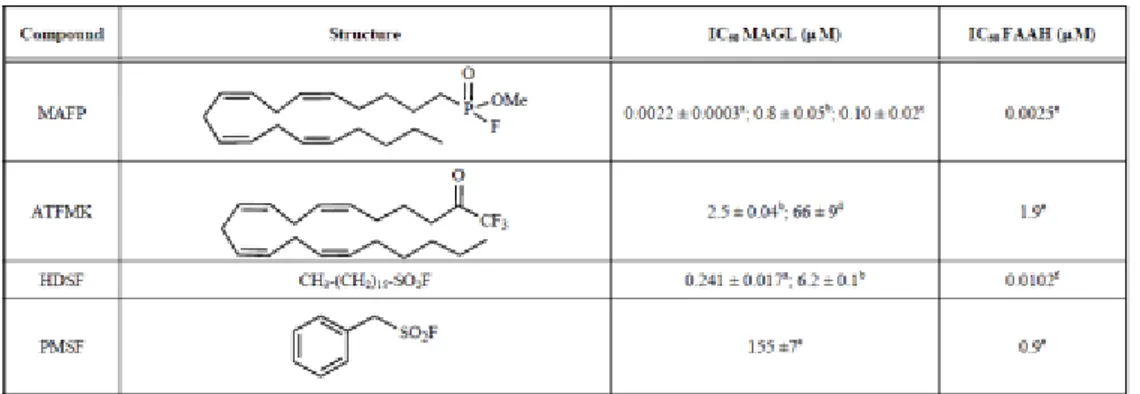
Since mechanistically MAGL is a serine hydrolase, the first wide group of inhibitors are the general serine‐hydrolase inhibitors. This series of mechanistically based inhibitors binds in either a reversible or irreversible covalent manner to the nucleophilic serine, thus disrupting its catalytic activity. Chemically, we can distinguish three main reactive groups: fluorophosphonates,
39, 40 trifluoromethylketones and sulfonylfluorides (Table 1, Table 2).
As the most representative compound within each of these classes we can mention methyl arachidonylfluorophosphonate (MAFP), 41, 42 arachidonyltrifluoromethylketone (ATFMK) and henylmethylsulfonylfluoride (PMSF). All these compounds are not new to the ECS field, since they are old known FAAH inhibitors. Considering the similarity between the mechanism and the structure of the substrate between MAGL and FAAH, it is clear that one of the first aspects to consider when identifying potential MAGL inhibitors is the selectivity between the two hydrolytic enzymes FAAH and MAGL.
28
Tab. 1: Inhibition of 2-AG and AEA hydrolysis by general serine hydrolase inhibitors
Tab. 2: Inhibition of 2-AG and AEA hydrolysis by trifluoromethylketones
Further modification of the trifluoromethylketone moiety with the introduction of a sulfide (R‐S‐) group in the α position, structural motif known to inhibit esterases, has been also analyzed in a brief SAR study aimed at the search of new antitumor agents acting as MAGL inhibitors (Fig. 15). From the experimental values of this series of compounds, it is observed that the inhibition of degradation of 2‐AG and 2‐OG is increased with the length of the alkyl chain.
Fig. 15 Effect of the introduction of a α‐sulfide group (R‐S‐) in trifluoromethylketones on 2‐AG hydrolysis and invasion of prostate cancer cells.
29
4.2 Inhibitors Inspired by the Endogenous Substrate
4.2.1 2-AG Analogues
In the structure of 2‐AG we can distinguish three main parts susceptible to modifications: fatty acid chain, linker and glycerol moiety (Fig. 16).
