The contents of this report is strictly confidential, as
arguments protected by law as confidential. Therefore all those
who take knowledge are required, even sanctioned
fees to Articles 325 and 623 of the Criminal Code, and not to disclose
not use the information gained
.Some chapters of this thesis referred to as "omitted" will still be part of the
final academic defence and will be presented.
(These terms are in accordance with regulations of the University of Pisa in
relation to the protection of intellectual property patents.)
Chapter 1. Metzincines
1.1. Introduction.
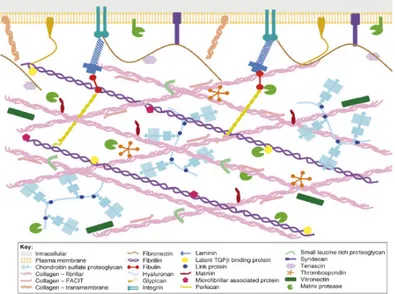
The extracellular matrix (ECM) is a complex dynamic network of secreted macromolecules and proteolytic enzymes. The ECM plays a vital role in the structure and development of tissues; it provides mechanical strength, it acts as a template for cell growth and it influences cell behaviour, such as migration, proliferation, differentiation and adhesion. The ECM is composed by two basic molecular types (Fig. 1.1). Structural molecules, like collagens and laminins, form a basic integral network, connecting the matrix to the surrounding cells. Glycoproteins endow additional properties to the ECM, for example integrins promote cell-signalling, while elastin and fibronectine-like molecules provide flexibility. Proteoglycans, through their high water-binding capacity provides turgor and stabilize the matrix; in addition they form a collagen network which is fundamental to support compressive forces and to store cytokines and grow factors. The dynamism of the extracellular matrix is pivotal to provide suitable environments for growth, development and tissue remodelling, while disruption of the ECM balance is frequently related to many human diseases. This make some ECM molecules attractive druggable targets.
Figure 1.1. Diagramatic representation of the extracellular matrix1
Various types of proteinases are implicated in ECM remodelling and many of them are zinc-based metalloproteinases, belonging to the so called “Metzincins superfamily”2. This superfamily of endopeptidases includes Matrix Metallo Proteinases (MMPs),
snapalysins, leishmanolysins and pappalysins3. Metzincins catalytic domains share a zinc ion and a zinc binding consensus motif and a methionine containing a Met-turn. Metzincins are able to cleave proteins of the extracellular matrix participating both to unspecific protein degradation and to specific cleavage events, as enzyme activation. It’s evident that the deregulation of their degrading potential could lead to various serious pathologies.
1.2. Matrix Metalloproteinases (MMPs).
Matrix Metalloproteinases (MMPs) are secreted or membrane-bound proteinases. Their vertebrate collagenolytic activity was firstly discovered 47 years ago by Gross and Lapiere in tadpole tissues4. MMPs are found mainly in higher mammals, but related sequences have been found also in fish, amphibians, insects, plants, prokaryotes and viruses. Due to their attitude of degrading ECM elements, MMPs are also called
matrixins. Human genome has 24 matrixin genes which encode for 23 enzymes.
Through turnover of extracellular matrix proteins, MMPs are involved in tissue resorption, remodelling, repair, as observed during embryogenesis and development, branching, and organ morphogenesis, and angiogenesis. However, their potent proteolytic potential or its absence may also lead to pathologies, such as inflammation, ulcers, rheumatoid arthritis and osteoarthritis, periodontitis, heart failure and cardiovascular diseases, fibrosis, emphysema, cancer, metastasis. MMPs altered activity has been also investigated in many neurodegenerative diseases, such as Alzheimer disease and multiple sclerosis.
Matrix metalloproteinases are multidomain zinc endopeptidases. Basing on their domain organization and substrate preference, they have been grouped into
collagenases, gelatinases, stromelysins, matrilysins, membrane-type (MT)-MMPs and
other more recently discovered (Table 1.1).
Following phylogenetic relationship of the human MMP protease domain sequences, six evolutionary sub-groups have been proposed: sub-group A (MMP-19, -26 and -28); sub-group B (MMPs -11, -21 and -23); sub-group C, (MMP-17 and -25); sub-group D, (MMP-1, -3, -8, -10, -12, -13 and -27); group E, (MMP-14, -15, -16 and -24); sub-group F, (MMP-2, -7, -9 and -20)5.
Enzyme MMP
Collagenases
Interstizial collagenase; Collagenase-1 MMP-1
Neutrophil collagenase; Collagenase-1 MMP-8
Collagenase 3 MMP-13 Gelatinases Gelatinase A MMP-2 Gelatinase B MMP-9 Stromelysins Stromelysin 1 MMP-3 Stromelysin 2 MMP-10 Stromelysin 3 MMP-11 Matrilysins Matrilysin 1 MMP-7 Matrilysin 2 MMP-26 Membrane-Type MMPs (A) Transmembrane Type
MT1-MMP MMP-14 MT2-MMP MMP-15 MT3-MMP MMP-16 MT5-MMP MMP-24 (B) GPI-Anchored MT4-MMP MMP-17 MT6-MMP MMP-25 Others Macrophage elastase MMP-12 - MMP-19 Enamelysins MMP-20 - MMP-21 CA-MMP MMP-23 - MMP-27 Epilysin MMP-28
1.2.1. Mammalian MMPs: Domain constituents and substrates.
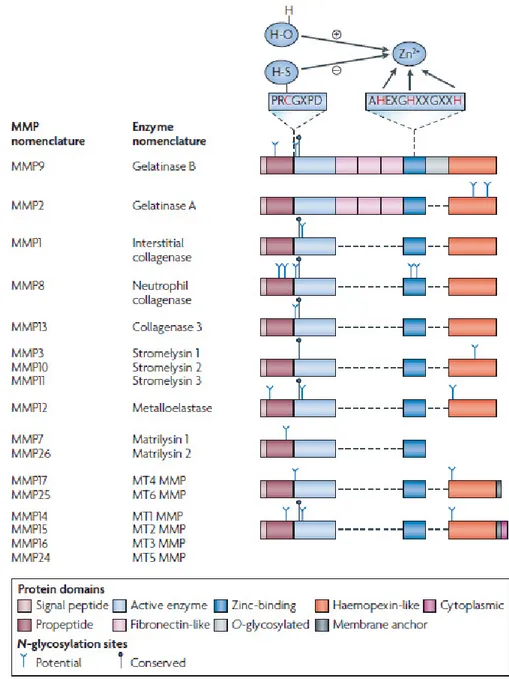
Matrixins are multidomains extracellular proteins, but recent studies have indicated that MMP-1, MMP-2, MMP-11 are also found intracellularly and may act on intracellular proteins. A typical MMP consists of a propeptide of about 80 aminoacids, a catalytic metalloproteinase domain of about 170 aminoacids, a linker peptide of variable lengths (hinge region) and a Hemopexin domain (Hpx) of about 200 aminoacids (Figure 1.2). Exception to this are MMP-7, MMP-26, MMP-23 that lack the linker peptide and the Hpx domain. MMP-23 has a unique cysteine-rich domain and an immunoglobulin-like domain after the metalloprotease domain. Two gelatinases, MMP-2 and MMP-9, have in the metalloprotease domain three modules of fibronectin type II motif. The zinc binding motif HEXXHXXGXXH in the catalytic domain and the cysteine switch motif PRCGXPD in the propeptide are common structural signature. The catalytic domain also contains a conserved methionine, forming the “Met-turn” eight residues after the zinc binding motif, creating the active site cleft around the zinc ion (Figure 1.3).
Figure 1.2. Basic architecture of MMPs.
Collagenases (MMP-1, MMP-8, MMP-13) cleave fibrillar collagene I, II, III into
characteristic ¾ and ¼ fragments, but they are able to digest other ECM proteins. Collagenolytic activity is due to a cooperation between the hemopexin and the catalytic domains.
Gelatinases (gelatinase A, MMP-2 and gelatinase B, MMP-9) with the help of the three
repeats of a fibronectin type II motif, degrade denatured collagens, gelatine and a number of ECM molecules including native types IV, V and XI collagens, laminin, fibronectin, and aggrecan core protein. MMP-2, but not MMP-9 also digest collagens I, II, III similarly to collagenases, but in a weaker manner.
collagens. MMP-3 and MMP-10 digest ECM substrates and participate to enzyme activation, while the role of MMP-11 is quite unknown. Moreover while MMP-3 and MMP-10 are secreted as inactive pro-MMPs, MMP-11 is activated intracellularly by furin convertase.
