1. Introduzione
Tumori ed infiammazione vanno di pari passo nel senso che i processi infiammatori spianano la via alla crescita e alla progressione del cancro. L’ipotesi avanzata circa 20 anni fa e allora considerata eretica, oggi ha ricevuto il battesimo ufficiale del mondo scientifico.
Fu Rudolf Virchow il primo a notare, nel 1863, la presenza di leucociti nei tessuti neoplastici e a mettere in relazione infiammazione e cancro, suggerendo che l’infiltrato linforeticolare potesse riflettere l’origine del tumore in siti di infiammazione cronica (1).
Nel corso dei decenni si è poi fatta strada l’idea secondo cui i leucociti presenti a livello delle masse tumorali potessero svolgere la consueta funzione di difesa per il nostro organismo, sovvertendo così il possibile legame tra la progressione del cancro e i processi infiammatori. Sostenere tale ipotesi, considerata “eretica” in effetti, era come dire che l’immunità innata, quella più primitiva, rappresentata da macrofagi e linfociti, viene sovvertita dai tumori e indotta ad agire non per contrastare queste formazioni estranee ma addirittura per favorirne la crescita e l’espansione.
Le conoscenze accumulate negli ultimi dieci anni a questo proposito hanno fornito un sostegno importante all’idea di Virchow
somministrazione di farmaci destinati non solo a rallentare o ad arrestare la crescita delle cellule pretumorali ma addirittura a farle regredire. E’ stato infatti dimostrato che l’azione mitogenica delle molecole dell’infiammazione si esplica in diversi modi, tra cui la promozione dell’angiogenesi, la stimolazione della produzione di radicali liberi che agiscono sul DNA e l’azione favorente sul diffondersi di metastasi (2). Da tali considerazioni si può dedurre che
è possibile controllare il tumore bloccando alcune vie infiammatorie.
In linea con tale principio, è stato dimostrato, per esempio, che l’inibizione della ciclossigenasi 2 (Cox-2) rappresenta un’efficace strategia chemiopreventiva nei confronti di una condizione preneoplastica, la poliposi familiare del colon (Fig.1).
Figura 1: Incidenza di casi di tumore del colon-retto in Italia
Un grande numero di studi clinici in corso è quindi basato sul tentativo di bloccare alcune molecole dell’infiammazione con lo scopo di aggredire i tumori. Anche la costituzione genetica
influenza le caratteristiche delle singole molecole pro-infiammatorie, alterandone il significato patogenetico. Ad esempio, i geni che codificano per le citochine hanno un alto grado di polimorfismo e ciò significa che esistono diverse versioni della medesima citochina funzionalmente diverse ed alcune di queste varietà aumentano la suscettibilità ai tumori. Infatti, individui con un determinato tipo di TNF (TNF 308) sono 14 volte più a rischio degli altri di sviluppare un tumore della prostata.
1.1
Infiammazione e tumore
I legami tra cancro e infiammazione sono stati per la prima volta studiati nel XIX secolo sulla base di evidenze sperimentali che fanno supporre che i tumori spesso nascono in siti di infiammazione cronica e le cellule infiammatorie sono presenti sono presenti in campioni biologici di tumori. Studi epidemiologici, infatti, hanno mostrato che l’infiammazione cronica predispone gli individui a vari tipi di cancro ed è stimato che le risposte infiammatorie sono legate per il 15-20% a tutte le morti da cancro nel mondo.
Ci sono molte cause all’origine dell’infiammazione cronica che aumentano il rischio di sviluppare un cancro. Tra queste cause ritroviamo infezioni microbiche (ad esempio, l’infezione da
tumore prostatico) (3). I vari segni caratteristici che fanno mettere in relazione cancro e infiammazione includono l’infiltrazione di cellule infiammatorie nei tessuti tumorali che a loro volta producono mediatori dell’infiammazione come citochine (TNF, IL-1, IL-6 e IL-8) chemochine (CCL6, CCL7, CCL8, CCL12) e
prostaglandine (PGE2, PGF2, PGD2) ossia molecole che orientano la
crescita, la migrazione e la differenziazione delle cellule tumorali . I mediatori e gli effetti cellulari dell’infiammazione sono importanti costituenti delle condizioni locali del tumore. In certi tipi di cancro, l’infiammazione è presente prima che avvenga il cambiamento in maligno. Contrariamente in altri tipi di cancro un cambiamento oncogenico induce un ambiente infiammatorio che promuove lo sviluppo di tumori. Noncurante delle sue origini l’infiammazione comunque, nell’ambiente tumorale, ha l’effetto di promozione.