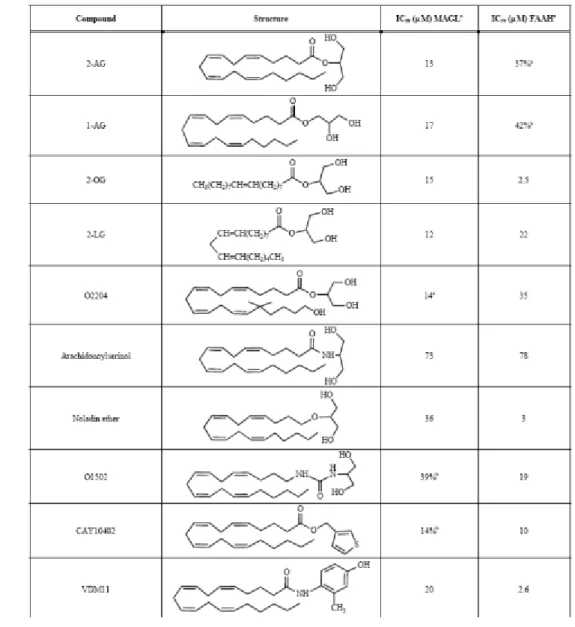
Based on this, the abilities of a series of analogues of 2‐AG have been examined (Table 3) in their ability to inhibit cytosolic MAGL activity. With respect to the fatty acid chain, the experimental data reveal that both isomers 2‐AG and 1‐AG are equipotent in disrupting MAGL activity (IC50 = 13 and 17
μM, respectively).38
30
Tab. 3 Inhibition of MAGL and FAAH by 2-AG analogues
4.2.2 1-AG Homologues
Since 1‐AG shows comparable potency to 2‐AG as MAGL inhibitor (IC50 (1‐AG) = 17 μM and IC50 (2‐AG) = 13 μM, Table 4) but it is more stable in biological solutions, a group of analogues of 1‐AG have been examined as MAGL inhibitors. The structural study (Table 4) includes variations in length (14‐22 carbons) and number of unsaturations (0‐5) of the fatty acid chains. The main conclusions of this study indicate that for cytosolic MAGL the number of unsaturations does not affect significantly the interaction as the IC50 values range between 4.5 and 21 μM.
31 However, a decay of inhibition is observed for the monounsaturated compounds (O3908 and O4066) and a total loss of activity for the saturated C‐20, 1‐arachidinoylglycerol. In contrast, shorter fully saturated compounds such as 1‐palmitoylglycerol (C‐16) and 1‐myristoylglycerol (C‐14) inhibit MAGL.42
32
4.3 Organophosphoric (OP) Derivates as MAGL Inhibitors
Organophosphorus (OP) derivatives with general structure showed in figure 17, as reported in literature, are another class of potential MAGL inhibitors and could interact with several components of the cannabinoid system.42, 43, 44, 45
Fig. 17 General structure of OP derivates
In particular Casida et al. indicated that some OP derivatives inhibit MAGL activity in vitro and that this inhibition leads to elevation of 2‐AG levels in mice brain9, furthermore they showed that most OP compounds possess little or no
selectivity for MAGL versus FAAH. One of the most representative compounds of this class of derivatives is UP101 which showed, as we can see from table 6, a high inhibition activity of the ECS enzyme but not selectivity towards MAGL. Other OP derivatives which did not showed high selectivity for MAGL versus FAAH are CPO and Paraox reported in figure 18. 46
33
Fig. 18
Moreover Nomura et al. demonstrated that several of the non‐cholonergic OP effects are mediated by the ECS and that the inhibition of the metabolic enzyme by OP compounds is due to the phosphorylation of the catalytic site serine (S122 for MAGL and S241 for FAAH). 41
34
4.4 Inhibitors Targeting the Essential Sulfhydril Group of MAGL
The presence of some thiol groups close to the catalytic site of the enzyme led to the design of NEM analogues as MAGL inhibitors (Table 6). Maleimide analogues behave as Michael acceptors toward thiol residues, binding irreversible to the enzyme.39, 40
35
1.4: De novo Inhibitors
Two MAGL inhibitors whose structures (Table 7) do not resemble any endogenous cannabinoid have been recently described. URB754 was reported to inhibit 2‐AG degradation through a non-competitive and irreversible mechanism with an IC50 value of 200 nM, measured in rat brain MAGL expressed in HeLa cells. Besides, it showed a high degree of selectivity towards FAAH of about 150 fold (IC50 = 31.8 μM).