Figure 1.3. Domain structures of MMPs6
Matrilysins (MMP-7 and MMP-26) are characterized by the absence of the hemopexin
domain. MMP-7 is synthesized by epithelial cells and secreted apically. Besides ECM components it processes cell-surface molecules, such as pro-α-defensin, Fas-Ligand, Pro-Tumour necrosis factor-α, and E-cadherin. MMP-26 is expressed in normal cells such as dose of endometrium and in some carcinomas. It digests several ECM
Membrane-Types MMPs in mammals includes four types I transmembrane proteins
(MMP-14, -15, -16, and -24) and two glycosylphosphatidylinositol-anchored proteins (MMP-17 and -25). They are all activated intracellularly by furin. All MT-MMPs except MMP-17 join to the activation of MMP-2 from its proform. Moreover MMP-14 has collagenolytic activity on collagens I, II and III.
Seven matrixins have not been included in the above categories, though some of them (MMP-12, -20, and -27) are strictly related to Stromelysins sub-family.
Metalloelastase (MMP-12) was originally found in macrophages. It degrades elastin and it is fundamental in macrophage migration. MMP-19 digests many ECM molecules including the components of basement membranes. It is also called RASI (rheumatoid arthritis synovial inflammation) as it is found in the activated lymphocytes and plasma from patients with rheumatoid arthritis and it is also recognised as an autoantigen in patients with rheumatoid arthritis and systemic lupus erythematosis. It is, however, widely expressed in many organs including proliferating keratinocytes in healing wounds.
Enamelysin (MMP-20) is expressed in newly formed tooth enamel and digests amelogenin. MMP-21 was originally found in Xenopus and more recently in mice and humans. It is expressed in various fetal and adult tissues and in basal and squamous cell carcinomas. It digests gelatin, but information about the action on ECM components is not known. MMP-23 (Cysteine Array-MMP) is a unique member as it has unique C-terminal cysteine-rich immunoglobulin-like domains instead of a hemopexin domain. The propeptide lacks a cysteine switch. It is proposed to be a type II membrane protein having a transmembrane domain at the N-terminal of the propeptide, but the enzyme is released from the cell as the membrane anchored propeptide is cleaved by a proprotein convertase.
MMP-27 was first found in chicken embryo fibroblasts. Chicken MMP-27 digests gelatin and casein and causes autolysis of the enzyme, but little information is available on the activity of mammalian enzyme. Epilysin (MMP-28) is expressed in many tissues such as lung, placenta, heart, gastrointestinal tract and testis. The enzyme expressed in basal keratinocytes in skin is considered to function in wound repair. It is also elevated in cartilage from patients with osteoarthritis and rheumatoid arthritis.
1.2.2. MMPs Tridimensional structure.
In the last 15 years X-ray crystallography and NMR studies have revealed the tridimensional (3D) structures of MMPs. Full length MMP X-ray structures of proMMP-1 and proMMP-2 revealed that the propeptide domain consists of three α-chains and connecting loops. The susceptible “bait region” is located between helix 1 and helix 2. A cysteine of the prodomain, chelating the catalytic zinc ion with its sulfhydryl group, keeps the enzyme in its latent form. During the activation, disruption of the Cys-Zn interaction, allows a water molecule to bind the zinc ion. The “cysteine switch” motif (PRCGXPD) lies in the substrates binding site, but the orientation of this segment is opposite from that of a peptide substrate.
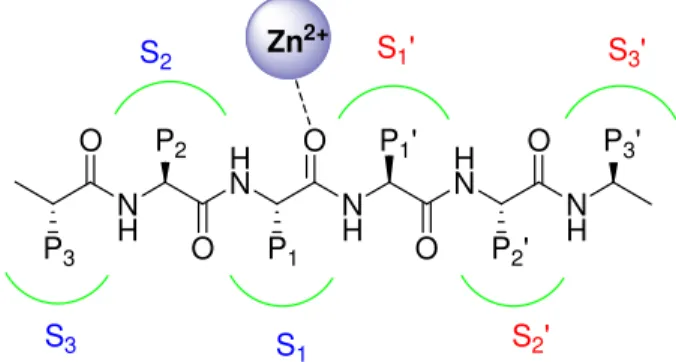
The polypeptide folds of MMP catalytic domains are essentially superimposable. The domain consist of a 5-stranded β-pleated sheets, four parallel and one anti-parallel, three α-helices and a connective loop. Two zinc ion are included in the active site, one catalytic and the other with structural function, and 1-3 calcium ions which stabilize the structure. The catalytic zinc is coordinated by three histidine residues of the conserved sequence HEXXHXXGXXH with the distance between the ε-nitrogen and the zinc varying between 2.0 and 2.2 Å. The glutamic acid residue facilitates the catalysis. In absence of a substrate or inhibitor, a water molecule acts as additional zinc ligand, keeping the zinc in a tetra-coordinated state. MT-MMPs have additional 8 residues between β-strand I e II which are critical for the activation of proMMP-2. Another basic feature of the MMP active site is the presence of three substrate-binding subsites to the left (unprimed side) and right (primed side) of the catalytic zinc (Figure 1.4). These sub-sites accommodate the side chains of the peptide to be cleaved, and the local structural characteristics and electrostatic environment of the individual sub-sites effectively determine substrate specificity.
N H H N N H H N N H P3 O P2 O P1 O P1' O P2' O P3' S3 S 1 S2' S2 Zn S1' S3' 2+
Of these pockets, the S1' pocket varies the most among the different MMPs in both the amino acid makeup and depth of the pocket, whereas the shallower S2' and S3' pockets are more solvent exposed. For this reason S1’ is also named “specificity pocket”. Basing on the size of the S1’ pocket MMPs can also be described as being shallow, intermediate, or deep pocket enzymes. MMP-1 and MMP-7 have a shallow pocket, MMP-2, MMP-9, and MMP-13 have an intermediate pocket, while MMP-3, -8, -12 have a deep S1’ pocket9.
Gelatinases fibronectin type II domains are settled in the loop between the fifth β-strand and the catalytic site helix and they consist of two antiparallel β-sheets connected with a short α-helix and stabilized by two disulfide bonds.
The linker region connects the catalytic domain and the hemopexin domain, and mutations at this level dramatically decrease collagenolytic activity, thus suggesting its importance in protein folding.
The hemopexin domain has 4-bladed β-propeller structure. The center of the propeller generally contains one calcium ion and chloride.
1.2.3. Peptide hydrolysis.
The mechanism of peptide hydrolysis is shown in Figure 1.5. The carbonyl group of the scissile peptide bond points towards the catalytic zinc and therefore can be polarized. The catalytic water molecule intercalates between this carbonyl group and the catalytic glutamate. The water molecule itself becomes polarized and is the nucleophile that attacks the carbonyl carbon of the scissile peptide linkage, giving rise to a pentacoordinate transition state. Proton transfer to the peptidic nitrogen, again possibly facilitated by the catalytic glutamate completes the catalytic reaction, enabling release of cleavage products and free enzyme10.
R1' NH O R1 O O H H O C O Zn2+ R1' NH O R1 O O H H O C O Zn2+ R1' NH3 O R1 O O C O Zn2+ O His His His
His His His His His His
1.2.4. Regulation of MMPs activity.
MMPs are responsible for the turnover and degradation of the ECM. However this is not the only function of MMPs because they also process other non matrix proteins, such as cytokines, chemokines, receptors and more. The MMPs proteolytic activity needs to be finely regulated. As for all secreted proteinases, the catalytic activity of MMPs is regulated at four points: gene expression, compartmentalization, proenzyme activation and enzyme inactivation, and it is further controlled by substrate availability and affinity.
For the most part, the production of MMPs is regulated at the level of transcription by specific signals. Some constitutive or homeostatic MMP genes possess simple promoter enhancer regions with cis-acting elements for basal transcription and are switched on in most cells under steady-state conditions. Other MMPs have complex promoter regions. The expression of these MMP genes is regulated by various agonists.
Once transcripted, MMPs are synthesized as pre-proenzymes. The signal peptide is removed during translation and proMMPs are generated. Activation of zymogens can occur by cystein-switch or by furin activation. Regarding the cysteine-switch, the interaction between the thiol of the cysteine of the propeptide and the catalytic zinc can be broken by three mechanisms:
1) cleavage of the prodomain, which could be mediated by another proteinase (also by other active MMPs);
2) oxidation of the free thiol by oxidants; 3) allosteric perturbation of the zymogen.
Oxidation of the thiol and allosteric perturbation would lead to inter or intramolecular autolytic cleavage of the prodomain .Regardless of how the cysteine-Zn++ interaction is disrupted, proteolysis of the prodomain is considered to be the final step in MMP activation (Figure 1.6).
Oxidant generated by leukocytes or other cell can both activate (via oxidation of the prodomain thiol) and subsequently inactivate MMPs (via modification of aminoacids critical for catalytic activity). In vitro, a number of proMMPs are activated by reactive oxygen species (ROS). For instance hypochlorus acid (HOCl) a product of neutrophil myeloperoxidase (MPO) activates pro-MMP7. However the role of ROS as in vivo activators has to be more deeply investigated.
