Sindromi
mielodisplastiche
prof. Gian Matteo Rigolin
WHO classification of myeloid neoplasms and
acute leukemia
1. Myeloproliferative neoplasms (MPN)
2. Myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA,
PDGFRB, or FGFR1, or with PCM1-JAK2
3. Myelodysplastic/myeloproliferative neoplasms (MDS/MPN)
4. Myelodysplastic syndromes (MDS)
5. Acute myeloid leukemia (AML) and related neoplasms
6. Blastic plasmacytoid dendritic cell neoplasm
7. Acute leukemias of ambiguous lineage
8. B-lymphoblastic leukemia/lymphoma
9. T-lymphoblastic leukemia/lymphoma
MDS: definizione
Gruppo eterogeneo di disordini clonali della cellula staminale,
contrassegnati da citopenia periferica e nella maggior parte dei
casi da un midollo ipercellulato con evidenti alterazioni
maturative (displasia).
Le MDS presentano un aumentato rischio di evoluzione in LAM.
MDS incidenza
Zeidan et al. Blood Reviews 2018
Incidence of MDS or AML in the
USA (Surveillance, Epidemiology,
and End Results data, based on
the Nov 2017).
MDS: incidenza
Zeidan et al. Blood Reviews 2018
Incidence of MDS or AML in the
USA (Surveillance, Epidemiology,
and End Results data, based on
the November 2017 submission).
Sopravvivenza a 5 anni
Five-year overall survival of cancer patients in the United States (Surveillance, Epidemiology, and End Results data, based on the November 2017 submission).
Eziologia
• La causa delle MDS è di solito sconosciuta.
• In alcuni casi una MDS può svilupparsi dopo
l’esposizione a radiazioni, ad alcuni tossici
ambientali quali il benzene o dopo trattamenti
con alchilanti o inibitori delle topoisomerasi II per
una precedente neoplasia
MDS
• The cause is known in only 15% of cases
• Inherited predisposition
– is evident in a third of paediatric cases, including in children with Down’s syndrome, Fanconi’s anaemia, and neurofibromatosis. – In adults, inherited predisposition is less common but should be
investigated in young adults or in families with other cases of MDS, AML, or AA.
• Environmental factors include previous use of
chemotherapy,especially of alkylating agents and
purine analogues,radiotherapy,and tobacco smoking.
• Recognised occupational factors include exposure to
benzene and its derivatives,
7and an excess of cases is
reported in agricultural and industrial workers.
Pathogenesis of
MDS
Citogenetica
• Anomalie citogenetiche sono
riscontrabili del 40-70% delle
MDS de novo e nel 95% delle
forme secondarie a
chemioterapia (therapy-related)
•
Ogawa S. Blood. 2019 Mar 7;133(10):1049-1059.
Common driver
alterations in MDS and
other myeloid
Ogawa S. Blood. 2019 Mar 7;133(10):1049-1059.
Common driver
alterations in MDS and
other myeloid
Ogawa S. Blood. 2019 Mar 7;133(10):1049-1059.
Major driver genes
in MDS
Nucleosomes consist of DNA (black line) wrapped around histone octomers (purple).
Post-translational modification of histone tails by methylation (Me), phosphorylation (P) or acetylation (Ac) can alter the higher-order nucleosome structure.
Nucleosome structure can be regulated by ATP-dependent chromatin remodellers (yellow cylinders), and the opposing actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs). Methyl-binding proteins, such as the methyl-CpG-binding protein (MECP2), target methylated DNA (yellow) and recruit HDACs.
a. DNA methylation and histone deacetylation induce a closed-chromatin configuration and transcriptional repression. b. DNA demethylation and histone acetylation relaxes chromatin, and allows transcriptional activation.
Clonal hematopoiesis (CH)
• WGS of 11.262 Icelanders reveals that CH
is very common in the elderly.
• Somatic mutation of some genes is
strongly associated with CH, but in most
cases, no driver mutations were evident
(12.6%, 177/1403).
• CH is associated with increased mortality
rates, risk for hematological malignancy,
smoking behavior, telomere length,
Y-chromosome loss, and other phenotypic
characteristics.