The other MAGL inhibitor identified, URB602, showed a lower capacity to disrupt MAGL hydrolyzing activity (IC50 = 28‐75 μM depending on the source of enzyme) but did not affect the FAAH catalytic activity.47, 48
Tab. 7 Inhibitory potencies of URB754 and URB602 towards MAGL and FAAH
Another inhibitor of this class of compounds is SAR629, a derivative of the triazolocarboxamide series (Fig.19). It shows a nanomolar activity range, and its mechanism of inhibition mimics the pathway of 2‐AG MAGL-mediated hydrolysis by making a relatively stable carbamate adduct with the catalytic serine instead of the relatively labile ester adduct (Fig. 20).37
36
SAR629
Fig. 19 SAR 629: De novo inhibitor
Fig. 20 a) hydrolysis of 2‐AG by MAGL. b) hydrolysis of SAR 629 by MAGL (37: J. Mol. Biol. (2010) 396, 663–673)
In the analysis of model in which SAR629 and MAGL bound covalently, the compound adopts a Y shape with its two fluorophenyl moieties pointing toward opposite directions and perpendicular to each other. These moieties are perpendicular to the piperidine linker that adopts a chair conformation. In addition to the covalent bond with Ser122, SAR629 interacts with MAGL essentially by hydrophobic interactions and few polar interactions. Water‐mediated interactions
37 occur between the inhibitor nitrogen piperazine or oxygen atoms of its carbamate function and His269 side‐chain or Ala61 main‐chain carbonyl oxygen (Fig. 21).37
Fig. 21 A) Docking study of SAR629 into the active site of the enzyme, B) van der Waals interactions of the
piperazine moiety (37: J. Mol. Biol. (2010) 396, 663–673)
The role of the piperazine is to orient the other moieties toward pharmacophoric points, and it could probably be replaced by other moieties.37
The reported MAGL inhibitor JZL184 49, 50 has a chemical structure very close
to that of SAR629 (Fig. 22). Its mechanism of inhibition and its mode of recognition should be similar (Fig. 20).37
Fig. 22 Pharmacophoric model of SAR629 and JZL184. (Dark green elements depict flat and hydrophobic
pharmacophoric points, whereas the orange element depicts a polar pharmacophore. Ar, aromatic) (37: J. Mol. Biol. (2010) 396, 663–673)
38
INTRODUCTION TO THE
EXPERIMENTAL PART
39 Monoacylglycerol lipase (MAGL) is a membrane-associated cytosolic serine hydrolase with the highest expression levels in the brain, white adipose tissue and liver. MAGL catalyses the hydrolysis of monoacylglycerols into fatty acids and glycerol and in particular the endocannabinoid 2-arachidonoylglycerol (2-AG) is one of its main substrates The lipid messenger 2-AG is synthesized by phospholipase C (PLC)-mediated cleavage of membrane phospholipids, resulting in the release of diacylglycerol (DAG) that is then hydrolysed by diacylglycerol lipase (DAGL) (fig. 23).
Fig. 23 2-arachidonoylglycerol biosynthesis
2-AG acts primarily by binding to both cannabinoid G protein-coupled receptors CB1R and CB2R, and it is either synthesized on demand or likely
associated to binding proteins in spatially separated pools (fig.24).
Fig. 24 Schematic of retrograde endocannabinoid signaling in the nervous system (12: Pharmacol Rev
40 In the brain, MAGL is responsible for about 80% of the hydrolysis of 2-AG while the remaining 20% arises mainly from the activity of other two serine hydrolases, α,β-hydrolase-6 (ABHD-6) and -12 (ABHD-12). The pharmacological inhibition of MAGL activity in vivo leads to a strong accumulation of 2-AG, in particular in the brain which triggers a higher activation of CB1R and CB2R
resulting in analgesic, anxiolytic, antidepressant, sleep-enhancing and anti-inflammatory effects. Recently, it has been shown that in some tissues, such as brain, liver and lung, 2-AG acts as a precursor for the arachidonic acid production. Arachidonic acid is mainly oxygenated by cyclooxygenase-2 to generate pro-inflammatory eicosanoids such as prostaglandin-E2 and –D2. Therefore, the analgesic and anti-inflammatory effects induced by MAGL inhibition can be dependent on two underlying mechanisms: the first by directly increasing 2-AG levels and the second, by lowering the arachidonic acid formation as previously described in brain (figure 23). Thus, MAGL could represent the link between two lipids signalling pathways: the endocannabinoid and the eicosanoid systems. For these reasons, the modulation of MAGL activity can lead to several beneficial effects through either enhancing the tone of the endocannabinoid system or lowering eicosanoids production. Furthermore in cancer, MAGL plays a distinct role in controlling global FFAs (free fatty acid) levels that serve as the building blocks for synthesis of pro-tumorigenic signaling lipids such as PGE2 and lysophosphatidic acid (LPA) (figure 25).
41
Fig. 25 Lipid signaling pathways coordinately regulated by MAGL.