Ten pro-MMPs, including all MT-MMPs, possess a recognition sequence RX[K/R]R between the pro- and the catalytic domain, which serves as a target sequence for furin or proprotein convertase. Furin is a type 1 membrane, subtilisin-like serine protease present in the trans-Golgi network and thus, MMPs with a furin cleavage site are processed intracellularly before secretion. For the other MMPs, the mode of activation is more presumed than proved, and the in vivo mechanism for activation of most non-furin cleaved proMMPs is still unknown11.
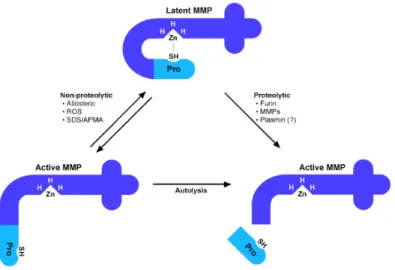
Figure 1.6. Latency of MMPs is maintained by the interaction between the thiol of a conserved cysteine
of the prodomain with the catalytic Zn. MMP could be activated by proteolitic cleavage of the propeptide mediated by Furin, by other MMPs or by plasmin. The activation can occur also in a non proteolytic way mediated by ROS, by allosteric modification, and in lab by organomercurials, which induce autolysis of the propeptide11.
Matrix Metalloproteinases activity is largely regulated by endogenous inhibitors: α-macroglobin and Tissue Inhibitors of Metalloproteinases (TIMPs) .
Human α-macroglobin is a glycoprotein of 725 kDa consisting of four identical subunits of 180 kDa. It is a general proteinase inhibitor and is found in the blood and in tissue fluids. It inhibits proteinases by entrapping the proteinase within the macroglobulin, and the complex is rapidly cleared by the low density lipoprotein receptor-related protein-1-mediated endocytosis. MMPs activity in fluids is mainly regulated by α-macroglobin and related proteins.
TIMPs (Tissue Inhibitors of Metalloproteinases) were originally found as collagenases inhibitors in serum and in the conditioned medium from fibroblast cultures. Four TIMPs have been currently characterized in humans and designated as TIMP-1, -2, -3, -4. They are expressed by a variety of cell types and they are present in most tissues and body
denaturation and proteolytic degradation12. TIMPs differ in many aspects, including structure, solubility, interaction with the proenzymes (proMMPs), and regulation of expression.
TIMP-1 and TIMP-3 are glycoproteins, while TIMP-2 and TIMP-4 do not contain carbohydrates. TIMP-1, -2, -4, are present in soluble forms while TIMP-3 is tightly bound to the extracellular matrix. Whereas TIMP-2 expression is constitutive and widely expressed through the body, TIMP-1, -3, -4 expression is inducible, and often exhibits tissue specificity: TIMP-1 is enriched in reproductive organ systems, TIMP-3 in heart, kidney, and thymus, while the expression pattern of TIMP-4 suggests that this tissue inhibitor may be an important tissue-specific regulator of extracellular matrix remodelling. TIMP-4 mRNA are localized in brain, heart of adult humans, ovary and skeletal muscle.
TIMPs have also the ability to form complexes with the proMMPs, so regulating the MMP activation processes. TIMP-1 forms a preferential complex with proMMP-9, while TIMP-2 binds to proMMP-2 and facilitates enzyme activation. TIMP-3 binds both proMMP-2 and pro-MMP-9. TIMP-2, -3 are also effective inhibitors of membrane-types MMPs. TIMP-3 can also inhibit some members of the ADAM family such as ADAM-17 and ADAM-10 and aggrecanases.
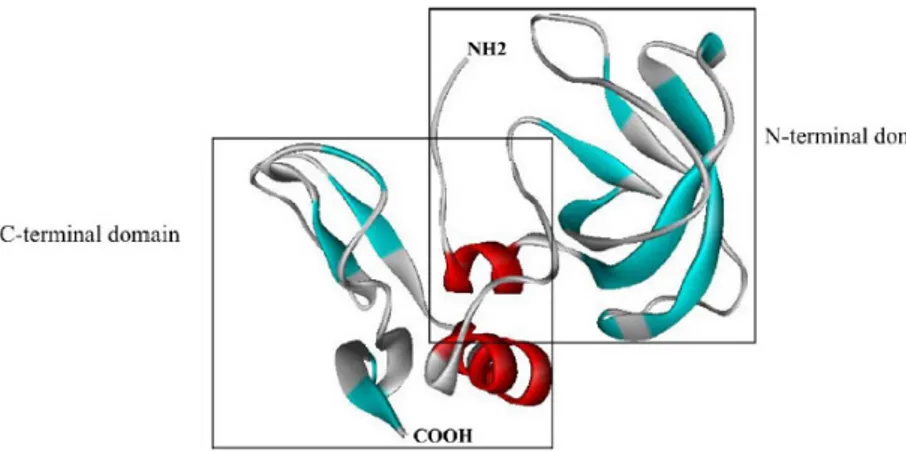
TIMPs are 21-34 KDa proteins of 184-194 aminoacids. They all possess 12 conserved cysteine residues forming six disulfide bonds, that fold the protein into two domains, the N-terminal and the C-terminal domain (Figure 1.7). The N-terminal domain is responsible for the MMP inhibition. It contains the VIRAK sequence conserved in all TIMPs and it is fully functional alone for MMP inhibition.
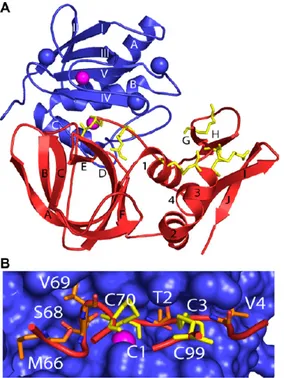
The exact mechanism of inhibition was understood in 1997 from the TIMP-1/MMP-3 complex13 (Figure 1.8A). The overall shape of the TIMP molecule is “wedge-like” and the N terminal residues Cys1-Thr-Cys-Val4 and the residues Glu67-Ser-Val-Cys70, that are linked by a disulfide bond, slot into the active site of MMPs. The catalytic zinc ion is bidentately chelated by the N-terminal amino group and the carbonyl group of Cys1, which expels the water molecule, bounds the zinc atom (Figure 1.8B). The crystal structure of TIMP-2 with MMP-14 revealed a similar complex.
Figure 1.8.(A) Tridimensional diagrams of TIMP-1/MMP-3 complex. (B) TIMP-1 reactive residues
(red) in MMP-3 active site (blue). Disulfide bonds are shown in yellow, zinc ion in pink8.
Several other proteins have been reported as endogenous MMP inhibitors, such as the secreted form of β-amyloid precursor protein that inhibits MMP-2 and RECK (a GPI-anchored glycoprotein that inhibits MMP-2, MMP-9, MMP-14). Nevertheless the inhibition mechanisms of these proteins are still unknown.
1.3. The ADAM metalloproteinases.
The ADAMs (a disintegrin and metalloproteinase) belong to the metzincines superfamily and together with snake venom metalloproteinases and ADAMTS form the adamlysin subfamily. ADAMs is a family of transmembrane and secreted proteins of approximately 750 amino acids in length, with functions in cell adhesion and proteolytic processing of the ectodomains of diverse cell surface receptors and signal molecules. ADAMs have been linked to many biological processes, such as sperm-egg interactions, cell fate determination in the nervous system, cell migration, axon guidance, muscle development and diverse aspect of immunity.
In humans ADAM-17 drives immune and inflammatory responses by activation of pro-TNF-α, and it is also essential in activating membrane-associated ligands of epidermal growth factor receptor (EGFR). ADAMs play an important protective role in Alzheimer’s disease, by participating in non amyloidogenic processing of amyloid-β precursor protein (APP). In cancer ADAMs contribute to enhance malignant aspects of tumour behaviour by stimulating cell proliferation via EGFR receptor activation.
For their multiple roles in pathogenesis ADAMs have emerged as attractive targets in drug development. 14,15
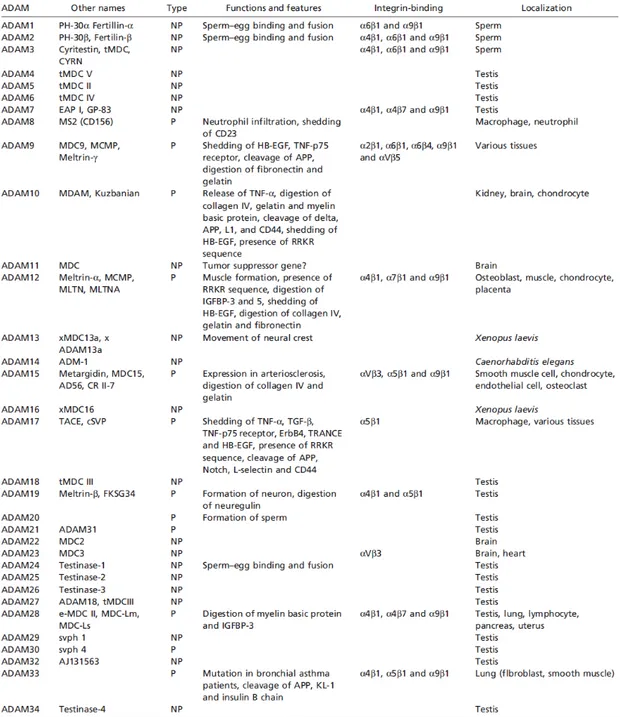
Human genome contains 25 ADAM genes of which four appear to be pseudogenes. Several ADAM genes display alternative spliced transcripts that give rise to variant proteins. For instance ADAM12 encodes both a long form (ADAM-12L), that has the typical transmembrane structure, and ADAM-12S which is a soluble form without the transmembrane and the cytoplasmatic domain.