Struttura del TNFα
Citochine e fattori di crescita, in particolare, possono promuovere lo sviluppo del tumore stimolando la proliferazione cellulare,
l’adesione, la vascolarizzazione e la formazione di metastasi (Fig.2).
Figura 2: Relazione tra infiammazione e tumore
Inoltre, altri elementi cellulari quali neutrofili, eosinofili e cellule mononucleate producono specie reattive dell’azoto e dell’ossigeno (ROS, RNS) che possono danneggiare DNA, RNA, lipidi e proteine.
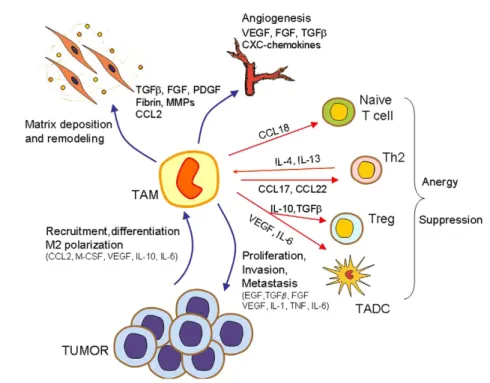
Il 50% della massa tumorale è costituita da leucociti, linfociti e macrofagi, e questi ultimi in particolare hanno un ruolo chiave nello sviluppo e nella progressione del tumore. Infatti, i macrofagi, sono cellule della linea di difesa primaria dell’organismo detta immunità
In ciò si distinguono dalle cellule dell’immunità specifica (linfociti T e B) che attivano efficacemente le loro risposte contro un determinato antigene in seguito ad un precedente incontro con esso. Questi due compartimenti dell’immunità cooperano fra di loro per sviluppare una risposta immune efficace. Ad esempio in presenza di pericoli i macrofagi lavorano per promuovere l’immunità specifica richiamando e attivando i linfociti. Alcune delle loro funzioni principali sono la produzione di molecole infiammatorie e quello di fagocitare eventuali microrganismi al fine di digerirle e eliminarle. Una volta fagocitato l’elemento estraneo, “illustrano” le caratteristiche del nemico ai linfociti attraverso alcuni segnali specifici, consentendo lo sviluppo di una reazione immunologia “su misura” e più efficace. Purtroppo questo dialogo essenziale viene alterato da tumori. Infatti sappiamo che i macrofagi vengono attivati nel tessuto tumorale e “riprogrammati” con il risultato di venire disarmati delle loro funzioni antitumorali e dirottati a contribuire alla crescita e diffusione delle cellule malate. Quando un macrofago entra all’interno di un tumore perde la sua funzione “killer” e la capacità di attivare la risposta antitumorale specifica. Acquisisce invece una serie di funzioni che favoriscono pericolosamente lo sviluppo delle cellule maligne (4).
Molti tumori solidi hanno una caratteristica in comune: una scarsa vascolarizzazione. In particolare, nella parte più interna della massa tumorale, non arrivando ossigeno a sufficienza, si formano delle aree di tessuto necrotico e i macrofagi vanno a posizionarsi proprio in queste regioni. Vengono richiamati dal sangue periferico e si
localizzano nelle zone dove c’è maggiore scarsità di ossigeno. Qui subiscono un adattamento metabolico e cominciano a favorire la crescita, la vascolarizzazione dell’area (neoangiogenesi), la formazione di metastasi e la loro diffusione in altri distretti del corpo.
