UNIVERSIT
À
DEGLI STUDI DI URBINO “CARLO BO”
DIPARTIMENTO DI SCIENZE PURE E APPLICATE (DiSPeA)Corso di dottorato di ricerca in:
SCIENZE DI BASE E APPLICAZIONI
Curriculum:
SCIENZE CHIMICHE E SCIENZE FARMACEUTICHE XXX CICLO
Exploiting Alcohols as Alkylating Agents of
Heterocyclic Nucleophiles through the “Borrowing
Hydrogen” Process
SSD: CHIM/06RELATORE DOTTORANDO
Chiar.mo Prof. Giovanni Piersanti Dott. Giovanni Di Gregorio
i
Contents
Abbreviations iii
1. Introduction 1
1.1 Introduction. 1
1.2 Alcohols in alkylation reactions. 1
1.3 The Borrowing Hydrogen Strategy. 2
1.4 General Activation pathway via Borrowing Hydrogen. 6
1.4.1 C-N Bond Formation-Amination of Alcohol. 7
1.4.2 C-C Bond Formation. 18
1.4.3 Consecutive C-C, C-N bond formation. 31
2. Aim of the Thesis 36
3. Results and Discussions 40
3.1 Observations concerning the synthesis of tryptamine homologues and branched tryptamine derivatives via the borrowing hydrogen process:
synthesis of psilocin, bufotenin, and serotonin. 40
3.2 Divergent Borrowing Hydrogen Approach from Oxindoles and
Ethanolamine: Synthesis of C3-Substituted Oxindoles and Diarylamine. 45 3.3 Iron-Catalyzed Direct C3-Benzylaion of Indoles with Benzyl Alcohols
through Borrowing Hydrogen. 51
4. Conclusions 57
5. Experimental Data 59
5.1 Materials and Methods 59
5.2 Compounds Characterizations and Synthetic Methods 60
5.2.1 Observations concerning the synthesis of tryptamine homologues and branched tryptamine derivatives via the borrowing hydrogen process:
synthesis of psilocin, bufotenin, and serotonin. 60
5.2.2 Divergent Borrowing Hydrogen Approach from Oxindoles and
ii
5.3.3 Iron-Catalyzed Direct C3-Benzylaion of Indoles with Benzyl Alcohols
through Borrowing Hydrogen. 74
iii
Abbreviations
Ac acetyl acac acetylacetonate BHT butylated hydroxytoluene BINAP (2,2'-bis(diphenylphosphino)-1,1'-binaphthyl) BIPHEP 2,2'-Bis(diphenylphosphino)biphenyl Bn benzyl cod 1,5-cyclooctadiene cot 1,3,5- cyclooctatriene Cp* 1,2,3,4,5-Pentamethylcyclopentadiene DCM dichloromethane dppf 1,1′-Ferrocenediyl-bis(diphenylphosphine) dppp 1,3-Bis(diphenylphosphino)propane Ms mesylate MS molecular sieves MW micro waveNMR nuclear magnetic resonance Nu nucleophile
Pc phthalocyanine Py pyridine
THF tetrahydrofuran Ts tosylate
1
1. Introduction
1.1 Introduction.
The extreme growth of global economy has led to a massive exploitation of fossil reserves both for energy need end for the production of chemicals. Nowadays these requests are partially provided from renewable sources. The passage form fossil-based economy to a more sustainable economy has become a top priority. This transition from fossil economy to a sustainable economy is necessary to meet the urgent environmental concerns in favour to a sustainable development, preserving the integrity, stability and beauty of natural biotic system. The use of renewable alternatives and sustainable sources has a number of advantages in comparison with the use of fossil sources, for example, reduction of air pollution, global warming, and dependence from petroleum.1 Chemists, scientists, and engineers have a key role in
the developing processes that exploit renewable sources. In recent years, there is an enormous interest in the usage of alcohols as starting materials, as they are readily available by a variety of industrial processes, inexpensive because they can be obtained renewably via fermentation or catalytic conversion of lignocellulosic biomass, relatively nontoxic, and easy to use.2 The work presented in this thesis is focused on the use of alcohols as alkylating agents to develop new synthetic methodology exploiting the borrowing hydrogen process, as activation strategy that allow the preparation of valuable classes of molecules of industrial and pharmaceutical interest according to the guide lines of green chemistry.
1.2 Alcohols in alkylation reactions.
Alcohols are versatile, organic compounds since they undergo a wide variety of transformations. Considering the use of alcohols in alkylation reactions, they are able to react only prior activation. Conventionally the activation consists in the conversion of OH group into a good leaving group. To increase the electrophilicity at C1 of the alcohol and generate a good leaving group allowing the reactivity toward the nucleophilic attack the simplest method would be the protonation. Despite the protonation looks attractive as it produces only water as byproduct, this method can deactivate the nucleophile as occur in acid conditions, reducing drastically the scope and efficiencies of the reaction. Therefore, alcohol activation as alkylating agent is afforded by the interconversion of OH into alkyl halide, tosylate or mesylate. These kind of activations from a green metrics point of view does not respect the sustainability and the efficiency of the reaction. Not only an extra step is required but also both for the activation and the subsequent reaction a stoichiometric amount of
2
chemical waste is produced, reducing the safety due to the intrinsic mutagenicity and toxicity of the halide. Alternatively, also the in situ nucleophilic substitution of alcohols operated through a Mitsunobu reaction, has the problem of the stoichiometric use of the derivatizing reagents. Dialkyl azodicarboxylate (e. g. diethyl azodicarboxylate, DEAD) and triphenylphosphine, are also unsafe and toxic (Scheme 1).
Scheme 1 Traditional activation of alcohols.
Despite these facts, the direct substitution/alkylation of alcohols has been developed. Through the use of Lewis acids and Bronsted acids, π-activated alcohol such as allylic and benzylic can undergo the direct substitution of the hydroxyl group, via formation of a carbocation.3 However, many of these methods have some drawbacks,
even if the problem of the stoichiometric use of Lewis acids or Bronsted acids is bypassed by the catalytic variation/form. Low yields of the products, long reaction times, harsh reaction conditions, tedious work-ups and relatively expensive reagents, the requirement for an inert atmosphere, and formation of a significant amount of side products, are still present. Only one example of the use of less-activated alcohols was reported, nevertheless the scope continues to be mainly restricted to π-activated alcohol.4 For all these reasons aforementioned, the development of efficient and
sustainable methodologies for the activation of alcohols is needed. Among the strategy developed for the direct use of alcohols as alkylating agents, the catalytic activation of alcohols by hydrogen borrowing has attracted significant attention in recent years as a powerful and sustainable solution for addressing some of the contemporary goals of pharmaceutical and chemical industries. As highlighted during the ACS GCI Pharmaceutical Roundtable the direct nucleophilic substitution of alcohols is one of the most important and challenging priorities both for academia and industry.5
1.3 The Borrowing Hydrogen Strategy
The increasing demand to have more green processes in chemistry has focused the attention to bond construction strategies that promote atom economy and avoid mutagenic reagents.6 The borrowing hydrogen or hydrogen autotransfer is a catalytic
fashion way to exploit the potential of alcohols to be ideal green substitute of halides or (pseudo)halides. Alcohols are ideal candidates for green chemistry because they have low-molecular-weight leaving group (HO, 17 u.m.a.), low environmental impact factor of the leaving group (H2O), they are inexpensive and sustainable (most of them
3
come from biomasses and renewable sources1) and they are easy to handle and store
(safely).7 The more general description of the borrowing hydrogen process can be
given as follow: an inert substrate after the dehydrogenation operated by a metal catalyst become reactive (oxidized species), this new reactive oxidized substrate will form the reacted substrate, and finally the metal catalyst makes the hydrogenation to form the final product. In this cycle, the catalyst act both as oxidant (dehydrogenation) and as reductant (hydrogenation) – Globally no net oxidation or reduction takes place in the catalytic cycle itself (Scheme 2).