Of the functional 21 presumed ADAM genes, 13 encodes proteins that have the characteristic reprolysin-type active site (HEXGHXXGXXHD) in the metalloproteinase domain, followed downstream by the “Met-turn”, signing them as Metcinzin enzymes. ADAMs can be thus divided in two subgroups, the first, having the typical metzincines features, includes ADAMDEC1, ADAM8, 9, 10, 12, 15, 17, 19, 20, 21, 28, -30, -33. The second group, which comprises ADAM-2, -7, -11, -18, -22, -23, -29 and 32 lacks one or more critical elements in the Zn-active site, suggesting that the metalloprotease domain in these proteins may play roles in protein folding and in protein interaction rather than catalysis.
Table 1.2. The Disintegrin and Metalloproteinase (ADAM) family16.
In humans and other vertebrates, ADAM2, 7, 18, 20, 21, 29 and 30 are expressed primarily in the testis, consistent with their involvement in spermatogenesis and sperm function. Of the remaining genes, ADAM9, 10, 12, 15, 17 and 19 are expressed quite broadly in somatic tissues, while ADAM28 and 33 show a more restricted tissue range and ADAM8 is primarily active in hematopoietic cell types.
Noteworthy, the expression of ADAM11, ADAM22 and ADAM23 is predominantly in the central and peripheral nervous systems, and ADAM12 is expressed in placenta and in mesenchymal or adult stem cell populations.
1.3.1. ADAMs structure and domain functions.
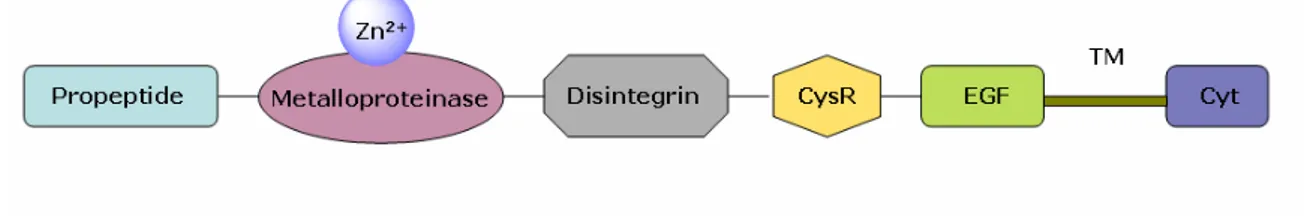
Similarly to MMPs, ADAM proteinases are multidomain proteins. As represented in Figure 1.9 starting from the N-termini they are composed by the prodomain, which is followed by the metalloprotease and by the disintegrin-like domain, which give the name to this class of enzymes. A cysteine rich region is also present, that is generally followed by an EGF-like domain, which is absent in ADAM-17 and ADAM-10. Protein are anchored to the plasmatic membrane through the transmembrane segment (TM) which links the ectoprotein to the cytoplasmatic tail.
Figure 1.9. ADAMs domain organization.
After synthesis and translocation in the endoplasmatic reticulum, ADAMs are delivered and further matured in the Golgi compartment, where the prodomain is recognized at a consensus RX(R/K)R motif and cleaved by proprotein convertase 7 or by furin. As for some MMPs, activation of the enzyme occurs through a cysteine switch mechanism. The prodomain of some ADAMs may act as intramolecular chaperone, facilitating the correct protein folding and maintaining the proteinase in a latent state. Isolated prodomains of ADAMs may be selective inhibitors of the mature and active forms of the enzymes.
The catalytic metalloprotease domain is quite similar to MMPs. The catalytic zinc is chelated by three histidine residues of the zinc binding consensus sequence (HEXGHXXGXXHD). A conserved methionine forms the Met-turn in the active site helix, creating the active site cleft. In the globular shape the active site is divided into two subdomains, with the active site cleft running between the two. The upper N-terminal subdomain consists of a highly twisted, 5-stranded-β-pleated sheet, the strands
helices. The lower C-termini domain is substantially constituted by an α-helix. The catalytic zinc lies at the bottom of the groove between the two sub-domains. The groove is also divided in some sub-sites (S3, S2, S1, S1’, S2’, S3’) which accommodate specific amino acid sequences (P3, P2, P1, P1’, P2’, P3’) of the substrate. The topology of this groove and in particular the depth and the lipophilicity of S1’ is critical for substrate specificity. ADAM metalloproteinases domain is also stabilized by three disulfide bonds and by a structural Ca2+, which is not present in ADAM-17 and ADAM-10. The disintegrin domain contains a 14 amino acid stretch known as the “disintegrin loop” which is implicated in integrins binding. The disintegrin domain can be divided into two sub-domains, which have been described as the shoulder (Ds) and the arm domain (Da). Following this nomenclature, the Cysteine-rich domain has been named the “wrist” (Cw) which is followed by an “hand” (Ch) segment that contains an hypervariable region (HVR) critical for substrate recognition. In ADAMs devoid of metalloprotease activity the HVR region could be functional to modulate cell-cell- and cell-matrix interactions (Figure 1.10).
Figure 1.10. MDC substrate cleaving. Membrane-anchored substrate molecule ‘X’ is directly recognized
and captured by the HVR on the membrane-bound ADAM molecule. The distance between the center of the HVR and the catalytic zinc ion is about 3.5 nm17.
This unusual denomination refers to the overall shape of the metalloprotease/disintegrin/cystein rich domain (MDC) which results to be a C-shaped molecule, where part of the Cys-rich domain is close and faces toward the
rich domain recognize the substrate, warranting the substrate specificity by the HVR of the Cys-rich domain, and offer the substrate to the metalloprotease domain for cleaving.17
In membrane-bound ADAMs, the EGF-like domain (~60 aa) follows the Ch-domain and presumably works as a rigid spacer connecting the MDC-domains with membrane-spanning region.
The transmembrane domain connects the ectodomains to the cytoplasmatic tail. The cytoplasmatic tail varies widely in length and sequence, ranging from 11 residues of ADAM-11 to 231 residues of ADAM-19. It may be involved in the regulation of protease function in response to intracellular signal events, or it can undergo to conformational changes after ectodomains-substrate binding, leading to recruitment of signalling molecules and adapters. Indeed the cytoplasmatic tail contains binding site motifs and Proline-rich motifs that are potential sites for phosphorylation by diverse kinases or potential binding sites for SH3 domain-containing proteins.
1.3.2. Control of ADAMs activity.
ADAMs activity can be controlled at different levels. After transcription, the first level involves removal of the prodomain mediated by protein convertases (PCs) which recognize and cleave at the consensus RX(R/K)R sequence. Since the autocatalytic process is autoregulated, in some cases retention of the cleaved prodomain in complex with the PC is a potential regulation point of ADAM expression at the cell surface. The isolated prodomains can act as potent selective inhibitors of the active mature forms of the enzymes, as has been reported for ADAM-10 and -1718’19. In the case of ADAM-17, the prodomain seems to inhibit the enzyme not via cysteine switch, but acting as a chaperone to protect the enzyme from degradation in the secretory pathway.
After zymogenes activation, further proteolytic processes may significantly affect protein conformation and function.
Other potential control points for ADAM activity include the trafficking of enzymes to particular sites within the cell, the activation of enzyme activity following cell stimulation, and the subsequent removal of active enzyme by internalization or other mechanisms. Recent data indicate that ADAM-19 and mature, active ADAM-17 are sequestered within cholesterol-rich membrane microdomains, otherwise known as lipid rafts or detergent-resistant microdomains (DRMs)20. Numerous substrates of ADAM-17
As MMPs, ADAMs activity is remarkably controlled by TIMPs. Among four TIMPs, several ADAMs are inhibited exclusively by TIMP-3. The catalytic domain of ADAM-10 is inhibited both by TIMP-1 and TIMP-3, whereas ADAM-8, -9 and -19 are insensitive to TIMP inhibition. In contrast, ADAM-12S has recently been shown to be inhibited by TIMP-3 and TIMP-2 in preference to TIMP-1.