Therefore, MAGL inhibitors may represent an attractive therapeutic approach for the treatment of pain, inflammation, anxiety, neurodegeneration, cancer and other disorders. Moreover, regarded the anti-inflammatory activity, because MAGL inhibitors do not exert control over arachidonic acid and prostaglandin pathways in the gastrointestinal system, they also do not induce the gastrointestinal bleeding associated with the use of dual COX1/COX2 inhibitors.
Since mechanistically MAGL is a serine hydrolase, the first wide group of inhibitors are the general serine-hydrolase inhibitors that bind in either a reversible or an irreversible covalent manner to the nucleophilic serine. Chemically, these compounds are distinguished in three main series on the base of reactive group: fluorophosphonate, trifluoromethylketone and sulfonylfluoride.
The second group of inhibitors was inspired by the endogenous substrate 2-AG and then several 1-AG derivatives were studied.
Finally, de novo inhibitors were developed, whose structures do not resemble any endogenous cannabinoid. They are characterized by general structure is
42 characterized by flat structure with a hydrophobic pharmacophoric group and a linker connected with polar pharmacophore.
One of these derivatives, compound URB 602, was reported as a selective MAGL inhibitor with relatively low potency (IC50 MAGL = 75±7 µM) (figure
26).
Fig. 26 URB-602
Despite its low activity, URB-602 is able to attenuate nociception in rodent models of acute, inflammatory, and neuropathic pain.
Starting from these knowledge, during the first period of my thesis work compounds structurally similar to URB-602, with general structure A, B and C (fig. 27), were designed and synthetized.
Fig. 27 General structures of compounds A, B and C
The novel compounds are characterized by a pyridine nucleus with carbamate (A), amide (B) and urea (C) functionality (fig. 27), that were found to be essential for the mechanism of MAGL inhibition (fig. 28). In addition, substituents at the 6-position of pyridine ring were chosen on the basis of the substituent present at the same position on the central phenyl ring of URB-602.
43
Fig. 28 Mechanism of MAGL inhibiton by carbamate, X = O or NH
Compounds with general structure A were obtained as reported in Scheme 1.
Scheme 1
Reagents and conditions. (i): anhydrous CH2Cl2, pyridine, phenyl cloroformate.(ii): toluene,
tetrakis, Na2CO3, benzenboronic acid or p-methoxyphenylboronic acid, MW (biotage), 135 °C, 30
min.
To a solution of phenyl chloroformate in anhydrous CH2Cl2 was added
pyridine and the resulting mixture was stirred at 0 °C for 15 minutes. After that a solution of 2-ammino-6-bromopyridine (1) in anhydrous CH2Cl2 was added
dropwise, under a nitrogen atmosphere. The mixture was stirred for 30 minutes at R.T. to give the desired carbamate 2 which was purified by flash chromatography using n-hexane/EtOAc 11:1 as eluent (yield ≈ 50%) (Scheme 1).
44 Carbamate derivatives 3 and 4 were synthetized starting from the 2-amino-6-bromopyridine (1) which reacted with the proper boronic acid (benzenboronic acid or 4-methoxyphenylboronic acid), via a Suzuki-type cross coupling, using tetrakis as catalyst and aqueous Na2CO3 as base (Scheme 1). The reaction was
heated in a microwave reactor at 135 °C for 30 minutes. The desired compounds 5 and 6 were obtained pure, in high yield, after purification by flash chromatography, using hexane/EtOAc 5:1 as eluent (Scheme 1).
The reaction of compounds 5 and 6 with phenyl chloroformate was performed at the same conditions used to obtain the derivative 2, and provided the desired carbamate derivatives 3 and 4 in ≈ 50% yield, after purification by flash chromatography using n-hexane/EtOAc 9:1 as eluent.
The amide and the urea derivatives with general structure B and C respectively (fig. 27) were synthetized according to synthetic pathway reported in Scheme 2.
Scheme 2
Reagent and conditions. (i): benzoyl chloride, TEA, 4h..(ii): anhydrous 1,4-dioxane, phenyl
isocyanate, R.T., 4h.
The 2-aminopyridine derivatives 1, 5 and 6 were treated with benzoyl chloride and triethylamine, at room temperature, for 4 hours to give amide derivatives 7, 8
45 and 9 in good yields (Scheme 2). These compounds were purified by flash chromatography using, as eluent, petroleum ether/EtOAc 6:1 for compound 7 and petroleum ether/EtOAc 9:1 for compounds 8 and 9.