Clonal Hematopoiesis and Blood-Cancer Risk
Inferred from Blood DNA Sequence
• whole-exome sequencing of DNA in PB cells from 12,380
persons, unselected for cancer or hematologic
phenotypes from Swedish national patient registers.
• Clonal hematopoiesis with somatic mutations was
observed in 10% of persons older than 65 years of age
but in only 1% of those younger than 50 years of age.
• Clonal hematopoiesis was a strong risk factor for
subsequent hematologic cancer (HR, 12.9; 95%
confidence interval, 5.8 to 28.7).
G eno ve se e t al , N Eng l J Med 20 14 ;3 71 :2 47 7 -87 .
Clonal hematopoiesis of undetermined potetial (CHIP)
Steensma et al, Blood 2015;126:9
ICUS: idiopatic cytopenia of undetermined significance CCUS: clonal cytopenia of undetermined signficance
Clonal evolution of CHIP
• Investigators have focused on
various MDS- and
AML-associated phenomena that may
aid and abet clonal evolution.
• The primary driver of
progression of CHIP to overt
neoplasia has been assumed to
be acquisition of new mutation in
a clonal cell with self-renewal
properties.
David P. Steensma, ASH 2018
CHIP as a precursor state for hematological neoplasms.
Molecular abnormalities in del(5q) MDS
Decreasing RPS14 expression enhances other ribosomal proteins (RPs) such as RPL11 or RPS19, which sequester MDM2, an E3 ubiquitin ligase that also negatively regulates TP53.
Elevated TP53 activation leads to enhanced TP53-dependent apoptosis of erythroid progenitors.
Molecular pathogenesis
of 5q-syndrome
CK1α: casein kinase 1α,
Molecular pathogenesis
of 5q-syndrome
LEN decreases expression of CK1α.
Normal cells treated with LEN can survive because 50% of CK1α expression remains.
In del(5q) cells, CK1α is at 50% due to haploinsufficiency, so LEN treatment is lethal to
cells by completely losing CK1α.
If del(5q) MDS cells acquire TP53 loss-of-function mutations, they would become
resistant to LEN because complete loss of CK1α could not induce TP53-dependent
Mohamedali and Mufti, BJH 2008;144:157
Ribosomal biogenesis and BM falilure syndromes
Valutazione morfologica sangue periferico e midollare
• Valutazione morfologica sangue periferico per orientamento
diagnostico
– diagnosi differenziale, segni di displasia, presenza di blasti
• Valutazione morfologica midollare:
– Riscontro di segni di displasia
– La valutazione morfologica dei blasti
• La % di blasti valutata su almeno 500 cellule (almeno 100 cellule non eritroidi)
• Non raccomandata la valutazione citofluorimetrica
Caratteristiche morfologiche di displasia
filiera Nucleare Citoplasmatica eritroide Multinuclearità, carioressi,
mitosi anomale, megalobastosi
Vacuoli, difetti di emoglobinizzazione, sideroblasti ad anello
granulocitaria Forme Pseudo-Pelger,
ipersegmentazione, nuclei ad anello, forme giganti, clumping cromatinico, granulociti binucleati
Ipogranulaione, corpi di Dohle, vacuolizzazioni, difetti di mieloperossidasi
megacariocitaria Micromegacariociti, forme mononucleate,
megacariociti con nuclei dispersi
Asincronia nucleo/citoplasmatica, piastrine giganti, piastrine ipogranulate o granulate
monocitaria Ipersegmentazione, nuclei con forme bizzarre
Aumentata basofilia citoplasmatica, granulazioni prominenti
Displasia eritroide
Displasia eritroide
Displasia eritroide
Erythroid karyorrhexis in myelodysplasiadisgranulopoiesi
Blasti e pseudo Pelger
Corpi di Dohle
Displasia megacariocitaria
Sideroblasti
Haematologica 2008; 93:1712-1717.