ADAM-33 is moderately inhibited by TIMP-3 and TIMP-4, but weakly by TIMP-2 and is insensitive to TIMP-1. In addition to TIMPs, REversion-inducing Cysteine-rich protein with Kazal motifs (RECK), an unrelated protein that also displays MMP inhibitory capability, has been shown to be a physiological inhibitor of ADAM-10 in development of the nervous system.
ADAM-mediated shedding is both constitutive and inducible and ADAM activity can be also regulated by diverse stimuli, like G-protein coupled receptor (GPCR) activators, PKC activators, Ca ionophores, and other experimental or natural stimuli. The mechanisms are poorly understood, though involvement of the cytoplasmic domain is an obvious possibility. Phosphorylation or other modifications of the cytoplasmic domain may influence the interactions of ADAMs with adapter or trafficking proteins that direct substrate interactions or location in membrane sub-domains or other specific cellular locations. Following stimulation, ADAM-17 rapidly disappears from the cell surface via endocytosis which represents another potential control point for regulation of ADAM function.
1.3.3. Biological functions of ADAMs.
ADAMs have been long studied for their ability of cleaving a large number of membrane-bound proteins, generally at a stalk region close to the plasma membrane, solubilising a part of each target from its trans-membrane precursor. This mechanism has been termed ectodomain shedding. In Table 1.3 are listed potential ADAM substrates, whose shedding releases cytokines, grow factors, and chemokines.
Shedding of membrane anchored elements can mediate cell signalling in autocrine, paracrine and in juxtacrine fashion. Indeed the ligand may interact with a receptor on the same cell (autocrine) or on a neighbour cell (paracrine) or may activate a ligand that remains membrane associated (juxtacrine).
Table 1.3. Potential substrates of the major ADAM proteinases21.
The ADAMs mediated proteolysis is not only essential for providing extracellular signals, but also represents a prerequisite for intracellular signalling by regulated
intramembrane proteolysis (RIP). RIP can be defined as a sequential proteolytic
cascade, which involves an initial ectodomain sheddase action on a transmembrane protein that promotes a further proteolysis of the transmembrane segment, which leads to intracellular release of soluble fragments. Soluble intracellular domains are in most cases intermediates that are destined to degradation, but some “RIPped proteins” can translocate to the nucleus and act as transcription factors21 (Figure1.11).
Notch, CD44, ERBB4, an amyloid precursor protein (APP) cytoplasmatic domains translocate to the nucleus and interfere with transcription.
Notch is a transmembrane protein which undergoes three proteolytic cleavages in its lifecycle. The first proteolysis occurs in the trans-Golgi network by a furin-like convertase, generating the active heterodimeric proteins. The binding with membrane-anchored ligands activates Notch-receptor, causing a conformational change which is followed by a proteolytic process. In vitro experiment reveals that ADAM-10 and ADAM-17 are responsible for this second proteolysis. A further hydrolysis generates a Notch Intracellular Domain (NICD) which translocates to the nucleus and modulates signal transduction of delicate mechanisms, like cell fates and developmental processes. ADAMs also participate to transactivation of EGFR by G-protein coupled receptors (GPCRs), a mechanism termed GPCR-EGFR cross talk or Triple Membrane-Passing
Signal (TMPS). After Src kinase activation, GPCR activates phosphatidyl
inostol-3-kinase (PI-3K) that in turn activates phosphatidyl inostol-dependent inostol-3-kinase (PDK1) that through phosphorilation of the citoplasmatic tail activates ADAM. ADAM becomes thus able to shed EGFR ligands, making them interacting with the EGFR receptors (Figure 1.12). The ADAMs involved are mainly ADAM-17 and ADAM-10, although ADAM-9, -12 and ADAM-15 seem to be implicated. The integration of different signal pathways and the EGFR seems to be crucial in different types of tumour.
CD44, E-cadherin and N-cadherin, which could have profound effects on cell–cell interactions and cell migration, as well as downstream signalling pathways in tumours. To date, the implications of adhesion molecule proteolysis by ADAMs and other proteinases have largely been considered within the context of inflammation, notably leukocyte–endothelial cell interactions and in neurobiology. Nevertheless, similar events are likely to be important in both tumorigenesis and angiogenesis.
Chapter 2. ADAM-17
2.1. Structural features of ADAM-17.
ADAM-17 (or TACE, Tumor Necrosis Factor-α Converting Enzyme or CD156q) was first identified in 1997 by Black from Immunex Corporation 22 and Moss from Glaxo-Wellcome23 as the metalloproteinase responsible for the release of Tumor Necrosis Factor-α (TNF-α) from its membrane-bound precursor. The involvement of TNF-α in a variety of diseases associated with inflammation, made this discovery of a great pharmacological relevance. Currently, ADAM-17 proteolytic activity has been linked to a wide substrate range, making the landscape of cell mechanisms in which ADAM-17 is implicated more and more interesting.
ADAM-17 is a member of ADAM proteinases. Like most ADAMs, ADAM-17 is a multi-domain type I transmembrane protein that contains, in addition to the signal peptide, four functionally distinct extracellular domains followed by a transmenbrane region and a cytoplasmatic tail. As in other metzincines the prodomain contains a cysteine that, interacting with the active-site zinc, keeps the protein in its latent state. The prodomain also acts as a chaperone protecting the enzyme from degradation during transport through the secretory pathway.
The catalytic site contains the characteristic reprolysine-type active sequence (HEXGHXXGXXHD) involved in coordinating the zinc ion. The tridimensional structure of ADAM-17 catalytic site was first determined by Maskos24 et al. in 1998. The catalytic site has the shape of a flattened ellipsoid with a cleft that divides a small lower subdomain from an upper subdomain. The catalytic site is constituted by five stranded β-pleated sheets flanked by α-helices which protrude from the centre. The active-site cleft of TACE is relatively flat on the left-hand (non-primed) side but becomes notched toward the right. The catalytic zinc is penta-coordinated by the three imidazole Nε2 atoms of His-405, His-409, and His-415, by carboxylate oxygen of the catalytic Glu-406 and by a water molecule in the active enzyme. This zinc-imidazole ensemble is placed above the distal ε-methyl-sulfur-moiety of the strictly conserved Met-435, found in the Met-turn characteristic of the metzincin clan2. Immediately to the right of the catalytic zinc invaginates the medium-sized hydrophobic S1’ specificity
entrance. S3’ is a deep pocket, and the S1’-S3’bend is an hallmark of ADAM-17 because it is L-shaped.
Comparing to the other adamlysins, ADAM-17 lacks the adamalysin-like calcium-binding site and the charge pattern in and around the primed subsites is inverted.
Bioinformatic analysis between TACE and MMPs active sites revealed that zinc microenvironment in ADAM-17 is more polar than in MMP-2,-1, -3, -7, -925.
Notwithstanding the obvious differences in secondary structure, the active-site cleft of ADAM-17 bears some similarity to that of the MMPs, with the flat, nonprimed side, and the narrow primed side centering around the S1’pocket of varying size. This subsite, similarity to the MMPs, explains the observed sensitivity of a TACE-like activity to synthetic hydroxamic acid inhibitors originally designed for MMPs.
As in the other ADAMs, the disintegrin domain is implicated in integrin binding, while the Cysteine-Rich region is fundamental for substrate recognition. Cys-Rich domain is required for the shedding of Interleukin-1 receptor II shedding, but not for the shedding of TNF-α.
TACE cytoplasmatic tail is 130 aminoacids long, and it binds to a variety of intracellular signalling molecules including Protein Kinase C (PKC), Extracellular signal-regulated kinase (ERK), and mitotic arrest deficient 2 (MAD2). The exact functional role of these interactions is largely unknown, but it is sought to influence the catalytic activity.
2.2. ADAM-17, a sheddase with a wide degradome.
In the last decade ADAM-17 degradome has grown immensely and more than fifty substrates have been described (Table 2.1). The substrates of ADAM-17 are structurally heterogeneous and include membrane-anchored growth factors and cytokines, receptors, cell adhesion molecules and ectoenzymes. In many cases ADAM-17 has been shown to regulate the function of a given substrate leading to its activation, inactivation or modulation of the activity.