Scheme 2 General scheme of Borrowing Hydrogen Process.
The substrates involved in the borrowing hydrogen activation are alkanes,8 alcohols,9 and amines.10 Regarding the catalytic cycle of borrowing hydrogen for alcohol activation mainly consists of two or three steps: The first step is the alcohol dehydrogenation run by the catalyst, then the correspondent carbonyl compounds derived from the dehydrogenation of alcohol can carry out its typical reactivity, and lastly in the third step the abstracted hydrogen is usually returned back to the intermediate. It is also possible to intercept the intermediate for a further functionalization or block the reaction and have the hydrogenation of the sacrificial intermediate or that the intermediate can react again with additional reagents or the abstracted hydrogen is further returned and incorporated to the final product, hence the reaction name (Scheme 3). In other words in some specific cases, the borrowing process can be truncated to the reactive intermediate using a hydrogen acceptor or H2
gas removal. The result in these cases is a reactive final compound that might be ready for further functionalization/ additional cascade transformation.
4
Scheme 3 General Borrowing Hydrogen Process for Alcohols Activation.
From a mechanistic point of view the borrowing hydrogen process (activation via dehydrogenation) operated by homogenous catalyst depends from the type of the complex, the substrates as well as the conditions of reaction. Considering that the catalytic cycle is based on three steps. (i) Dehydrogenation (ii) functionalization and (iii) hydrogenation and since functionalization in step (ii) is usually a classical organic transformation such us a nucleophilic addition and there is no catalyst needed as it can act probably coordinating the carbonyl compound enhancing the electrophilicit, it is possible to assume no participation of the catalyst in this step. The more significant and central steps are the first (i) and the third (iii), assuming reversibility between they are possible to discuss only the first (i). The mechanistic possibilities for the step (i) of transfer hydrogenation are mainly classified as: Inner sphere, intermediate sphere, outer sphere.11 In the inner sphere, after the alcohol
coordination to the metal centre trough an insertion, the correspondent carbonyl compound is formed via -hydride elimination both for dihydride and monohydride mechanisms (Scheme 4).
5
Scheme 4 Inner Sphere Transfer Hydrogenation Mechanisms.
The intermediate sphere mechanism consists in a transfer of the hydride from the donor to the acceptor where the metal hold together the two species like in the Meerwein-Ponndorf-Verley (Scheme 5).
Scheme 5 Intermediate Sphere - Mechanism of the Meerwein-Pondorf-Verley Reduction and
Oppenauer Oxidation.
The outer sphere mechanism is plausible when the metal complex have no open site for binding the substrate. This kind of mechanism is for all the catalysts that have a hydride in position cis respect to the protonic hydrogen, typically in a form coordinating R2NHligand, like in the Noyori’s Catalyst (Scheme 6).
6
Scheme 6 Outer sphere transfer Hydrogenation of Noyori’s Catalyst.
The mechanism of dehydrogenative activation via homogeneous transition-metal complexes varies greatly, depending on the substrate and catalyst employed as well as the conditions chosen. The two H atoms extruded can be delivered to the metal complex or directly to a hydrogen acceptor. The pathways for stepwise mechanisms generally consist of two separate components: association of the substrate with the catalyst followed by cleavage of a C-H bond. A saturated molecule must first coordinate in some manner to a transition-metal complex. This step often requires direct activation of a C-H bond for alkanes, or an O-H bond for alcohols, or an N-H bond for amines. Alcohol or amine binding to the metal catalyst is typically followed by a deprotonation event. The resulting metal alkoxide or amine complex then undergoes to a C-H bond cleavage, resulting in a metal hydride and a dehydrogenated organic species. Following the dehydrogenative step, a functionalization occurs. This process can be catalyzed by the same catalyst employed for dehydrogenation, catalyzed by a different species added to the reaction mixture for this purpose, or simply occurs without the need for a catalyst. Once generated, the resulting intermediate can react with additional reagents and be oxidized or reduced depending on the specific reaction sequence.
1.4 General Activation pathway via Borrowing Hydrogen.
The general reactivity of alcohols both as nucleophiles and as electrophiles is very limited and some kind of activation results necessary. As describe before, OH group is transformed to alkoxide to enhance the nucleophilicity, on the other end, electrophilicity is ideally enhanced adding an acid. This description involves the classical activation of alcohols. Another possibility is to convert alcohols transiently in to the correspondent carbonyl compound. In this way is possible to exploit the reactivity of carbonyls that in comparison to the alcohol, as functional groups, allow more transformation, and lastly, after the reaction, the intermediate product is reduced to the original oxidation state of the alcohol. This process become catalytic, when, after the oxidation of the alcohol (activation), the formation and the reaction of the carbonyl compound, the intermediate is reduced under the reaction conditions. Now a detailed discussion on the reactivity of the carbonyl compound follows. After
7
the oxidation, we can have aldehyde or ketone respectively from primary and secondary alcohol, that in term of reactivity they show almost the same reactivity. As shown in (Scheme 7) carbonyl compound reacts at C1 in nucleophilic addition reactions, and in C2 with electrophile exploiting the possible enol-enolate chemistry, and alkene formation in Wittig type reaction (ylide addition to C1).
Scheme 7 Alcohol Activation Pathways via Borrowing Hydrogen.
1.4.1 C-N Bond Formation-Amination of Alcohol: alcohol amination is
traditionally achieved using alkyl halide agents. The alkyl halide strategy for the formation of amine by alkylation may have control limitation like the over alkylation. For example, the alkylation of a primary amine could results in the formation of a mixture of mono, di-alkylated amine and also the quaternary ammonium salt. This over alkylation simply happens because the more the amine is substituted, the more it becomes nucleophilic and reactive in the substitution reaction with alkyl halides. Methods for the controlled alkylation of primary amine have been reported, and in some instances, is possible to have mono alkylation under appropriate conditions. At the beginning Gabriel synthesis was introduced for the synthesis of primary amines12 and many other methods was developed. For examples, more recently a potassium iodide catalyzed method for the selective N-monoalkylation of amines with alkyl halides and alkyl tosylate under microwave irradiation has been described. A simple method for the N-alkylation of primary amines was developed using ionic liquids as solvent in order to prepare secondary amines selectively. However, these approaches have some drawbacks even if they are selective they continue to employ mutagenic and toxic reagents. The direct amination via borrowing hydrogen resolve both the problems, over alkylation and the use of mutagenic and toxic reagents. The sequence of alcohol amination consists in: alcohol dehydrogenation operated by the metal catalyst, the resulting carbonyl compound react in an addition reaction with the
8
ammine generating an imine fially is reduced to amine by the catalyst, with the hydrogen borrowed from the alcohol in the dehydrogenation step. In this alcohol activation process the over alkylation is controlled, due to the initial secondary amine formed by alkylation of primary amine, the intermediate doesn’t tend to react further because this would require the unfavorable formation of an iminium cation. The borrowing hydrogen activation process is a good alternative to the reductive amination. In other words, it could be considered the catalytic version of reductive amination employing alcohols rather than aldehydes that in some case result instable like -amino aldehyde13 that can react itself or lose the optical purity/stability. These
problems are overcome with the borrowing hydrogen because the aldehyde is transiently formed and consumed soon after. Several examples of C-N bond formation via borrowing hydrogen have been reported, and among them, a selection of examples follows. The first alkylation of amine using alcohols via the borrowing hydrogen route date back to 1932 with a work where a heterogeneous nickel catalyst was employed.14 The first report of N-alkylation with alcohols of primary and secondary amine via homogeneous catalysis was by Grigg and co-workers in 1981.15 A screening with different catalysts based on rhodium, iridium and ruthenium was presented (Scheme 8).