The reaction of 2-aminopyridine derivatives 1, 5 and 6 with phenyl isocyanate , was performed in anhydrous 1,4-dioxane, under nitrogen, at room temperature, and afforded, after 2 hours, urea derivatives 10, 11 and 12 in quantitative yield (Scheme 2).
In the second part of my thesis, in order to evaluate the influence of thet position of the substituents on the pyridine nucleus on MAGL inhibitory activity, compounds with general structure D, E and F (fig. 29) were synthesized. These compounds present the same substituents selected for compounds A, B and C, but shifted at the 5 position of the heterocyclic nucleus.
Fig. 29 D) carbamate derivatives, E) amide derivatives, F) urea derivatives
The carbamate derivatives D were obtained following the synthetic route reported in the Scheme 3.
46 Scheme 3
Reagent and conditions. (i): anhydrous CH2Cl2, pyridine, phenyl cloroformate.(ii): toluene,
tetrakis, Na2CO3, benzenboronic acid or p-methoxyphenylboronic acid, MW (biotage), 135 °C, 30
min.
Similarly to the synthetic pathway used to obtain thecarbamate derivatives 2, 3 and 4, a solution of phenyl chloroformate and pyridine in anhydrous CH2Cl2 was
stirred at 0 °C for 15 minutes, and then, to the obtained suspension, a solution of 2-ammino-5-bromopyridine in anhydrous CH2Cl2. was added dropwise, under a
nitrogen atmosphereThe reaction mixture was stirred at room temperature for 30 minutes to afford the desired carbamate 14 purified by flash chromatography using n-hexane/EtOAc 11:1 as eluent.
The carbamate derivatives 15 and 16 were obtained starting from the 2-amino-5-bromopyridine (13) which reacted with the proper boronic acid (benzenboronic acid or 4-methoxyphenylboronic acid), via a Suzuki-type cross coupling, using tetrakis as catalyst and 2 M Na2CO3 as base (Scheme 3). The reaction was heated
in a microwave reactor at 135 °C for 30 minutes (5 bar, stirring on, very high adsorbition). The desired 5-substitued pyridine derivatives 17 and 18 were obtained pure, in high yield, after purification by flash chromatography, using hexane/EtOAc 5:1 as eluent. . Treatment of compounds 17 and 18 with phenyl chloroformate, at the same conditions aforementioned for compounds 2, 3 and 4,
47 afforded the desired carbamate 15 and 16 after purification by flash chromatography, using, as eluent, n-hexane/EtOAc 5:1 for compound 15 and petroleum ether/EtOAc 5:1 for compound 16 (Scheme 3).
As reported in Scheme 4, the reaction of the 5-substituted pyridines 13, 17 and 18 with benzoyl chloride was performed at 85 °C for 2 minutes in a microwave reactor (CEM, power 250 W), to afford the amide derivatives 19, 20 and 21. These compounds were purified by flash chromatography, using as eluent, petroleum ether/EtOAc 5:1 for compound 19, petroleum ether/EtOAc 4:1 for compound 20 and petroleum ether/EtOAc 6:1 for compound 21.
Finally, the reaction of 2-aminopyridine derivatives 13, 17 and 18 with phenyl isocyanate , was performed in anhydrous 1,4-dioxane, under nitrogen, at room temperature, and afforded, after 4 hours, urea derivatives 22, 23 and 24 in quantitative yield (Scheme 4).
Scheme 4
Reagent and conditions. (i): benzoyl chloride, MW (CEM), 85 °C, 250W, 2 min..(ii): anhydrous
48 In order to evaluate their inhibitory activity, the synthesized compounds were tested using a HPLC-UV based assay, previously developed in our research laboratory. This assay employs human recombinant MAGL as enzyme and 4‐nitro phenylacetate (4‐NPA) as its enzymatic substrate. The 4-NPA was reported to be a good and safe MAGL substrate, and its interaction of with the enzyme leads to the production of p-nitrophenol (PNP) as metabolite (Fig. 30). 51
Fig. 30 Hydrolysis of 4‐NPA and release of PNP.
The assay is based on the chromatographic separation and subsequent absorbance measurement of PNP, that is detected at the optimal wavelength of 315 nm (fig. 31). MAGL activity was related to PNP concentration, whereas inhibitory activity of the tested compounds was telated to residual enzymatic activity.