Many soluble growth factors and cytokines are synthesized as transmembrane proteins and processed by ADAM-17. This is an excellent example of how metalloproteinases control signal pathways. This regulatory system is faster than the transcription-translation system because in response to environmental stimuli a pathway can be switched on or off. The role of TACE in shedding TNF-α has been confirmed by in
mice are deficient in release TNF-α. Most of the TACE∆Zn/∆Zn mice show developmental defects, such as failure of eyelids to fuse and specific hair and skin defects observed in the embryos closely resembling the defects of TGF-α deficiency. Tumour necrosis factor-α is a pleitropic cytokine produced by monocytes, macrophages, neutrophils, T-cells, mast T-cells, epithelial T-cells, osteoblasts and dendritic T-cells, with potent immunomodulatory and proinflammatory properties. Excessive or prolonged production of TNF-α is a feature of septic shock and several important autoimmune diseases like rheumatoid arthritis, Crohn’s disease and multiple sclerosis. TNF-α is initially expressed as an active 26 kDa membrane-bound protein (pro-TNF-α), which is cleaved by TACE between Ala 76 and Val 77 to form the 17 kDa soluble cytokine. Other MMPs, such as MMP-1, -2, -3, -7, -9, cleave pro-TNF-α, but only MMP-7 and TACE cut at the natural cleavage site, the latter with a specificity 30 fold higher than MMP-7. Also the release of Transforming Grow Factor-α (TGF-α), a ligand for the Epidermal Growth Factor Receptor (EGFR) necessary for normal mammalian development, is another example of this straightforward activation process. Among EGFR ligands, ADAM-17 also processes other substrates such as amphiregulin, epiregulin, epigen, heparin-binding (HB)-EGF and neuregulins. These ligands bind the ErbB receptors, whose aberrant activity has been implicated in the development and progression of a variety of tumour types.
Table 2.1. Substrates processed by ADAM-17.
Abbreviations: APP, amyloid precursor protein; CNS, central nervous system; CX3CL1, CX3C-chemokine ligand 1; EGF, epidermal growth factor; GPCR, G-protein-coupled receptor; GPI,
glycoprotein I; GPIb-IX-V, glycoprotein Ib-IX-V; (GP)VI, platelet collagen receptor; IBD, inflammatory bowel disease; ICAM-1, intercellular adhesion molecule 1; 1Ra, interleukin 1 receptor antagonist; IL-15Ra, interleukin 15 receptor antagonist; IL-6R, interleukin 6 receptor; JAK, Janus kinase; LAG-3, lymphocyte-activated gene 3; NgR, Nogo receptor; p75NTR, p75 neurotrophin receptor; PAR1, proteinase-activated receptor 1; Pref-1, preadipocyte factor 1; SorLA, neuronal sorting protein-related receptor; STAT, signal transducer and activator of transcription; TGF-α, transforming growth factor α; TNF, tumor necrosis factor; TNFR, TNF receptor; VCAM, vascular cell adhesion molecule 1.26
ADAM-17 is involved in proteolysis of APP (Amyloid Precursor Protein). APP is a transmembrane protein which undergoes to two cleavages mediated by α- and β- secretases. α-Secretases generate soluble extracellular fragments non amyloidogenic while secretases, like BACE, lead to the release of small neurotoxic peptides called β-amyloid peptides, whose accumulation in cerebral cortex is a key step in Alzheimer’s disease. ADAM-17 behaves like α-secretase, and a further cleavage by γ-secretase generates an APP intracellular domain which undergoes to the RIPping events.
ADAM-17 is also responsible for the inducible release of many cytokines, as interleukins IL-6 and IL-15. IL-6 acts as growth and survival factor and also as pro-inflammatory cytokine. Upon binding to its specific transmembrane receptor (IL-6R) the cytokine activates the transmembrane signal-transducing molecule gp130 which
initiates the signalling process. IL-15 is considered a pro-inflammatory cytokine as it induces the production of inflammatory cytokines (IL-6 and TNF-α).
In addition to ligands, ADAM-17 degradome includes many receptors, since the cleavage of this type of molecules offers an additional way to regulate the response of the cell to growth factors and cytokines. Indeed a shed-receptor not only is unable to transmit signals to the cell, but it can act as a ligand sequestering scavenger. As represented in Figure 2.1 TACE is not only able to shed TNF-α, but it can also process TNF-α receptors TNFRI and TNFRII. TNFRI is constitutively expressed in most tissues while TNFRII is typically found in cells of immune system. Cleavage of TNFRs leads to downregulation of cellular responsiveness to TNF-α and additionally generates a soluble TNF-α antagonist that binds soluble TNF-α and prevents interaction with its membrane expressed receptors26.
Figure 2.1. Processing of TNFR and TNF- α by TACE26.
In addition to EGFR ligands, ADAM-17 proteolitically regulates ErbB receptors. ADAM-17 mediates shedding of ErbB4/HER4 receptors which have essential functions in heart and neural development27.
Among receptors ADAM-17 processes also the Notch receptor. On the cell surface Notch is an heterodimer, which is activated from its membrane-bound ligand Delta 1. This interaction is required in determining neuroepithelial development of mainly neuronal and hemapoietic cell population. ADAM-17 and ADAM-10 cleave the extracellular domain of Notch, while the so called “stub”, consisting of the transmembrane and the cytoplasmatic domain, becomes a substrate for γ-secretase, that
Highlighting the importance of ADAM-17 in the regulation of cell-cell interactions, ADAM-17 was shown to act on a large group of cell adhesion molecules. Shedding of cell adhesion molecules causes weak cell-cell interactions increasing cell motility, and in case of cancer, improving the spreading of tumour cells. In addition, the shedding of certain cell adhesion proteins is relevant in signal transduction. An example is CD44 which acts as a binding molecule for hyaluronan and mediates migration and invasion of tumour cells. ADAM-17 is involved in induced shedding of CD44, which has been correlated to an increased metastatic potential of tumour cells. This can be attributed to a reduced adhesiveness of the extra-cellular matrix via CD44. Furthermore, CD44 shedding is followed by intra-membranous cleavage via γ-secretase, activating the RIP cascade. Among cell adhesion molecules TACE sheds VCAM-1 (Vascular cell adhesion molecule-1) which is expressed on lympohocytes, monocytes, and eosinophils. Elevated levels of soluble VCAM-1 have been detected in different types of cancer but also after infections and inflammatory diseases28.
Recently has been discovered that ALCAM is another substrate for ADAM-17. ALCAM (Activated Leukocyte Cell Adhesion Molecule) mediates cell-cell clustering through homophilic (ALCAM-ALCAM) and heterophilic interactions (ALCAM-CD6). ALCAM is expressed at the cell surface and can be trimmed from the cell membrane by proteolysis leading to the generation of soluble ALCAM form (sALCAM) that is constituted by the main part of the ectodomain. Shedding of ALCAM has been documented to be related to an aggressive behaviour of some type of cancer, like Epithelial ovarian cancer (EOC)29.
2.3. ADAM-17 as therapeutic target in multiple diseases.
Despite the wide TACE degradome, to date the most of the proposed pathological roles of ADAM-17 are related to just few substrates, namely TNF-α, EGFR ligands and Amyloid Precursor Protein (APP). However, given the central role of these molecules in many biological processes, the number of diseases in which ADAM-17 has been suggested to be implicated has grown exponentially in the last years. The involvement in EGFR pathways and in CAM processing makes ADAM-17 a target for cancer, while dysregulation of TNF-α is connected to inflammatory disorders such as rheumatoid arthritis, osteoarthritis, inflammatory bowel disease, stroke, and multiple sclerosis. Moreover, participating to APP processing, TACE seems to be an interesting target also
EGFR is a family of four receptors (HER1/Erb1, HER2/Erb2, HER3/Erb3 and HER4/Erb4) that are activated by EGFR ligands that are synthesized as transmembrane elements. HER receptors are clearly involved in cancer progression and EGFR ligands, shedded by ADAM-17, are frequently overexpressed in cancer. High protein levels have been detected in the majority of human lung cancers, and they have been correlated to HER3 activity with subsequent promotion of cell growth, migration and resistance to apoptosis. In breast cancer HER2 receptor is overactivated and overxpression of ADAM-17, due both to transcriptional as well as post-transcriptional mechanisms, is associated with tumour progression and metastasis. ADAM-17 levels are elevated also in human colorectal cancer, in pancreatic ductal carcinoma, in prostate cancer and in ovarian cancer.
The TNF-α signal pathway is over-activated in many inflammatory diseases. Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic systemic inflammation of joint, which results in loss of function. Human RA cartilage displays upregulated ADAM-17 mRNA expression, indicating that the metalloproteinase is responsible for the activation of the TNF-α pathway. Moreover the poor oxygen condition of inflammation seems to upregulate the transcription of ADAM-17 in synovial cells through HIF-1 (Hypoxia Inducible Factor). ADAM-17 is involved in other inflammatory diseases as ulcerative colitis and Crohn’s disease, collectively known as inflammatory bowel diseases (IBD), peritonitis, polymyositis, dermatomyositis, psoriasis and others.
ADAM-17 can be also a potential target in Alzheimer’s disease. Immunohistochemical studies showed that ADAM-17 is intimately associated with neurons, senile plaques, and neurofibrillary tangles. Indeed it is known that ADAM-17 takes part in APP shedding, acting as α-secretase and furthermore the reduction of ADAM-17 substrates, such as TNF-α, TGF-α and L-selectin, induced by enzyme inhibition, seems to have neuroprotective effects30.