Scheme 8 N-alkylation via Borrowing Hydrogen with Rh Ir Ru Catalyst.
Watanabe has reported a study about the selectivity for N-alkylation of primary amine demonstrating that different ruthenium catalysts and adopted conditions show different selectivity between mono and di-alkylation for the same substrates (Scheme 9). 16
9
Scheme 9 Selectivity: Mono-Alkylation vs Di-Alkylation of Amines.
A method for converting primary alcohols in primary amines using a PNP pincer complex of ruthenium and reasonable pressure of ammonia has been reported by Milstein and co-workers (Scheme 10).17
Scheme 10 Direct Formation of Primary Amine from Ammonia and Alcohol via Borrowing
Hydrogen.
Alkylation of primary amines with primary alcohols has been accomplished by Williams and co-workers, an alternative combination of phenylethanol and tryptamine or tryptophol and phenylethanamine give the secondary amine product in good yields (Scheme 11).18
10
Taddei and co-workers reported an interesting example of C-N bond formation at low temperature, via borrowing hydrogen. The alkylation of arylamines has been achived using stoichiometric amounts of aliphatic and benzylic alcohols in the presence of
tBuOK, the reaction was carried out at 55 °C using ruthenium with low catalyst
loading. (Scheme 12).19
Scheme 12 Alkylation of different aniline under mild condition.
The catalytic enantioselective synthesis of chiral amines via borrowing hydrogen is possible and represents a highly efficient way to prepare chiral amines from simple alcohols. Zhao reported the cooperative catalysis by iridium and a chiral phosphoric acid where different amines and alcohols have been employed for the enatioselective synthesis of chiral amines (Scheme 13).20
Scheme 13 Catalytic Enantioselective Synthesis of Chiral Amine.
Enantioselective version of C-N formation has been used for the formation of -amino-alcohols from different diols and secondary amines. The reaction is catalyzed by ruthenium complex/JOSHIPHOS and proceeds mainly through amino ketone intermediates that are converted into the corresponding optically active β-amino alcohols (Scheme 14). 21
11
Scheme 14 Enantioselective Synthesis of β-Amino Alcohols.
Dynamic kinetic resolution (DKR) for the amination of alcohols was recently presented by Zhao.22 Applying the borrowing hydrogen concept, a cooperative
catalysis by an iridium complex end chiral phosphoric acid, -substituted alcohol that exist as a mixture of four isomers was converted in disastero- and enantio-pure amine. More precisely as shown in (Scheme 15) the initial racemic alcohol is converted in to the enantioenriched amine through a borrowing hydrogen pathway where a DKR take place. Dehydrogenation of the alcohol to ketone, condensation of ketone to form imine followed by transfer hydrogenation of amine and tautomerization /racemization of the two imines allow the formation into the amine that come from the fastest asymmetric hydrogenation.
12
Scheme 15 Dynamic Kinetic Asymmetric Amination of Alcohols and Mechanism.
A recent related work has been presented by the same group, an acid-assisted Ru-catalyzed enantioselective amination of 1,2-diols through borrowing hydrogen. In this reaction, the involved mechanism is borrowing hydrogen combined with dynamic kinetic resolution, and from a preliminary mechanistic study the beneficial effect of
13
achiral Brønsted acids on the enantioselectivity of the reaction may come from an accelerated tautomerization/racemization. Several β-amino alcohols have been prepared through monoamination of readily available racemic diols (Scheme 16).
Scheme 16 Enantioselective Amination of Alcohols with Secondary Amine.
Nitrogen containing heterocycles are target of great interest in organic synthesis due to the common occurrence in natural products and pharmaceutical active compounds. The amination via borrowing hydrogen has been extended with success to the formation of nitrogen containing heterocycles. As strategy, amino alcohol cyclisation has been employed as shown in (Scheme 17), ruthenium and rhodium complexes are able to provide the reaction in good yield. 23
Scheme 17 Cyclization Reaction of Aminalcohols.
Annulation reaction employing diols and primary ammines is another disconnection affordable via borrowing hydrogen strategy for the preparation of nitrogen containing heterocycles. With a suitable catalyst is possible to promote a double alkylation of the primary amine, with the second step being an intramolecular alkylation (Scheme 18).
14
Scheme 18 Annulation Disconnection for the Formation of N-heterocycles from Primary Amine.
With this strategy Koten and co-workers have prepared piperazine (4) which is a potent serotonin agonist from aniline (1) and diol (2) using a ruthenium pincer complex (3).24 Watanabe and co-workers have converted primary amine into piperidines (5) morpholines (6) and piperazines (7) by condensation with suitable diols in presence of ruthenium phosphine complex (Scheme 19).25
Scheme 19 N-Heterocyclization from Primary Amine via Borrowing Hydrogen.
15
Iridium complex [IrCp*Cl2]2 has been used for this heterocyclization process.
Representative examples include as primary amine the use of aliphatic amine, benzylamine, and aniline with different diols in good yields (Scheme 20).26
Scheme 20 Nitrogen Heterocycles Formation Catalyzed by [IrCp*Cl2]2.
Moreover, an efficient method for the formation of 5-, 6-, and 7-membered cyclic amines from tryptamine and suitable diols has been presented employing an iridium complexes show in (Scheme 21).27
16
Scheme 21 Cyclic Amines from Tryptamine.
Another interesting N-heterocycles construction has been reported by Taddei and co-workers. In particular starting from 2-aminobenzyl alcohols and 1,2-aminoalcohols, 2,3,4,5-tetrahydro-1H-1,4-benzodiazepines (THBDZ) can be prepared through a one-pot ruthenium-catalyzed reaction encompassing two consecutive borrowing hydrogen cycles (Scheme 22).28
Scheme 22 Ru Catalyzed Benzodiazepine Derivatives Synthesis Reported by Taddei.
Stereoselective version of N-heterocycle construction has been reported by Fujita and Yamaguchi for the preparation of 2-substituted piperidines. Starting from racemic diols and enantioenriched amine the major diastereoisomer was formed in 92 % de. via reduction of the iminium intermediate, and 86% ee. The slight loss of enantiopurity is explained by the isomerization of intermediate (Scheme 23).29
17
Zhao reported the stereoselective construction of 2-methyl-tetrahydroquinolin, using iridium complex as catalyst and chiral phosphoric acid as additive (Scheme 24).19
Scheme 24 Stereoselective Intramolecular Amination of Alcohols.
Urea motifs with glycol as diol can form the correspondent cyclic product dihydroimidazolone,30 whereas employing the same catalytic system based on
ruthenium and still using ethylene glycol with a primary amine, symmetric formation of piperazine has been reported (Scheme 25).31
18
1.4.2 C-C Bond Formation: In general, the carbon-carbon bond formation via
borrowing hydrogen is affordable in two different manners. After the dehydrogenation of alcohol and formation of correspondent carbonyl compound, one is the attack of C1 of the carbonyl with a carbon nucleophile, usually methylene active compounds, electron rich aromatic compounds, and stabilized or non-stabilized phosphorus ylides. The other possibility is the -functionalization with an electrophile, or a mixed case where the alcohol act as electrophile and nucleophile in the same reaction. Indirect Wittig reaction of alcohols via borrowing hydrogen has been described by Williams employing [Ir(cod)Cl]2. After the dehydrogenation step
of the benzylic alcohol the correspondent carbonyl compound readily undergo olefination with the ylide and then alkane is formed by hydrogenation completing the catalytic cycle (Scheme 26).
Scheme 26 Indirect Wittig Reaction of Alcohols by Borrowing Hydrogen Activation.