Il Working Group ha definito 3 tipi di sideroblasti:
– Tipo 1: meno di 5 granuli di ferro nel citoplasma;
– Tipo 2: 5 o più granuli di ferro, ma non in una distribuzione perinucleare;
– Tipo 3 o sideroblasti ad anello: 5 o più granuli in posizione perinucleare, che circondano il nucleo o interessano almeno un terzo della
circonferenza nucleare.
Nel conteggio dei sideroblasti ad anello, occorre valutare almeno 100 precursori eritroidi nei vari stadi maturativi. La percentuale di sideroblasti ad anello ai fini della
classificazione rimane il 15% come per la classificazione FAB e WHO.
blasti e promielociti nelle MDS
Aspetti
cellulari non granulato Blasto granulato Blasto Promielocito normale Promielocito displastico Nucleo Centrale di forma variabile Centrale di forma variabile Ovale, rotondo, indentato Centrale od eccentrico Ovale, rotondo, indentato in posizione eccentrica
Cromatina fine fine Fine od intermedia Fine o grossolana
Nucleolo 1-2 1.2 Ben riconoscibile Ben visibile
Zona Golgi Non evidente Non evidente Ben visibile Presente ma poco sviluppata Granuli Non visibili Presenti (talora
corpi di Auer)
Azzurrofili
uniformemente dispersi
irregolare presenza e distribuzione Citoplasmaa basofilo basofilo basofilo Basofilia ridotta ed
irregolare
Criteri diagnostici minimi nelle MDS
A. Prerequisiti
1. Citopenia costante in una o più delle seguenti filiere: Hb <11 g/dL, ANC < 1500 uL o PLT <100,000 uL 2. Esclusione di tutti gli altri disordini come causa della citopenia/displasia
B. Criteri decisivi correlati alla MDS
1. Displasia in almeno il 10% di tutte le cellule o >15% di sideroblasti ad anello 2. 5–19% di cellule blastiche nello striscio midollare
3. Anomalie cromosomiche tipiche (citogenetica o FISH)
C. Co-criteri (per i pazienti che soddisfano i criteri A ma non quelli B) 1. Anomalo fenotipo mediante citometria a flusso
2. Anomalie molecolari (gene chip profiling, o mutazioni puntiformi (RAS, etc) 3. Anomalie colturali dei progenitori midollari e/o circolanti (CFU-assay)
• La diagnosi di MDS può essere formulata quando entrambi i prerequisiti ed almeno un criterio
decisivo sono soddisfatti.
• Se nessun criterio decisivo è soddisfatto, ma è molto probabile che il paziente sia affetto da una neopasia mieloide clonale, i co-criteri devono essere applicati e possono aiutare nel raggiungimento della diagnosi di MDS o di una condizione definita ‘fortemente sospetta di MDS’.
Citopenia idiopatica di incerto (indeterminato) significato (ICUS)
Definizione
Citopenia in una o più delle seguenti filiere (per più di 6 mesi):
Hb < 11 g/dL; neutrofili <1500 uL; piastrine <100,000 uL
Esclusa una MDS
Escluse tutte le altre possibili cause di citopenia Indagini iniziali richieste per la diagnosi di ICUS
Anamnesi dettagliata (farmaci, tossici, mutageni, etc.)