ADAM-17 also has a crucial role in stroke. Indeed, despite the complexity and heterogeneity of the disease, stroke is partly based on inflammatory disorder involving TNF-α and ADAM-17. ADAM-17 has been also studied as target in cardiovascular and respiratory diseases, in chronic renal diseases, in metabolic disorders and in other pathologies31.
2.4. Regulation of ADAM-17.
The regulation of ADAM-17 activity is poorly understood. The shedding rate of ADAM-17 rapidly increases by addition of cell activators like phorbol esters (PMA) thereby irreversible activation of protein kinase C. Dephosphorylation by intracellular phosphatases can also regulate ADAM-17 function. Inhibitors of Mitogen Activated Protein Kinase (MAPK) cascade block the increase in shedding rate in a number of case where TACE is the primary sheddase, even if the mechanism of action of MAPK is not clear. As ADAMs, ADAM-17 physiological activity is susceptible to the action of TIMPs. ADAM-17 is selectively inhibited by TIMP-3. Recent studies using stopped-flow-X-ray spectroscopy showed that interaction between TIMP-3 and ADAM-17 ectodomain implies communication between distal sites of the enzyme and the catalytic core which in turn undergoes to a dynamic charge transition.
2.5. ADAM-17 Synthetic Inhibitors.
Given the panel of substrates processed by ADAM-17 and its central role in inflammatory diseases, a great interest in developing synthetic TACE inhibitors has grown in the last years.
Similarities between ADAM-17 and MMP active site have driven the starting design of early inhibitors. As the relative MMP inhibitors, ADAM-17 inhibitors generally contain a group able to bind the catalytic zinc (Zinc Binding Group, ZBG), a peptide or peptidomimetic backbone that lies the primed (right-hand) site, an H-bond acceptor portion and a lipophilic substituent which is directed into S1’pocket.
In MMPIs many functional groups, with the ability of chelating the active zinc, thus competing with natural substrates, have been developed32,33. Common MMPI ZBGs are
scaffold Hydrophobic residue H bond acceptor ZBG
hydroxamates, carboxylates, thiolates, phosphonates, phosphinates, pyrimidintriones (Figure 2.2).
Figure 2.2. MMPI ZBGs classified by mode of binding to the active zinc(II) ion32.
Up to now ADAM-17 inhibitors reported in literature bear almost exclusively the hydroxamate, which is the most potent ZBG (100-2000 fold compared to carboxylate). As occurs in MMPs inhibition, the zinc ion, bound to the hydroxamate in a bidentate fashion and linked to three histidines of the active site, is penta-coordinated with a bipyramidal trigonal geometry (Figure 2.3).
Figure 2.3. Representation of binding between a peptidomimetic hydroxamate and the zinc ion in
ADAM-17 active site32.
Early broad spectrum MMP inhibitors like succinyl hydroxamate Marimastat (BB2516)
understanding biological functions of ADAM-17 and as starting point for the development of new synthetic inhibitors. Like many other broad spectrum MMP inhibitors, 2.1 and 2.2 failed in clinical trials as they showed dose-limiting musculoskeletal side effects. Though the exact reason for this side effect is unknown, their toxicity seems to be due to the ability to inhibit MMP-1 and/or MMP-14.
N H HO O OH O H N N H O N H HO O N S O2 OMe N 2.1 2.2 Ki ADAM-17: 22 nM ADAM-9: 274 nM MMP-1: 1 nM MMP-3: 68 nM MMP-9: 1 nM MMP-13: 0.1 nM Ki ADAM-17: 54 nM ADAM-9: 1nM MMP-1: 11 nM MMP-3: 16 nM MMP-9: 3 nM MMP-13: 5 nM
Figure 2.4. Early broad spectrum ADAM/MMP inhibitors.
Hence researchers focused their efforts in the development of synthetic inhibitors with enhanced selectivity towards ADAM-17, sparing MMPs that are also involved in many physiological processes34,35.
2.5.1. Succinate-based inhibitors.
As previously stated early succinate-based MMP inhibitors like marimastat 2.1 were also TACE inhibitors. Succinic scaffold was derivatized by many companies with the aim of improving selectivity or pharmacokinetics features of these compounds. Compound 2.3 with a phenyl guanidine group was developed by Daichii Fine chemicals in order to improve its water solubility. It is able to inhibit TNF-α synthesis in LPS stimulated model in THP cell with an IC50 of 130 nM, but it is also active on MMP-1 (IC50=6 nM) and MMP-13 (IC50=0.1 nM). Compounds 2.4a-b, developed by Novartis, showed to be potent broad spectrum TACE inhibitors effective in various models of lung inflammation. The replacement of P2’ aminoacidic portion with a substituted hydrazide group led to compound 2.5, which had good TACE activity (IC50=3 nM) and 100-fold of selectivity for TACE over most MMPs. Unfortunately its development has been stopped because of its poor bioavailability.
N H HO O O H N N H O N H NH2 NH 2.3 N H HO O O H N N H O HO 2.4a: R=-OMe 2.4b: R=-Me R N H HO O O H N N O2 S 2.5 N H O H N N H O 2.6 OH O NH HN NH NO2 S N N H O H N N H O 2.7 OH O N
Figure 2.5. Succinate-based ADAM-17 inhibitors.
Also succinate reverse hydroxamates have been exploited as TACE inhibitors. An example is the N-hydroxyformamide GW4459 (2.6) which is a good TACE and MMPs inhibitors (ADAM-17 Ki=4 nM). GW4459 reduced TNF-levels in the pleural fluids of zymosan treated rats. Related compounds like 2.7 (GW3333) with a methyl substitution at the P2’ arginine side chain were synthesized, with the aim of endowing the compound of stability and oral bioavailability. Compound 2.7 resulted a potent inhibitor of TACE (IC50=40 nM) and MMPs, able to reduce TNF release in mice subjected to LPS administration. Despite its potency and its long half life (T1/2>12h) in dog and rats, it failed because it was poorly oral bioavailable.
Succinyl hydroxamates with a bridge between P1 and P2’ showed to be good TACE inhibitors. This cyclic systems have been designed with the purpose of reducing potency towards MMP-1 and consequently adverse side effects. Various P1’ groups have been introduced. Early compounds with small P1’ were active, but with a large profile of activity also over MMPs. The insertion of the biphenyl moiety instead of the isopropyl group led to compound 2.10 which resulted 100-fold more selective towards TACE than over other MMPs, orally bioavailable and able to reduce TNF-α in vivo.
HO H N O N H O H N O N O O O O HN O 2.10 CF3 N H HO O HN O O N H O OMe OMe OMe 2.8 N H HO O O H N NH O 2.9 NH O
Figure 2.6. Macrocyclic Succinate-based hydroxamates.
2.5.2. Sulfonamido-based inhibitors.
Starting from the structure of the broad spectrum MMP inhibitor CGS27023A (2.2), several sulfonamido-based inhibitors have been disclosed. The sulfonamide group is crucial not only to increase the enzyme-inhibitor binding forming H-bonds with the two oxygens, but also because it directs the hydrophobic substituent deeply into S1’36. On this basis, several sulfonamidic inhibitors with different P1’ moieties able to explore the narrow and bended TACE pocket have been proposed.
Anthranilate derivatives like compounds 2.11-2.13 (Figure 2.7) showed a good TACE activity. Hydroxamate 2.11 inhibited TACE with a Ki value of 40 nM. The adjustment of the arylsulfonyl subsituent from methoxy to butynyloxy substituent 2.12 remarkably increases activity against TACE (Ki=15 nM), improving also selectivity over collagenase-1 and collagenase-3. The increase of activity is due to the butynyloxy substituent which fits perfectly into S1’ following the bend of the subsite. Derivative
2.13 is a good TACE inhibitor (IC50=25 nM) 10 times more selective towards TACE
than MMP-13 and 500 times than MMP-1. The oral activity of 2.13 was improved due to the introduction of basic piperazine moiety in position 3.
N H HO O 2.11 N Br Me SO2 OMe N N H HO O 2.12 N Br Me SO2 O N N H HO O N N SO2 O Br N 2.13
Figure 2.7. Anthranilic sulfonamido inhibitors.
The propargylic P1’ substituent has been largely exploited in the design of TACE inhibitors because of its ability to fit perfectly into S1’ subsite gaining in potency and selectivity (Figure 2.8). Among alkynyl derivatives, Levin and others developed the thiomorpholine analogue TMI-1 (2.14). This compound, proposed as dual MMP-13/ADAM-17 inhibitor for the treatment of rheumatoid arthritis, showed high activity in various in vivo models and currently it is in the Phase-II of clinical trials (IC50=8.4 nM on TACE). The same group developed Aprastat (2.15) which differs from the first for the presence of a free hydroxyl group on the alkynyl methylene. Notwithstanding its activity towards ADAM-17 and the absence of side effects, 2.15 was withdrawn from Phase II due to lack of efficacy. A wide SAR study led to discovery of a potent ADAM-17 inhibitor (IC50=2 nM) TMI-2 (2.16) with an hydroxyl chain in position α of the hydroxamate. Compound 2.16 was slightly selective in vitro (more than 250-fold selective over MMP-1, -7, -9, -14 and ADAM-10), and was able to reduce in cell-based assay TNF-α production in WBA (human whole blood assay) with IC50=1 µM. The in vitro efficacy was replayed in vivo in mice (ED50=3mg/kg) and in a rat model of arthritis, where administration p.o. of the inhibitor reduced joint arthritis scores.