Moreover, Williams and co-worker uses a Ru-NHC complex with great result in term of yields and more mild conditions in the Wittig Type process with alcohols activation (Scheme 27).32
19
Stereoselective version of indirect Wittig has been developed by Williams using [IrCp*Cl2]2 and a chiral phosphine BINAP, in the overall process the stereo-selection
is introduced during the hydrogenation step (Scheme 28).33
Scheme 28 Stereoselective Wittig Reaction Via Borrowing Hydrogen.
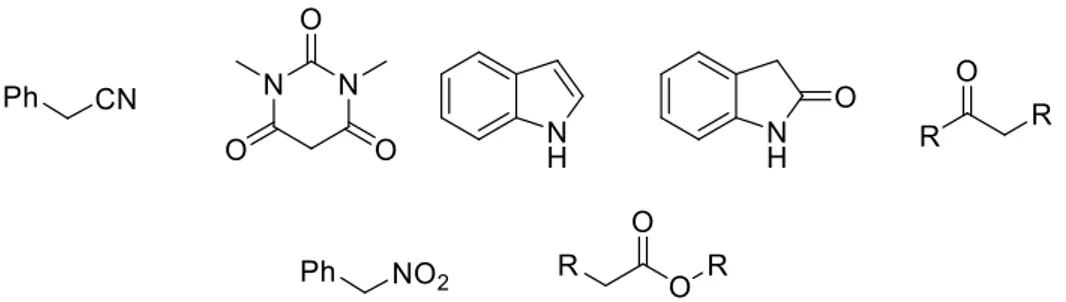
In the realm of C-C bond formation through borrowing hydrogen several nucleophiles has been employed, an overview follows in (Table 1).
Table 1 Representative Nucleophile used in Browning Hydrogen.
Grigg published seminal work on C-C bond formation via hydrogen borrowing with the monoalkylation of arylacetonitriles by alcohols, where the catalyst was prepared in situ from rhodium trichloride triphenylphosphine and sodium carbonate (Scheme 29).34
Scheme 29 First Example of Homogeneous Transition Metal Catalysed C-C Bond Formation via
20
More recently Grigg reported this transformation using [IrCp*Cl2]2 as catalyst and
catalytic amount of KOH as base. With this convenient and highly effective catalytic system has been achieved selective monoalkylation of arylacetonitriles with a wide range of aromatic, heteroaromatic, and aliphatic alcohols (Scheme 30).35
Scheme 30 Monoalkylation of Arylacetonitriles with Primary Alcohols.
Another methylene active compound studied, cyanoacetate, was reported by Grigg and Hisii in two distinct work. Grigg reported in his work the alkylation of T-butyl cyanoacetate with a variety of substituted benzyl and heteroaryl alcohols affording the corresponding -alkylated products from moderate to high yield.36 While Hisii
with different cyanoacetate as methylene active compound reported the iridium catalyzed alkylation with different primary aliphatic alcohols (Scheme 31).37
Scheme 31 Monoalkylation of Cyanoacetates Via Borrowing Hydrogen.
Catalytic alkylation of 1,3-dimethylbarbituric acid as methylene active compound with various alcohols under solvent-free microwave irradiation (MWI) conditions and [IrCp*Cl2]2 has been reported by Grigg (Scheme 32).38
21
Scheme 32 Alklyaltion of 1,3-dimethylbarbituric acid.
Indole is not a proper methylene active compound but can be considered an activated nucleophile. In particular, its electron rich system has been employed as nucleophile in C3, his behaviour is quite similar to enamine, and the resulting C3-nucleophilicity can be easily justified by resonance forms as shown in (Figure1).
Figure 1
Grigg reported the formation of substituted indoles with benzylic alcohol catalyzed by [IrCp*Cl2]2 (Scheme 33).
Scheme 33 Synthesis of Substituted Indoles Via Borrowing Hydrogen.
Inspired by the work of Grigg, Piersanti and co-workers reported the first iridium-catalyzed direct synthesis of tryptamines. Different substituted indoles (position 2, 4, 5, 6 and 7) were alkylated with different N-protected aminoles (Bn, dimethyl) using [IrCp*Cl2]2 as catalystend Cs2CO3 as base (Scheme 34). 39
22
Scheme 34 Selective C3-Alkylation of Indoles with N-Protected Ethanolamines Involving the
“Borrowing Hydrogen”
Since oxindole showns a pKa value of 18.2, and it is an interesting methylene active compound, has been exploited as nucleophile by Madsen. The use of RuCl3 · H2O,
PPh3, and NaOH has been proved as a very effective catalytic system for the site
selective mono 3-alkylation of unprotected and protected oxindoles with a range of aromatic, heteroaromatic, and aliphatic alcohols (Scheme 35).40
Scheme 35 Selective C3 alkylation o oxindoles with different alcohols.
Other interesting nucleophiles are ketones, as they are nucleophile in C2 and they undergo to indirect -alkylation with alcohols via borrowing hydrogen following the aldol reaction pathway as depicted in (Scheme 36).
Scheme 36 Oxidation/ Aldol Condensation/ Reduction Pathway.
In this reaction, the intermediate is the -unsaturated ketone, that can be transformed either in statured ketone or in the complete reduced form (alcohol) using an additional source of hydrogen or a high amount of alcohol (hydrogen source). For
23
example, ruthenium-catalyzed borrowing hydrogen of ketones and primary alcohols involving carbon-carbon bond formation has been developed by Cho and co-workers, acetophenone was alkylated with benzyl alcohol and butanol to obtain the correspondent alcohol, this when 3 equivalents of alcohol are used (Scheme 37).41
Scheme 37 Alkylation of Acetophenone with Benzylic Alcohol and Butanol.
The same ruthenium catalyst allows the formation of the correspondent carbonyl compound when only one equivalent of alcohol is used in a presence of dodecane as hydrogen acceptor (Scheme 38).42
Scheme 38 Alkylation of Acetophenone with Benzylic Alcohol.
Yus and co-workers used a related approach employing a catalyst system based on ruthenium, employing 1 equivalent of alcohol is possible to obtain the final product as alcohol while exploiting the same catalyst system in presence of 2 equivalents of alcohol and a triphenylphosphine the ketone was obtained (Scheme 39).43
24
Scheme 39 -Alkylation and Formation of Relative Ketone and Alcohol.
A one pot procedure was used by Nishibayashi to perform the condensation of an alcohol and a ketone in asymmetric way, where after the activation of alcohol operated by [Ir(COD)Cl]2 and subsequent aldol reaction the reduction of
-unsaturated intermediate is operated by an asymmetric complex of ruthenium (Scheme 40).44
Scheme 40 Asymmetric Alkylation of Ketone with Alcohols.
More recently asymmetric tandem α-alkylation via borrowing hydrogen of acetophenone with primary alcohols was demonstrated using ruthenium and enantioenriched amino acid as chiral ligand (Scheme41).45
25
Scheme 41 Tandem α-Alkylation of Acetophenone with Primary Alcohols, and Relative Schematic
Mechanism.
Finally, as methylene active nucleophile nitro alkane has been employed by Williams in nitro aldol reaction in the context of hydrogen borrowing with alcohols as electrophiles (Scheme 42).
Scheme 42 Indirect Nitro Aldol Reaction via Borrowing Hydrogen.
Still in the realm of enantioselective formation of C-C bond with the borrowing hydrogen strategy, enzymes were used as catalyst. -cyano-ketones were alkylated with different alcohols in the presence of Baker’s Yeast or C. Lunata as enzyme. In this biocatalysis example, the followed pathway is the borrowing hydrogen process (Scheme 43).
26
Scheme 43 Biocatalysis example of Borrowing Hydrogen.
-alkylation of Alcohols: When carbonyls compounds are formed transiently form alcohols they can undergo to enol-enolate chemistry as shown in (Scheme 44).
Scheme 44 - Functionalization of Alcohol via Borrowing Hydrogen.