Il tumore ottiene quindi un doppio vantaggio: riduce le nostre difese e le corrompe a suo favore. Per fare ciò nel microambiente tumorale vengono attivati una serie di circuiti molecolari e meccanismi che sfruttano la “plasticità funzionale dei macrofagi” che sono cellule in grado di esprimere diverse funzioni in risposta a diversi stimoli. E’ quindi il microambiente tumorale che decide e dirige le funzioni di queste cellule a suo favore. Ciò avviene agendo su una determinata famiglia di complessi proteici nucleari, i fattori trascrizionali, che sono simili a interruttori molecolari in grado di spegnere o accendere la cellula. In questo modo il tessuto tumorale riceve nutrienti dalla circolazione sistemica e, può progredire più facilmente. Ormai si sa che la presenza di un alto numero di macrofagi all’interno di un tumore corrisponde ad una prognosi negativa (5).
genomica e facilita l’azione di agenti genotossici (aflatossine, N-nitroso derivati) che inducono danni permanenti nel DNA. 2. L’infiammazione induce la formazione di perossido di
idrogeno (H2O2), che a sua volta può essere convertito in
specie reattive dell’ossigeno, che possono recare danni al DNA mediante l’ossidazione delle basi azotate che lo compongono.
3. Attivazione di NF-kB (nuclear factor-kappa B).
L’NF-kB è una proteina complessa che rappresenta un fattore di trascrizione. L’NF-kB si può trovare in tutti i tipi di cellule ed è interessata in tutte le reazioni delle cellule agli stimoli, quali stress, citochine, radicali liberi, irradiazioni con ultravioletti e attacco proveniente dagli antigeni, batteri, virus. L’ NF-kB gioca un ruolo chiave nella regolazione della risposta immunitaria all’infiammazione, di conseguenza una non corretta regolazione di esso è stata collegata al cancro del quale provoca l’iniziazione e progressione mediante la regolazione dell’espressione di geni implicati nel ciclo cellulare (5).
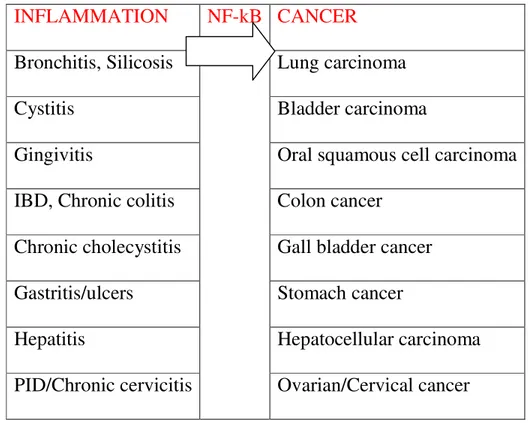
Circa il 15% dei tumori maligni hanno origine infiammatoria (Fig.3).
INFLAMMATION NF-kB CANCER
Bronchitis, Silicosis Lung carcinoma
Cystitis Bladder carcinoma
Gingivitis Oral squamous cell carcinoma IBD, Chronic colitis Colon cancer
Chronic cholecystitis Gall bladder cancer Gastritis/ulcers Stomach cancer
Hepatitis Hepatocellular carcinoma PID/Chronic cervicitis Ovarian/Cervical cancer
Figura 3: Ruolo NF-kB nella degenerazione da infiammazione a cancro
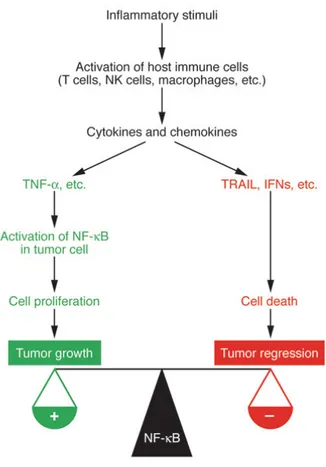
La connessione tra l’infiammazione e cancro infatti si può vedere costituita da due componenti principali: una intrinseca ed una estrinseca. La componente intrinseca è attivata da eventi genetici che causano la neoplasia. Questi eventi includono l’attivazione di vari tipi di oncogeni in seguito a mutazioni, riarrangiamenti cromosomici o amplificazioni e inattivazioni di geni soppressori dei tumori. Le cellule che sono trasformate in questo modo producono un microambiente infiammatorio nei tumori. Per quanto riguarda la
nell’attivazione di un fattore di trascrizione, appunto NF-kB, che è sintetizzato, in condizioni normali, in risposta all’infiammazione e, costitutivamente attivo nelle cellule tumorali dove coordina la produzione di mediatori dell’infiammazione incluse citochine e chemochine così come la produzione di Cox-2 e quindi di prostaglandine (6). Questi mediatori conseguentemente reclutano macrofagi , mastocellule, eosinofili, neutrofili. Si pensa quindi che NF-kB possa rappresentare un punto di collegamento tra questi due eventi (Fig. 4).