The butynyloxy substituent was also applied to quinoline hydroxamate 2.18 which was 50 fold more selective towards TACE than its precursor 2.17. The same inversion in selectivity was noticed for heteroaryl-fused pyridine derivatives. Compound 2.20 has a ADAM-17 IC50=6 nM and it gains affinity for ADAM-17 with respect to its precursor
2.19 which was selective for MMP-13. 2.20 inhibit 95% production of TNF-α in a
mouse model at a dose of 100 mg/kg after 1h of oral dosing. Some analogues with a benzodiazepine nucleus were synthesized (2.21-2.22). In this case the introduction of
N H HO O N 2.14 O2S S O N H HO O N O2S S O OH 2.15 N H HO O NH O2S O 2.16 OH N H HO O N O2S N O O 2.22 HO N N H HO O N O2 S N O N N H HO O OMe N O2 S O 2.17 2.18 N N H HO O N SO2 N 2.19 O N O N N H HO O N SO2 O N O 2.20 N H HO O N O2S N O O S 2.21
Figure 2.8. Development of butynyloxy derivatives.
Researchers from Wyeth Lab reported some β-sulfone-hydroxamates bearing butynyloxy group at P1’. Compound 2.23 is a potent TACE inhibitor (IC50=2 nM) and selective sparing some MMPs such as MMP-1. -2, -9, -13 and MMP-14 (Figure 2.9).
N H HO O S O2 O N O N 2.23
2.5.3. γ-Lactam inhibitors.
With the aim of improving biopharmacological properties such as good oral bioavailability and good inhibition of TNF in vivo models, many compounds were designed by inserting semirigid cyclic systems in the structure. This arrangement provided an appropriate orientation between the ZBG and the P1’ substituent. On this basis some classes of inhibitors were proposed, such as compound 2.24 (IM491), developed by Xue and colleagues, which was potent against ADAM-17 (IC50=6.2 nM) and selective towards other MMPs (IC50 in micromolar range) in vitro, and with an IC50 of 20 nM in TNF release in vivo in human WBA. TACE and MMP-3 active sites were proposed to be similar. Taking advantage from this comparison, a docking study of compound 2.24 in MMP-3 suggested that the hydroxamate binds to the catalytic zinc ion a bidentate fashion and the carbonyl group of the γ-lactam forms two important hydrogen bonds with the NH groups of Val163 and Leu164, while the aromatic moiety occupies the S1’ of the enzyme. Optimization of γ-lactam scaffold by insertion of different P1’ led to the discovery by Duan and others from Bristol-Myers-Squibb of compound 2.25 (IK682), characterized by the presence of [(2-methyl-4-quinolinyl)methoxy]phenyl as P1’ group. HO H N O N O 2.24 HO H N O N O 2.25 O N
Figure 2.10. Compound 2.24 in MMP-3 active site (white). Oxygen atoms of hydroxamate coordinate the
zinc ion (orange), the lactam carbonyl interacts with Ala165 and Leu164 and the phenyl ring is directed towards S1’.37
IK682 was tested on semipurified porcine TACE (pTACE) from porcine spleen, and resulted to be a potent inhibitor with excellent selectivity for TACE (Ki=0.56 nM)
µM37. Moreover from the pharmacokinetic point of view, IK682 showed rapid absorption, good oral bioavailability and long half life and it reached preclinical development.
Another excellent γ-lactam ADAM-17 inhibitor, BMS-561392 (DPC-333, 2.26), has been developed at Bristol-Myers-Squibb (Figure 2.11). This molecule resulted to be very active with an IC50 of 0.20 nM, and able to reduce TNF-α production in LPS stimulated model. Although it possesses good pharmacokinetics properties and it overcame Phase I it has been withdrawn in Phase II because it resulted to be hepatotoxic. HO H N O N O NH 2.27 O N Boc HO H N O N O NH2 2.26 O N
Figure 2.11. γ-Lactam hydroxamates.
γ-Lactam analogues with N-hydroxy-2-(2-oxo-3-pyrrolidynyl)acetamide scaffold with the [(2-methyl-4-quinolinyl)methoxy]phenyl have been developed. Compound 2.27 with a Boc-amino group in position α of the hydroxamate which is exposed to the solvent showed sub-micromolar inhibition in WBA (IC50=0.42 µM) and a selectivity of 700-fold towards TACE than MMP-1 and of 300-fold than MMP-2 and MMP-9.
2.5.4. β-Benzamido inhibitors
Some researchers at Bristol-Myers-Squibb discovered some succinate-hydroxamates as selective TACE inhibitors. Compound 2.28 showed excellent activity in in vitro assay (IC50=8 nM), but it was inactive in vivo. Its derivative 2.29 (IM-491), although with an IC50 on the same range of its precursor, was very potent in vivo (IC50=20 nM in WBA). Notwithstanding promising properties in vivo and in vivo and very good bioavailability of compound 2.29, it was suspended in clinical trials because of the mutagenic properties of [(2-methyl-4-quinolinyl)methoxy]aniline moiety. To overcome the toxicity of compound 2.29, scientists examined the possibility of reversing the central amide moiety to obtain a series of β-benzamido hydroxamates which could maintain the same interactions of succinyl hydroxamate IM-491, thus preventing toxicity. Inhibitor 2.30 is
WBA) and it was found to be 30,000 fold more active against ADAM-17 than MMP-1, -2, -13, -14, -15 and -16. Moreover in rats it demonstrated to be orally bioavailable (58%) with an ED50 of 3 mg/kg. The substitution of the tetrahydropyran ring with other 5-membered carbocycles led to the discovery of compound 2.31 which possesses in vitro IC50 equal to 0.14 nM and WBA IC50 of 109 nM. Also compounds with spirocyclic motifs have been proposed. Compounds 2.32-2.33 have WBA IC50 respectively of 108 nM and 143 nM, with selectivity of 2000-fold for ADAM-17 over MMP-1, -2, -9, and with a bioavailability of 31% and 35%.
To obtain more potent and selective inhibitors the P1’ group of β-benzamido scaffold, was optimized into benzimidazolemethylphenyl group. Compound 2.34 showed IC50 value of 1.4 nM and it was 10,000fold more selective for ADAM17 than MMP1, 2, -3, -7, -8, -9, -10, -1-3, -14, -15, and -16. 2.28 O N HN O O NHOH 2.29 O N HN O N O NHOH 2.30 O N O HN O O NH HO 2.31 O N O HN N O NH HO 2.32 O N O HN O NH HO O 2.33 O N O HN O NH HO O O HN O O NH HO N CF3
2.5.5. Non-hydroxamate inhibitors.
Similarly to what happened in MMPI research field, in the last few years many research group tried to develop synthetic ADAM-17 inhibitors with alternative zinc binding groups. Although the hydroxamate is the most potent ZBG, toxicity and metabolic instability of this chelating group has been debated. Often hydroxamate inhibitors are poorly bioavailable because of their very high renal clearance. Hydroxamates can undergo to hydrolysis leading to less active carboxylates and the mutagenic byproduct hydroxylamine can cause toxicity. Hydroxamates are also easily metabolized to glucoronides and sulfonates. Nevertheless it has been recently demonstrated that plasma stability of hydroxamates highly depends on inhibitor structure38.
To overcome the problems associated with hydroxamates, other ZBGs such as pyrimidinetriones, hydantoins, triazolones and imidazolones have been developed with good results (Figure 2.13).
Researchers from Bristol-Myers-Squibb replaced the hydroxamate with a pyrimidine-2,4,6-trione motif keeping the previously optimized [(2-methyl-4-quinolinyl)methoxy]phenyl scaffold. Compound 2.35 showed an IC50 of 1µM indicating that the pyrimidinetrione is an effective zinc binding motif. To improve affinity for TACE a spacer between the phenyl ring and the pyrimidinetrione was introduced. Compound 2.36 with an amido moiety between the phenyl ring and the ZBG resulted to be the best of this series with an IC50 of 26 nM and with a two order of magnitude of selectivity for ADAM-17 with respect to the MMPs. Nevertheless the promising results in vitro, none of these compounds resulted to be active in vivo assays.
2.35 O N HN HN O O O 2.36 O N HN O HN NH O O O 2.37 O N HN O NH HN O O O N NH O O NH HN O 2.38