In this reaction, different electrophiles can be employed, if the electrophile is a carbonyl compound, it can be directly added as regent in the reaction or can be formed in situ from another alcohol. Another possibility is that the initial alcohol can react with itself according to a Geurbet aldol pathway (Scheme 45).
27
Scheme 45 Guerbet Product Formation
Considering the reaction stoichiometry during an alcohol-alcohol coupling the product is an alcohol, except when the hydrogen is transferred to another spices or is liberated in atmosphere, otherwise results in -unsaturated carbonyl. While carbonyl and alcohol react together is possible to obtain an alcohol or a carbonyl, based on the amount of alcohol (hydrogen donor). In particular in the presence of an excess of alcohol (hydrogen donor) is possible to reduce the intermediate -unsaturated carbonyl all the way down to alcohol. Examples of the aforementioned chemistry follow. Indirect -bromination of alcohol has been presented by Williams, using aluminium alkoxide as catalyst and pyridinium tribromide as brominating agent. The reaction via temporary oxidation of the alcohol to a ketone, giving the -brominated product in good yield (Scheme 46).46
Scheme 46 Indirect Bromination of Alcohol via Borrowing Hydrogen Activation.
Self-condensation of alcohol (Guerbet Reaction) of primary alcohols leading to -alkylated dimer alcohols catalyzed by iridium complexes has been reported by Ishii in good yields (Scheme 47).47
28
Scheme 47 Guerbet Reaction of Primary Alcohols.
Two alcohols can react together forming a new C-C bond, usually a primary alcohol and a secondary alcohol are coupled. From a mechanistic point of view, both alcohols are oxidized to the correspondent carbonyl compounds (ketone, and aldehyde). These react together in aldol condensation giving an -unsaturated ketone, which undergoes reduction to give the saturated corresponding alcohol (Scheme 48).
Scheme 48 General Mechanism for Alcohol Indirect Aldol Condensation.
For example, butanol and methanol has been proved to react together employing different catalyst systems producing isobutyl alcohol. Ru(PPh3)3Cl2 has been used for
the coupling of a secondary alcohol with primary alcohols, using1-dodecene as hydrogen source. Yamguchi for the same reaction has reported a different catalyst system, based on iridium, enhancing the yield (Scheme 49).
29
Scheme 49 Examples of Alcohol-Alcohol Coupling.
As extension in the C-C bond formation, we can consider the functionalization of allylic alcohol, as they can be considered as homo version -functionalization of alcohols. Quintard and Rodriguez reported a dual catalyst system based on iron catalyst and an organocatalysts, for the enantioselective functionalization of allylic alcohol with Keto-ester as nucleophile (Scheme 50). 48
30
Scheme 50 Enantioselective Addition of Ketoester to Allylic Alcohols, and Relative Concept of
Dual Catalysis.
Interestingly, the chiral linear alcohols obtained in this transformation are in equilibrium with the closed lactol form. Building on this initial report, the same team reported the use of the same catalytic system for diketones instead of ketoesters leading directly to a linear 3-alkyl-pentanol scaffolds.49 This dual catalyst system allows the preparation of important synthetic intermediates that otherwise require long and tedious synthesis (Scheme 51).50
31
Scheme 51 Dual Catalysis: Enantioselective Addition of Diketones to Allylic Alcohols.
1.4.3 Consecutive C-C, C-N bond formation.
In the field of borrowing hydrogen, the last past year’s investigations have focused on the development of new catalytic systems more and more efficient with the goal to reach mild condition and low catalyst loading for the formation of C-C and C-N bonds. More recently considering the raising interest in the construction of complex structure motif in the most efficient possible way, and the interest in the formation of N-containing heterocycle due to their relevance from a medicinal point of view, the focus of borrowing hydrogen is addressed to the synthesis of sustainable consecutive formation of C-C and C-N bond for the preparation of N-heterocycles and more general to the combination of C-C and C-N bond formation in one pot process51.
Kempe and Co Worker reported the use of a PNP-type iridium-based catalyst for an efficient synthesis of pyrroles. They presented the formation of several substituted pyrroles, in particular: the formation of 2,5 and 2,3,5 di-and tri-substituted pyrroles, bicyclic pyrroles from cyclic secondary alcohols, dipyrroles from diols, and substituted pyrroles form amines and diols (Scheme 52).52
32
Scheme 52 Synthesis of Pyrroles via Borrowing Hydrogen.
Soon after Milstein presented another efficient, atom-economical, one-step synthesis of pyrroles, based on dehydrogenative coupling of -aminoalcohols with secondary alcohols, catalyzed by a ruthenium pincer catalyst. Alfa amino aldehyde is generated from the corresponding -aminoalcohols, and the ketone is generated from the secondary alcohol, these two activated species can undergo to coupling to form the correspondent pyrroles in the presence of base. Therefore, formation of pyrroles
33
through selective and consecutive C-C and C-N bond formations take place (Scheme 53).53
Scheme 53 Ruthenium Catalyzed Pyrroles Synthesis via Browning Hydrogen.
Saito reported the direct formation of pyrroles form 1, 2 amino alcohols and ketones instead useing the secondary alcohols as above described by Milstein, employing a ruthenium catalyst. In his work Saito demonstrated the preparation of 2, 5 and 2, 3, 5 di-and tri-substituted pyrroles and the synthesis of the core pyrrole of Lipitor (Scheme 54).54
34
Scheme 54 Saito Strategy for the Synthesis of Diverse Substituted Pyrroles via Borrowing
Hydrogen.
In the panorama of one pot multiple formation, an extra example outside the C-C and C-N bond follows. Beller reported the enantioselective synthesis of oxazolidin-2-ones from urea and diols. The synthesis of this heterocycle experiences the sequential formation of two different C-O and C-N bonds, in a domino process consisting of nucleophilic substitution and alcohol amination (Scheme 55).55
35
36
2. Aim of the Thesis
The work presented in this thesis is focused on the use of alcohol as green alkylating agents to develop novel synthetic routes using borrowing hydrogen methodology to synthesize small molecules and biologically active natural products. Initially, based on the recently reported protocol for the direct Ir-catalyzed alkylation of indoles with N-substituted ethanolamines (Scheme 1),37 the synthesis of substituted tryptamine
derivatives starting from indoles and amino alcohols via the borrowing hydrogen process were considered. / Initially was considered the synthesis of substituted tryptamine derivatives starting from indoles and amino alcohols via the borrowing hydrogen process based on the recently reported protocol for the direct Ir-catalyzed alkylation of indoles with N-substituted ethanolamines (Scheme 1).37
Scheme 1 C3-Alkylation of Indoles with N-substituted Tryptamine.
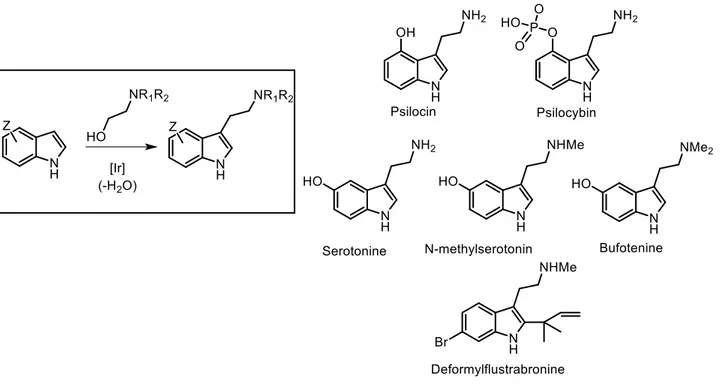
Tryptamine is commercially available through a variety of suppliers; however, when the indole ring of tryptamine is functionally substituted, the commercial availability of the resulting derivatives decreases while their cost increases, especially with 4 or 6-substituted tryptamine derivatives. Tryptamine motif occurs in a wide range of biologically active molecules, pharmaceuticals and naturally occurring compounds, such as those outlined in (Figure 1).