Attento esame clinico comprendente indagini radiologiche ed ecografia splenica
Emocromo con conteggio differenziale al microscopio e completa valutazione biochimica clinica Biopsia osteomidollare ed immunistochimica
Aspirato midollare e colorazione per il ferro. Citometria a flusso midollare e sangue periferico
Analisi cromosomica con FISH (pannello standard minimo: 5q31, CEP7, 7q31, CEP8, 20q,CEPY, p53) Analisi molecolare se appropriato
Esclusione di infezioni virali (HCV, HIV, CMV, EBV, altre) Indagini raccomandate nel follow-up
Emocromo con formula e biochimica clinica ad intervalli di 1–6 mesi In caso di evidente sospetto di MDS: esame midollare
Steensma et al, Blood 2015;126:9
CHIP: Clonal hematopoiesis of undetermined potetial ICUS: idiopatic cytopenia of undetermined significance CCUS: clonal cytopenia of undetermined signficance
Citogenetica
• La citogenetica ha un ruolo decisivo
nella diagnosi e nella definizione
della prognosi
• Anomalie citogenetiche sono
riscontrabili del 40-70% delle MDS
de novo e nel 95% delle forme
secondarie a chemioterapia
(therapy-related)
Anomalie cromosomiche e MDS
Recurrent cytogenetic abnormalities in MDS
Citogenetica e sopravvivenza
Karyotype
•Good: normal, -Y, del(5q), del(20q) •Intermediate: other abnormalities
2016 WHO classification of myeloid neoplasms and
acute leukemia
Evolution of MDS classification systems
RA
RARS
RAEB-1
RAEB 2
Sindrome da
5q-• Presentazione clinica
– Età avanzata
– Sesso femminile (F:M 7:3)
– Basso rischio di progressione in LAM – Buona prognosi
• Quadro ematologico
– Anemia macrocitica – Modesta leucopenia
– Normale/elevato numero di piastrine – ipoplasia eritroide midollare
– Megacariociti monolobati
– Delezione intestiziale braccio lungo del cromosoma 5 come singola anomalia – Blasti < 5%
MDS: clinical findings
• Clinical features are non-specific and mainly result from cytopenias.
• Anaemia, is symptomatic in many pts, leading to fatigue, poor quality of life, and destabilisation of underlying cardiovascular disease.
• Thrombocytopenia is commonly associated with platelet dysfunction, potentially leading to bleeding symptoms even in moderate thrombocytopenia.
• Infections (especially with gram-neg bacilli, gram-pos cocci, and fungi) can occur with only moderate neutropenia due to neutrophil function defects.
• Many patients have immune disorders, including relapsing polychondritis, vasculitis, and seronegative polyarthritis.
– The two disorders tend to be diagnosed almost simultaneously, which suggests a pathophysiological relation.
MDS: Differential diagnosis
• All other causes of cytopenias must be carefully excluded; – vitamin deficiencies – autoimmune disease, – Liver disease, – hypersplenism, – viral infections, – drug intake,
– exposure to environmental toxins, – aplastic anaemia,
– Acute leukemias
– Large granular lymphocytic leukemia – Hairy cell leukemia
– Myelofibrosis
– Paroxysmal nocturnal haemoglobinuria, – bone-marrow infiltration by malignancy,
therapy related MDS
• Rischio attuariale 0.25-1% per anno da 2 a 5-7 anni dalla fine della chemioterapia
• Rischio dose dipendente e aumenta esponenzialmente dopo i 40 anni
Survival MDS by subtype in the USA
(Surveillance, Epidemiology, and End Results data, based on the November 2017 submission).
Prognostic factors in MDS
International prognostic Scoring System
Risk stratification, scores, and median OS by each model.
Aziz Nazha, ASH 2018
• Current models can overestimate or underestimate the OS for some MDS pts. •
• Recognizing this limitation of current models is very important as the choice of therapy
and disease expectations are highly dependent on prognosis, and identifying the actual risk in these patients could alter their treatment recommendations
Comorbidities in MDS
MDS comorbidity score
terapia
• Trapianto di cellule staminali (nei pazienti fit e “giovani”)
– Allogenico
• Terapia di supporto
– Trasfusioni,
– Fattori di crescita: Epo, G-CSF
– antibiotici,
– etc
• Chemioterapia
• Terapia immunosoppressiva: ciclosporina, globulina antilinfocitaria (cariotipo
normale / trisoma 8)
• Farmaci immunomodulanti: lenalidomide (del5q)
• Agenti ipometilanti: 5 azacitidina, decitabina
Platzbecker U. Blood. 2019;133(10):1096-1107)
Priorities of
interventions in MDS
according to stage
Platzbecker U. Blood. 2019;133(10):1096-1107)
Historical time scale of registration of therapeutic
agents forMDS in the EU and United States.
P la tzb e c k e r U. Blood. 2 0 1 9 ;1 3 3 (1 0 ): 1 0 9 6 -1 1 0 7 )
Therapeutic
algorithm in
LR-MDS.