L’attivazione di NF-kB può avvenire mediante vari meccanismi. Una via metabolica è quella che ha origine dall’attivazione di Toll/IL-1, da cui originano due cascate di eventi: dipendente e indipendente. La via metabolica MyD88-dipendente è responsabile dell’attivazione del fattore di trascrizione AP-1, mentre la via MyD88-indipendente, induce la stimolazione di NF-kB attraverso le chinasi TBK1 e IKKi/ε e porta anche all’attivazione di IRF.
In questo modo NF-kB è disponibile ed è traslocato nel nucleo dove si lega a sequenze specifiche del DNA denominate “response elements” (RE). Il complesso DNA/NF-kB poi richiama altre proteine quali coattivatori e l’RNA polimerasi che trascrive DNA in mRNA, il quale, infine è esportato nel citosol e tradotto in proteina. Ciò porta ad un cambiamento delle funzioni della cellula. NF-kB viene attivato anche da TNFα e LTβ (7). Anche le specie reattive dell’ossigeno promuovono l’azione di tale fattore nucleare, mediante un processo IkBα-dipendente (Fig. 5).
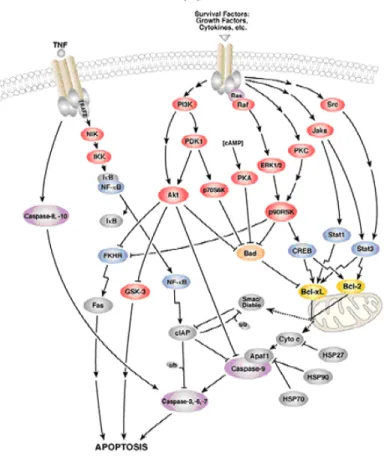
L’attivazione costitutiva di NF-kB in vari tumori solidi, è determinata da una mutazione cromosomica di IkB. Tutto ciò porta ad una elevata espressione di geni anti-apoptotici e quindi ad un aumento della sopravvivenza di cellule già trasformate e al mantenimento del fenotipo maligno (Fig. 6).
Figura 5: Ruolo cruciale di NF-kB
1.2 Farmaci antinfiammatori non
steroidei (FANS) e cancro
I FANS o farmaci antinfiammatori non steroidei sono anche detti “analgesici periferici” per il loro impiego come analgesici e antinfiammatori. Essi hanno dimostrato di possedere anche una spiccata attività antitumorale dovuta sia ad una rilevante attività chemiopreventiva, sia ad un effetto diretto verso il tumore. Il meccanismo d’azione dei FANS è quello di inibire gli enzimi ciclossigenasi, sia Cox-1 che Cox-2 che catalizzano la conversione da acido arachidonico a prostaglandine. L’inibizione dei tumori da parte dei FANS può essere mediata da diversi processi cellulari. Questi processi interessano la capacità dei FANS di riattivare l’apoptosi, indurre l’arresto del ciclo cellulare ed inibire l’angiogenesi. Poiché la Cox-2 aumenta fino al 90% in alcuni tumori, si presume che i FANS medino l’apoptosi attraverso l’inibizione della Cox-2 (8).
I FANS, inoltre, possono agire attraverso vie metaboliche Cox-2 indipendenti. L’effetto a livello di diverse vie metaboliche ed il blocco di bersagli diversi in fasi tumorali diverse, rende questa categoria di farmaci particolarmente interessanti in campo oncologico (Fig.7).