37
Figure 1 Synthesis of Biologically Interesting Tryptamine.
Tryptamine derivatives have attracted comprehensive and continuous interest from the chemical community.56 Moreover, tryptamine represent the basis for some condensed ring alkaloids, and it is a key starting building block for many intents and purposes, such as the total synthesis of polycyclic tryptamine-derived indole alkaloids.57 Simple synthetic approaches tryptamine derivatives are highly desirable considering their practical importance and because they avoid the lees sustainable approach.58 For this purpose efficient formation via borrowing hydrogen of
acetyl-protected branched tryptamines and homotryptamine derivatives, as well as N-methylated tryptamine cores of biological importance such as psilocin, bufotenin and serotonin, were taken in consideration.
Scheme 2 Synthesis of Tryptamine Derivatives by the Use of Borrowing Hydrogen Method.
Further oxindole, another scaffold that characterizes a large number of natural59 and synthetic compounds with important biological activities,60 will be explored in order
38
to show the importance and versatility of ethanolamines as alkylating agents in a different context and the power of iridium catalyst system already developed. Oxindoles have a wide range of applications and they show an extensive range of biological effects. Moreover, the biological activity of the oxindoles and their derivatives has made them very important in synthetic organic and medicinal chemistry. These characteristics of oxindoles prompt chemist to provide various applications of oxindoles and their derivatives. In particular using the already developed iridium catalyst37 it has been planned to present a divergent synthesis employing diversely substituted oxindoles: when N-acetyl-ethanolamine is used the C3-alkylation via borrowing hydrogen methodology has been expected, alternatively when N-benzyl ethanolamine is used a sequential/domino process, consisting of alkylation and transamidation processes, should took place providing α-substituted γ-lactams. When N-aryl oxindoles will be employed with N-benzyl ethanolamine, substituted diarylamines could be obtained, which represent an important class of compounds due to their wide applications and special pharmacological activities (Scheme 3).61
Scheme 3 Divergent Borrowing Hydrogen Approach from Oxindoles end Ethanolamines: Synthesis
of C3-Substituted Oxindoles and Substituted Diarylamines.
Though, in terms of sustainability, the use of precious transition metals should be substituted by more eco-friendly, inexpensive and earth-abundant metals. Among these metals, iron attracts significant attention and is considered as a valuable alternative.62 In the last decades, iron catalysts have increasingly been used in organic
synthesis in a number of reactions.63 Feringa and Barta, Wills and Zhao have reported
the alkylation of primary amines with alcohols to give secondary and tertiary amines by utilizing iron catalysts featuring functionalized cyclopentadienone or hydroxy cyclopentadienyl ligands based on Knölker’s complex or derivatives thereof.64
However, the borrowing hydrogen methodology using iron with alcohol in C-C- bond formation has been reported in few publications.65 Therefore, the focus was placed on
the search for an iron catalyst able to afford the selective iron C3-alkylation of indoles with different substituted benzylic alcohol. After a search for a suitable
39
catalyst system based on a commercial available iron complex, the reaction scope will be tested with different substituted primary and secondary benzyl alcohol and eterobenzylic alchol (Scheme 4).
Scheme 4 Iron Catalyzed Direct C3-Benzylation of Indoles with Benzyl Alcohols.
Mechanistic studies were expected to ensure that the catalytic process involves the borrowing hydrogen mechanism.
40
3. Results and Discussion
3.1 Observations concerning the synthesis of tryptamine homologues and branched tryptamine derivatives via the borrowing hydrogen process: synthesis of psilocin, bufotenin, and serotonin.
Initial studies were carried out using N-acetyl-protected propanolamine (2a) and butanolamine (2b) as representative suitable three-carbon and four-carbon nitrogen-containing electrophiles. Pleasingly, complete indole consumption and good yields of homotryptamine (3a) and dihomotryptamine (3b) were observed after 48 h at 150°C when powdered Cs2CO3 was used as the base and [Cp*IrCl2]2 was used as the
catalyst (Table 1, entries 1 and 2). We succeeded in carrying out the alkylation reaction even with longer carbon chain N-acetylated primary amino alcohols; for example, using (2c), (3c) has isolated in modest yield (Table 1, entry 3).
Entry N-Acetylamino Alcohol Product Yield (%)b
1 54
2 76
3 57
aReactions were carried out in a sealed vial at 150 °C for 48 h with indole (1 equiv.),
41
Table 1 Synthesis of N-Actyltryptamine Homologuesa.
Lower conversions and increased byproduct formation were generally observed when solvents such as toulene, tert-amylalcohol, and trifluoroethanol were employed. Apart from tryptamine analogues and homologues with linear side chains, derivatives with branched side chains, both in the - and -positions, are becoming increasingly studied in medicinal chemistry due to their ability to discriminate between the serotonin/melatonin receptor family subtypes. Recently, the first examples of branched tryptamines possessing pharmacologically interesting properties have been developed.66 Thus, we then turned our attention to more sophisticated substituted N-acetylethanol-amines, such as the homochiral primary N-acetyl-L-alaninol (4a) and N-acetyl-L-serine (4b), knowing their sensitivity toward racemization, once oxidised to the transient aldehyde. Interestingly, the methyl-substituted amino alcohol derivative (4a) performed poorly in this reaction, possibly due to the increased steric hindrance around the nitrogen atom that precludes effective ligand dissociation from the iridium centre. Notably, we did not observe any products that could arise from the potentially competitive four- membered ring cycloiridation pathway. Strong electron- withdrawing groups adjacent to the amine motif also failed to produce the desired product tryptophans in acceptable yields. In addition, the small amounts of products
(5a) and (5b) obtained were racemic, as expected (Table 2, entries 1 and 2). A
different outcome was obtained when we examined the applicability of the borrowing hydrogen reaction of indoles with racemic secondary N-acetyl ethanolamines, which proceeds via generally less electrophilic ketones (Table 2, entries 3 and 4). Therefore, we elected to examine the behaviour of indole in a borrowing hydrogen alkylation with N-acetyl-2-propanolamine (4c) and 2-acetamido-1-phenylethanol (4d). We found that iridium- catalyzed indole alkylation with branched alcohol (4c) afforded a superior yield to the parent linear congeners, and we ascribe this reactivity to the relatively lower energetic demand of secondary alcohol dehydrogenation compared to primary alcohols, as well as the faster elimination of water from the adduct-formed alcohol- containing products of ketone addition. Unsatisfactory results were obtained when chiral Ir complexes and/or chiral ligands were employed to attempt an enantioselective version of this approach, although good yields were confirmed.67 On
the contrary, the secondary alcohol (4d) gave a complex mixture of unidentified compounds, containing only trace quantities of a compound identified as the desired protected tryptamine by LC/MS. This latter result was quite surprising because secondary benzyl alcohols are known to be good substrates in redox-neutral alkylations, reflecting their more favourable oxidation relative to higher alcohols68.
Therefore, we suspected that the initial alcohol oxidation step had occurred but that alternative pathways (aldol-type reactions) prevented condensation (the resulting
42
conjugated ketone is notably less electrophilic) and the return of the hydrogen back to the desired indolenium intermediate, thus stalling the reaction.
Entry Amino alcohol Product Yield (%)b
1 37
2 15
3 78
43
5 72
aReactions were carried out in a sealed vial at 150 °C for 48 h with indole (1 equiv.), amino alcohol (3 equiv.),
[Cp*IrCl2]2 (2.5 mol%), and Cs2CO3 (1.1 equiv.). bIsolated yield.