G-CSF, granulocyte colony-stimulating factor;ATG, antithymocyte globulin; CSA, cyclosporine;
HMA, hypomethylating agent; LEN, lenalidomide;
LUSP, luspatercept; sEPO, serum EPO;
TPO-RA, thrombopoietin receptor agonist.
*Not presently approved.
P la tzb e c k e r U. Blood. 2 0 1 9 ;1 3 3 (1 0 ): 1 0 9 6 -1 1 0 7 )
Therapeutic
algorithm in
HR-MDS.
CTx, chemotherapy;IC, induction chemotherapy; BSC, best supportive care; TKI; tyrosine kinase inhibitor. *These could be IDH or
FLT3-inhibitors (not presently approved). †Consider posttransplant disease surveillance strategies
Platzbecker U. Blood. 2019;133(10):1096-1107)
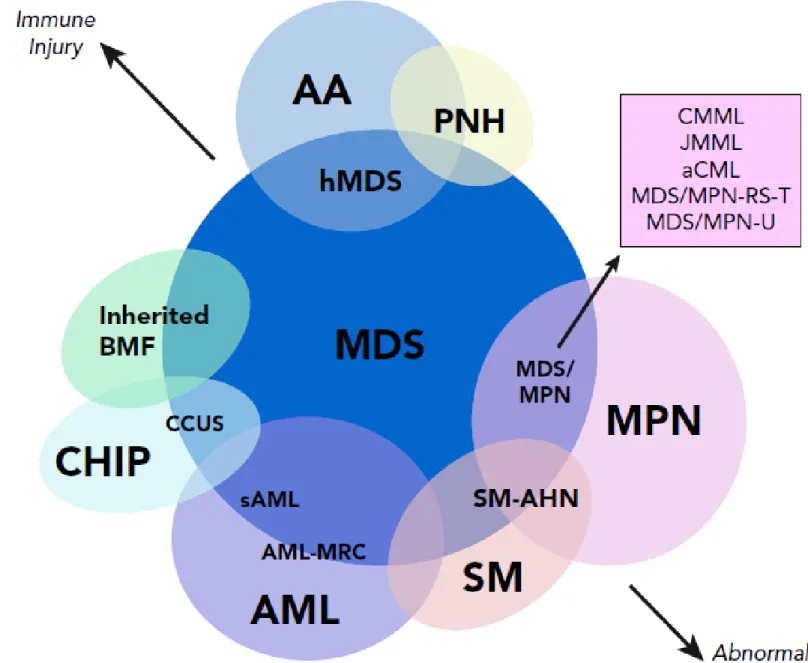
Diagram depicting myeloid
disorders with clinical and
genetic features shared
with MDS and the degree
to which they are driven by
proliferative and
immunologic mechanisms.
Diagnostic criteria for MDS/MPN with ring sideroblasts and thrombocytosis
• Anemia associated with erythroid lineage dysplasia with or without
multilineage dysplasia≥15% ring sideroblasts*, <1% blasts in PB and <5% blasts in the BM
• Persistent thrombocytosis with platelets ≥450 x 109/L
• Presence of a SF3B1 mutation or, in the absence of SF3B1 mutation,
no history of recent cytotoxic or growth factor therapy that could explain the myelodysplastic/myeloproliferative features†
• No BCR-ABL1 fusion gene, no rearrangement of PDGFRA, PDGFRB, or
FGFR1; or PCM1-JAK2; no (3;3)(q21;q26), inv(3)(q21q26) or del(5q)‡
• No preceding history of MPN, MDS (except MDS-RS), or other type of
MDS/MPN Schmi tt -G ra ef f A H, Ha e m at ol ogic a. 20 08 ; 93 :34
*At least 15% ring sideroblasts required even if SF3B1 mutation is detected. †A diagnosis of MDS/MPN-RS-T is strongly supported by the presence of SF3B1 mutation together with a mutation in JAK2 V617F, CALR, or MPL genes.
‡In a case which otherwise fulfills the diagnostic criteria for MDS with isolated del (5q)-no or minimal absolute basophilia; basophils usually ,2% of leukocytes.
CMML
T an aka T N , B lo o d . 2019; 133(10) :108 6 -109 5