49 The HPLC analysis conditions used in the developed method are:
Stationary phase: VARIAN 150x4.6 mm INTERSIL 5μ;
Mobile phase: composed of 53% MeOH and 47% 10mM ammonium acetate buffer at pH 4;
Flow: 1.0 mL / min;
Detector UV: set at wavelengths of 315 nm; Synthesized compound final concentration: 10 μM.
Moreover all the compounds need a pre‐incubation time, between the enzyme and inhibitor, of 30 minutes at 37 °C and an incubation time of 10 minutes at 37 °C after the substrate addition.
50
51
1 Equipments and Methods
Melting points were determined on a Kofler® hot stage apparatus and are uncorrected. Structure of synthesized compound was verified by 1H NMR analysis. NMR spectra were obtained with a Bruker 400MHz spectrometer. Chemical shift are reported in parts per million downfield from tetramethylsilane and referenced to the solvent residual peak. Coupling constants are reported in Hertz (Hz). Evaporation was performed in vacuo by a rotating evaporator. All reactions was monitored by thin-layer chromatography (TLC) using Merck aluminium silica gel (60 F254) sheets that were visualized under a UV lamp (254 nm). Silica gel flash chromatography was performed using silica gel (Kieselgel 40, 0.040-0.063 mm; Merck). Microwave assisted reactions were run in a CEM Discover® LabMateTM microwave synthesizer or in a Biotage initiator.
52
Phenyl N-(6-Bromopyridin-2-Yl) Carbamate (2)
Commercially available 2-amino-6-bromopyridine (1) (200 mg, 1.16 mmol) was dissolved inanhydrous CH2Cl2 (6 mL), and the resulting solution was added
dropwise under nitrogen at 0 °C, to a stirred suspension of phenyl chloroformate (150 µL, 1.16 mmol) and pyridine (190 µL, 2.31 mmol) in anhydrous CH2Cl2 (3
mL). The reaction mixture was warmed to room temperature over 30 min.
Then the reaction mixture was dried under vacuum and after co-evaporated several times with toluene to remove the remainder of pyridine. The solid residue was dissolved in CH2Cl2, washed repeatedly with water, dried over anhydrous
Na2SO4, filtered and concentrated under reduced pressure. The crude residue
obtained was purified by silica gel flash chromatography using n-hexane/EtOAc 11:1 as eluent to give pure compound 2 (269.8 mg,0.92 mmol) as yellow solid.
Yield: 74% Mp: 48 °C – 50 °C 1H-NMR: (CDCl 3) δ (ppm): 7.97 (d, 1H, J = 8.2 Hz, pyridine-H, H3), 7.78 (bs, 1H, -NH), 7.59 (t, 1H, J = 8.1 Hz, pyridine-H, H2), 7.46-7.41 (m, 2H, phenyl-H), 7.31-7.27 (m, 1H, phenyl-H), 7.24 (d, 1H, J = 7.7 Hz, pyridine-H, H1), 7.23-7.19 (m, 3H, phenyl-H)
53
Phenyl N-(6-Phenylpyridin-2-Yl) Carbamate (3)
Compound 5 (215 mg, 1.26 mmol) was dissolved inanhydrous CH2Cl2 (6 mL),
and the resulting solution was added dropwise under nitrogen at 0 °C, to a stirred suspension of phenyl chloroformate (160 µL, 1.26 mmol) and pyridine (200 µL, 2.53 mmol) in anhydrous CH2Cl2 (3 mL). The reaction mixture was warmed to
room temperature over 30 min.
After the solvent was removed under vacuum, and the residue co-evaporated twice with toluene in order to eliminate the remainder of pyridine. The solid residue was dissolved in CH2Cl2, washed repeatedly with water, dried over
anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude
residue obtained was purified by silica gel flash chromatography using n-hexane/EtOAc 9:1 as eluent to give pure compound 3 (176.7 mg, 0.61 mmol) as white solid. Yield: 48% Mp: 185 °C – 187 °C 1H-NMR: (CDCl 3) δ (ppm): 7.99-7.97 (m, 2H, phenyl-H), 7.93 (d, 1H, J = 7.8 Hz, pyridine-H, H3), 7.82 (bs, 1H, -HN), 7.86 (t, 1H, J = 8.1 Hz, pyridine-H, H2),
7.50-7.48 (m, 3H, pyridine-H, phenyl-H), 7.47-7.39 (m, 3H, phenyl-H), 7.29-7.20 (m, 3H, phenyl-H)