Table 2 Synthesis of α- and β-branched tryptamines and branched homotryptamines from indole
and amino alcohols.a
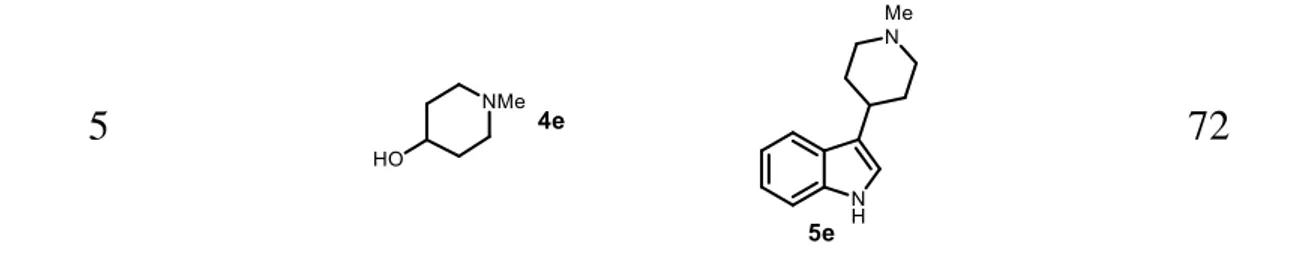
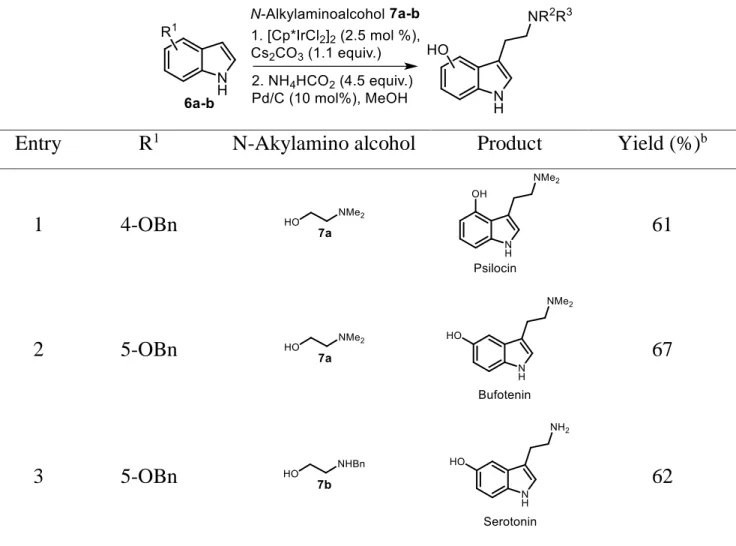
To confirm this hypothesis, we tested the viability of N-methyl-4-piperidinol (4e) as a secondary alcoholic substrate for hydrogen borrowing alkylation with indole, which allowed us to extend the methodology to branched homotryptamines (Table 2, entry 5). The reaction of indole with piperidine alcohol (4) under the same reaction conditions gave a good yield of 3-(N-methylpiperidyl) indole (5e), which is an important structural element of clinical candidates such as the antimigraine compound LY334370 and naratriptan (Fig. 1).69 Since most indole-based central nervous system drugs used in the treatment of migraines and cluster headaches, as well as some psychoactive natural product tryptamines, are tryptamine-based dimethylated amines,70 we used the commercially available and highly interesting amino alcohol N,N-dimethylethanolamine (7a) to further demonstrate the application of this methodology. We adopted this chemistry for the total syntheses of psilocin and bufotenin in a straightforward manner (Table 3). Thus, the reaction of known 4-benzyloxyindole (6a) and 5-4-benzyloxyindole (6b) with N,N-dimethylethanolamine
(7a) provided the desired products, which were debenzylated under hydrogenolysis
conditions to afford psilocin and bufotenin in 61% and 67% overall yields, respectively. The salient features of this method include the single step preparation of the tryptamine core through displacement of the alcohol without the need for prior activation and without resorting to a high dilution or the use of a protected amine or final reductive alkylation for the tertiary amine. Among the biologically important hydroxytryptamines, serotonin is perhaps the most important and best known as a neurotransmitter that modulates neural activity and a wide range of neuropsychological processes.71 Despite the fact that several syntheses and
biosynthesis of serotonin have been described,72 none seem practical or economical; thus, serotonin is still quite expensive on the market. Using our procedure of treating 5-benzyloxyindole with N-benzylethanolamine under Ir-catalyzed borrowing hydrogen conditions, followed by double debenzylation by catalytic reduction with palladium on carbon as the catalyst, serotonin was obtained in good overall yield.
44
Entry R1 N-Akylamino alcohol Product Yield (%)b
1 4-OBn 61
2 5-OBn 67
3 5-OBn 62
aReactions were carried out in a sealed vial at 150 °C for 48 h with indole (1 equiv.), N-alkylamino
alcohol (3 equiv.), [Cp*IrCl2]2 (2.5 mol%), and Cs2CO3 (1.1 equiv.), followed by hydrogenolysis with
NH4HCO2 (4.5 equiv.) and Pd/C (10%) in MeOH at 70 °C for 45 min. bIsolated overall yield.
Table 3 Syntheses of psilocin, bufotenin, and serotonin from N-alkylated
45
3.2 Divergent Borrowing Hydrogen Approach from Oxindoles and Ethanolamine: Synthesis of C3-Substituted Oxindoles and -Lactams.
Herein, we describe a convenient iridium-catalyzed process for the alkylation of 2-oxindoles with ethanolamines. The studies began with the investigation of the direct C3 catalytic alkylation of oxindole (1) with two aminoles (2) and (3). As catalytic system we used the commercially available trivalent iridium complex [Cp*IrCl2]2 in
the conditions previously developed in our laboratories.37 Essentially, we found that when the N-acetyl ethanolamine (2) reacts with oxindole (1) a direct alkylation of C-3 occurs giving the oxindole ethyl acetamide (4) in 61% yield. Unexpected result was obtained with aminol (3). Instead, to obtain the simply alkylated oxindoles in C-3, -substituted -lactam (5) was formed in 54% yield (Scheme 1).
Scheme 1 Divergent Reaction of Oxindoles with Ethanolamines Via Borrowing Hydrogen.
Therefore, we speculate on a possible pathway for the formation of -substituted -lactam. Sequential/domino process consisting of alkylation and intramolecular secondary amide transamidation process could take place as shown in (Figure 1).
Figure 1 Proposed Opening Ring Mechanism.
We can justify the different behaviour of ethanolamines considering the different availability of the lone pair on the ethanolamines nitrogen atom. In the case of the ethanolamine (2), the lone pair is delocalized due to the resonance with the acetyl
46
group resulting not enough nucleophilic to give the intermolecular addition/elimination. Instead, ethanolamine (3) have a strong nucleophilic nitrogen and could give the intramolecular addition/elimination on the carbonyl of oxindole
(1). After this finding, we have extended the scope of this divergent reaction studying
different substituted oxindoles. First, we investigated the reactivity of N-acetyl ethanolamine (2), with different substituted 2-oxindoles (Table 1). Through the treatment of 2-oxindoles (6), (8), (10) end (12) with N-acetylethanolamine (2) under the above-mentioned conditions, C3-amidoethyl oxindoles (7), (9), (11) and (13) were selectively achieved in 54–88% yield (Table 1, entries 1–4).73 High yields were also achieved using N-methyl and N-phenyloxindoles (14) and (16) (Table 1, entries 5–6). Given the enormous effort that has already been dedicated to the catalytic asymmetric construction of oxindoles bearing a C3-quaternary stereocenter from simple C3-monoalkylated oxindoles,74 which are then often converted into 2-oxindole
containing a 2-aminoethyl side chain (and subsequently cyclized under reductive conditions to generate the tricyclic pyrrolidinoindoline ring system), the C3-amidoethylated 2-oxindoles (7), (9), (11), (13), (15), and (17) can be considered as advanced starting materials for the more direct asymmetric synthesis of enantio-enriched 3,3-disubstituted oxindoles and some related natural products/drug candidates. Furthermore in this series, compound (4) can be readily converted to the natural product (±)-N-[2-(3-hydroxy-2-oxo-2,3-dihydro-1H-indol-3-yl)ethyl]acetamide, which is an antiproliferative compound from Selaginella
pulvinata, using a previously developed cobalt-catalyzed peroxidation reaction with
hydroperoxide and a hydrogenation reaction,75 whereas 2-oxo-2,3-dihydromelatonin (9), an oxidized analog of the neurohormone melatonin, is a valuable precursor for 2-substituted melatonin.76
47 1 57 2 54 3 70 4 88 5 99 6 54
aReactions were carried out in a sealed vial at 150 °C for 24 h with oxindole (1 equiv.),
N-Ac ethanolamine (3 equiv.), [Cp*IrCl2]2 (2.5 mol%), and Cs2CO3 (1.1) equivalent bIsolated yield.