Figura 7: Vie metaboliche Cox-1 e Cox-2
1.2.1 Meccanismi Cox-2 mediati
Nella classifica dei farmaci più prescritti e più impiegati in tutti i settori della pratica clinica, i FANS occupano una posizione di rilievo anche per la maggiore anzianità nella storia dei farmaci di sintesi: il salicilato di sodio fu sintetizzato nel 1875.
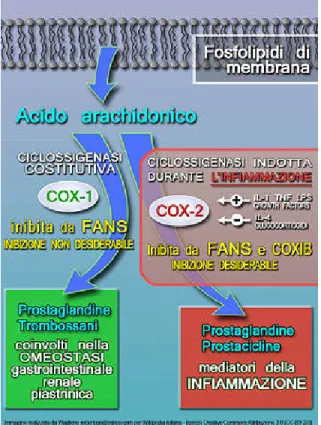
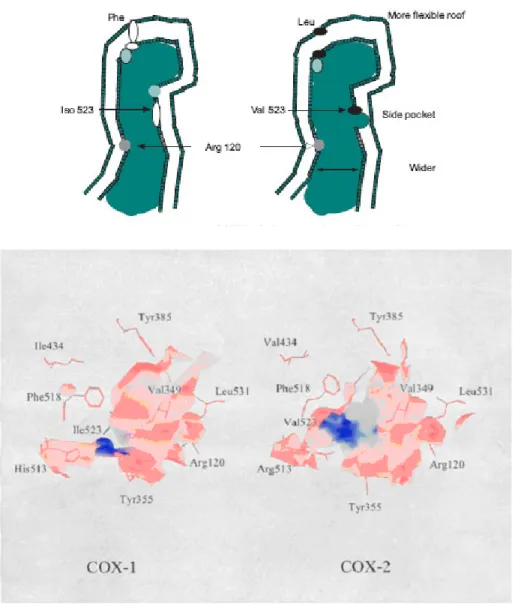
Nel 1971 si scoprì che l’effetto antinfiammatorio era legato all’inibizione di un enzima, la ciclossigenasi. Vent’anni dopo tale scoperta è stata inoltre dimostrata l’esistenza di due isoforme di questo enzima, codificate da geni situati in differenti cromosomi. La prima (Cox-1 costitutiva) è normalmente presente nei tessuti dove stimola la sintesi delle prostaglandine che regolano le normali attività cellulari contribuendo a processi fisiologici quali la citoprotezione gastroenterica, il flusso renale e l’aggregazione piastrinica. La seconda (Cox-2 inducibile), quasi assente in condizioni fisiologiche, viene espressa in corso di un’infiammazione sotto lo stimolo di fattori mitogeni e citochine e la sua produzione è indotta da agenti pro-infiammatori nelle cellule endoteliali, nei macrofagi e nei fibroblasti sinoviali (Fig 8).
Di particolare interesse, studi recenti hanno evidenziato che la Cox-2 è costitutivamente espressa nell’epitelio intestinale normale. Un aumento dei livelli di espressione della Cox-2 sembra essere in relazione con lo sviluppo neoplastico mediante la promozione della divisione cellulare, l’inibizione dell’apoptosi, l’alterazione del ciclo cellulare, la formazione di metastasi e la stimolazione della neovascolarizzazione pretumorale.
I dati di ricerca disponibili dimostrano con sufficiente certezza che gli effetti terapeutici dei FANS sono in gran parte dovuti all’inibizione della Cox-2 mentre gli effetti indesiderati (in
insorgenza di nausea, vomito, bruciori gastrici e diarrea. Se l’uso diventa abituale o prolungato, si può avere la formazione di vere e proprie ulcere della mucosa gastrointestinale con possibilità di emorragie che possono essere lievi ma anche di grave entità (9).