Table 1 Scope for the Direct C3-Alkylation of Substituted Oxindoles
respect to Acetyl ethanolaminea.
Then we investigated the reactivity of N-benzylethanolamine (3) with different substituted oxindoles (Table 2). In particular, this method was compatible with modification of the benzyl moiety of the 2-oxindole core, and substitution (e.g., Cl, OMe, and Me) was readily tolerated on the aromatic ring (Table 2, entries 1–6). Finally, we studied the domino alkylation/annulation of N-substituted 2-oxindoles such as N-methyl and N-phenyl oxindole (14, 16) with N-benzylethanolamine (3), and a decent yield was achieved with substrate 16 to afford ortho-substituted
48
diarylaniline 28 (Table 2, entry 8). Importantly, the diarylamine products of the type generated from the reactions described herein are highly valued motifs with important structural, electronic, and mechanical properties suitable for therapeutic applications. In addition, the amide of the γ-lactams can be cleaved to provide access to a variety of 2-(2-phenylamino)phenyl)ethanoic acid nonsteroidal anti-inflammatory agent derivatives, such as Lumiracoxib and Diclofenac.59
Entry Substrate Product Yield (%)b
1 37 2 42 3 35 4 48 5 46 6 72 7 35
49
8 68
aReactions were carried out in a sealed vial at 150 °C for 24 h with indole (1 equiv.),
N-Benzyl ethanolamine (3 equiv.), [Cp*IrCl2]2 (2.5 mol%), and Cs2CO3 (1.1) equivalent bIsolated yield.
Table 2 Scope for the Direct C3-Alkylation of Substituted Oxindoles
respect to Benzyl ethanolaminea.
Since the importance of pharmaceutical and biological activity of the diaryl amines, obtained with the strategy described above, where oxindoles are N-substituted with aril group, we extended and explore more structure with different substitution on aryl and heteroaryl group (Table 3).
Entry Substrate Product Yield (%)b
1 69
50
3 60
4 49
5 70
aReactions were carried out in a sealed vial at 150 °C for 24 h with indole (1 equiv.),
N-Acetyl ethanolamine (3 equiv.), [Cp*IrCl2]2 (2.5 mol%), and Cs2CO3 (1.1) equivalent bIsolated yield.
Table 3 Scope for the Direct C3-Alkylation of N-Aryl Substituted
Oxindoles respect to Benzyl ethanolaminea.
.
Transformations of N-aryloxindoles bearing a halogen atom, such as fluorine (29), afforded the corresponding product 30 in 69% yield (Table 3, entry 1). For N-aryloxindoles bearing an even stronger electron-withdrawing substituent, such as pyridil (36, 38), the reaction proceeded smoothly to afford the desired products 37, 39 in 49% and 70% yield, respectively (Table 3, entries 4, 5). Furthermore, when the alkylation/annulation was applied to N-aryloxindoles bearing an electron-donating substituent, such as a methoxy or dimethylamine group (31, 33), the corresponding products 32, 35 were afforded in 64% and 60% yield, respectively (Table 3, entries 2, 3).
51
3.3 Iron-Catalyzed Direct C3-Benzylaion of Indoles with Benzyl Alcohols through Borrowing Hydrogen
Inspired by the recent studies to use the metals of first row as catalysts, we report herein a green, economical, and efficient iron-catalyzed C3-selective alkylation of indole. This reaction employs iron(II) phthalocyanine (Fe(II)Pc), an inexpensive commercially available compound that is typically used as an industrial additive for ink as well as photonic and optical material manufacturing. Of note, to date, Fe(II)Pc complexes have not been applied as catalysts for the formation of new carbon−carbon bonds via the borrowing hydrogen process.77 The reaction of unsubstituted indole (1a) with benzyl alcohol (2a) was selected as the model reaction to establish the best
reaction conditions. Initially, the effect of various iron based catalysts was investigated (Table 1).
Entrya Catalyst Yield of 3b (%)
1 - - 2 FeSO4 c - 3 FeCl2d - 4 Fe(acac)3 - 5 Fe(II)Pc 99 6 Fe(II)Pc e 8 7 Fe(II)Pc f Trace 8 Fe-Knölker - 9 Fe-Knölker with PPh3 - 10 Fe-Knölker with Me3NO -
11 Fe(II)Pc in the dark 97
52
aReaction conditions: indole (0.5 mmol), benzyl alcohol (1 mmol), Cs
2CO3 (0.55 mmol), catalyst (1 mol%) at 140 °C
for 16 h. bIsolated yield. cOnly 4 was isolated in 13% yield. dOnly 4 was isolated in 21% yield eCs
2CO3 (0.05 mmol). fWithout base.
Table 1 Optimization of the Reaction Conditions for C3-Alkylation of Indole with Benzyl Alcohol.
The reaction did not proceed without catalyst in the presence of Cs2CO3, which
excluded the contribution of the base itself as a catalyst (Table 1, entry 1).78 The highest activity was observed with Fe(II)Pc and a stoichiometric amount of Cs2CO3
(Table 1, entries 5−7). Other iron salts were found to be ineffective and, not surprisingly, led to the formation of bis(indolyl)methane (4) in poor yields (Table 1, entries 2−4).79 Due to its inherent redox properties, the iron(0)tricarbonyl complex
such as the Knölker-type catalyst is widely known to activate inert substrates via dehydrogenation/hydrogenation reactions and has been reported previously for C−C bond formation.80 Neither alone (Table 1, entry 8) nor in the presence of 10 mol %
Me3NO oxidant (to form active catalyst) and PPh3 ligand did the alkylated product
form (Table 1, entries 9 and 10). This solvent-free reaction with Fe(II)Pc as the catalyst was excellent in terms of both yield and selectivity as compared with the corresponding reaction in toluene (1.0 M, 54% yield; 0.5 M, 10% yield) or tert-amyl alcohol (at reflux under air atmosphere for 16 h afforded the desired alkylated product 3a in 31% yield). The desired coupling product was also obtained in the absence of light as well as in the presence of radical scavengers such butylated hydroxytoluene (BHT), thus discarding the involvement of radical species in the reaction pathway, more precisely using TEMPO as a radical scavenger, only oxidative degradation of indole was observed. (Table 1, entries 11 and 12). Once the optimal reaction conditions were achieved, we examined the scope of the alkylation with respect to alcohols catalyzed by Fe(II)Pc, and these results are outlined in (Table2).
53
a Reaction conditions: indole (0.5 mmol), alcohol (1 mmol), Cs
2CO3 (0.55 mmol), Fe(II)Pc (0.055 mmol) at 140 °C for
16 h. bIndole (1mmol), alcohol (0.5 mmol). c1H-indole (0.5 mmol), 6-fluoro-1H-indole (0.5 mm0l),
2-(hydroxymethyl)phenol (0.5 mmol). d 140 °C for 36h.