Figura 8: Struttura ciclossigenasi 1 e ciclossigenasi 2
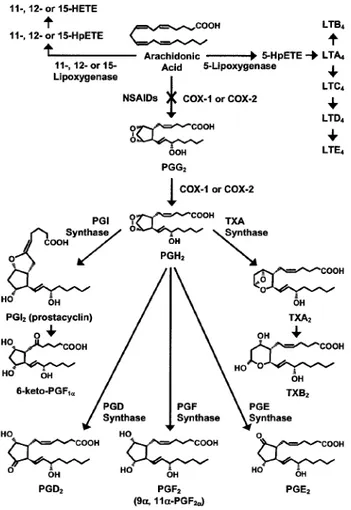
Le Cox sono enzimi coinvolti nella cascata dell’acido arachidonico, un acido grasso poli-insaturo, che all’interno della cellula è legato
ai fosfolipidi di membrana. L’azione delle fosfolipasi permette la liberazione dell’acido dalla membrana plasmatica e la sua bioconversione, a seguito dell’azione delle ciclossigenasi e delle lipossigenasi, nei mediatori classici dell’infiammazione che comprendono le prostaglandine, le prostacicline, i trombossani e i leucotrieni.
La biosintesi dei prostanoidi coinvolge numerosi processi biochimici tra cui la liberazione dell’acido arachidonico da parte della fosfolipasi A, l’ossidazione dell’acido arachidonico a PGH2,
grazie all’azione della Cox, e la successiva conversione di tale intermedio in PGD2, PGE2, PGF2, PGI2, TXA2 (Fig 9).
Figura 9: Cascata dell’acido arachidonico
I prostanoidi svolgono diverse funzioni biologiche importanti. In particolare sono coinvolti nella regolazione dell’infiammazione, nella protezione della mucosa gastrica, nella regolazione della temperatura corporea, nell’omeostasi renale, nella vasodilatazione e nell’aggregazione piastrinica.
I prostanoidi intervengono anche nella regolazione della progressione del tumore e la PGE2 è la prostaglandina ritenuta più
della PGES e della Cox determina un aumento rilevante dei livelli di PGE2 nei tumori.
La PGE2 si lega ai recettori di membrana EP presenti in adipociti,
utero, rene, mucosa gastrica, piastrine, macrofagi e sono denominati EP1, EP2, EP3 e EP4; la loro attivazione provoca un aumento dei
secondi messaggeri. L’attivazione di EP1, in particolare, determina
un aumento del calcio libero nel citoplasma, mentre l’attivazione di EP3 riduce i livelli di cAMP, che, invece aumentano per azione dei
recettori EP2 e EP4.
Il legame con i recettori EP è importante nella progressione tumorale. Ad esempio, la PGE2 regola la crescita tumorale e stimola
l’angiogenesi mediante il legame con il recettore EP2, mentre la
capacità di indurre metastasi è mediata dalla sovraespressione di EP4,soprattutto nel cancro intestinale. Attraverso il legame con EP,
la PGE2 può attivare le proteine G, ma anche le proteine Wnt, i
recettori nucleari PPARδ e il recettore per il fattore di crescita epiteliale, EGFR, che favorisce la migrazione e l’invasione delle cellule tumorali. Può inoltre indurre un aumento dei fattori di trascrizione c-myc, c-jun e della ciclina D.
La prostaglandina PGF2α è coinvolta nella contrazione della
muscolatura liscia vasale, nel controllo della pressione arteriosa e nella modulazione dell’infiammazione. La PGF2α ed il suo recettore
La PGI2, con il recettore IP distribuito nell’albero vascolare, è in
grado di attivare i PPARδ, condizione che favorisce la trasformazione neoplastica. Il trombossano TXA2 favorisce la
progressione del tumore, e la sua inibizione produce un blocco della migrazione delle cellule tumorali e favorisce l’apoptosi (10).
Un altro meccanismo d’azione dei Cox-2 inibitori è legato all’inibizione dell’HDAC. I nostri cromosomi sono costituiti da una grande quantità di DNA, al quale sono legate delle proteine dette istoni. Il DNA è avvolto intorno agli istoni e forma delle strutture che assomigliano a delle perline dette nucleosomi. La capacità di un particolare fattore di trascrizione di legarsi ad un determinato gene è in parte funzione delle possibili modificazioni che possono avvenire nelle proteine degli istoni. Gli enzimi istone acetil-trasferasi (HATs) sono in grado di alterare la struttura della cromatina (DNA-proteina) aggiungendo alle proteine dell’istone, corte catene di atomi di carbonio. Tale alterazione farà cambiare la struttura dell’interazione DNA-istone che perderà la forma la forma a “perlina”, favorendo la formazione del legame con i fattori di trascrizione. Un diverso gruppo di enzimi, istone deacetilasi (HDACs) è invece responsabile della rimozione dei gruppi acetile dagli istoni, bloccando il processo di trascrizione (11). Attività anomale da parte degli enzimi HDACs sono state evidenziate in molti tipi di cancro, come nel carcinoma colorettale. Quando questi enzimi agiscono in modo “scorretto”, possono impedire la trascrizione di geni fondamentali. Questo sembra essere una fase molto importante nel complesso processo di formazione di una
massa tumorale. L’iperacetilazione e l’ipoacetilazione promuovono rispettivamente l’espressione e la repressione genica. Uno dei meccanismi che favorisce la carcinogenesi è proprio la repressione dei geni oncosoppressori.
I membri della classe II HDAC 4, 5, 7 funzionano come corepressori trascrizionali che interagiscono con i fattori di trascrizione. Gli inibitori della HDAC sono capaci di dereprimere geni la cui espressione è bloccata da un’eccessiva ipoacetilazione della cromatina, favorendo quindi processi come l’apoptosi, la necrosi, la differenziazione e l’inibizione della proliferazione.
Inoltre, è importante sottolineare che nella linea cellulare tumorale di colon, HT-29, l’attivazione di citochine proinfiammatorie è sensibile agli effetti degli inibitori HDAC. Questi agenti riducono i livelli di espressione della Cox-2 indotti dal TNF-α. La riduzione della sintesi della proteina Cox-2 è determinata da un’azione del TNF-α che si realizza mediante la stabilizzazione dell’mRNA (12). Gli idrossammati sono una nuova classe di farmaci antitumorali inibitori selettivi della HDAC. L’interazione tra l’acido idrossammico e HDLP (histone deacetylase-like protein) avviene mediante l’interazione con lo ione zinco.
Il gruppo dell’acido idrossammico è importante per l’allineamento della molecola all’interno del sito attivo; coordina lo ione zinco attraverso i gruppi carbossilico e ossidrilico. Inoltre, forma legami
1.2.2
Effetti
collaterali
dovuti
all’inibizione delle Cox-2
I farmaci inibitori Cox-2 selettivi, hanno un profilo di sicurezza migliore rispetto ai FANS tradizionali. E’ da ricordare però che l’effetto collaterale principale dei FANS è la gastrolesività che comprende sintomi come la dispepsia, pirosi, disturbi addominali, lesioni della mucosa (erosioni ed ulcere) che a volte possono essere associate a gravi complicanze quali emorragie e perforazione gastrica. I FANS danneggiano la mucosa gastroduodenale principalmente attraverso due meccanismi: un’ alterazione della barriera della mucosa gastrica ed un inibizione dei meccanismi che la proteggono, che si esplica attraverso l’inibizione della Cox nel tratto gastrointestinale. Poichè le prostaglandine sono importanti mediatori della risposta infiammatoria, appare evidente come i FANS danneggiano la mucosa gastroduodenale con un meccanismo identico a quello con cui esercitano la loro azione antinfiammatoria. L’individuazione di due diversi isoenzimi della Cox ha suggerito che inibizioni selettive della Cox-2 potrebbero bloccare la sintesi delle prostaglandine nei siti di infiammazione senza interferire con la produzione di esse nella mucosa gastroduodenale e quindi esser privi di effetti tossici in questa sede. D’altro canto l’inibizione della Cox-2 può portare a edema periferico e ipertensione e può promuovere la trombosi. Nel 2000 in Italia sono stati introdotti in commercio nuovi farmaci antinfiammatori, celecoxib (Celebrex) e rofecoxib (Vioxx) che agiscono inibendo selettivamente la
ciclossigenasi 2 e che hanno messo in evidenza i primi rischi cardiovascolari legati ad un loro utilizzo.
N N CH3 NH2SO2 CF3 O S O O O Celecoxib Rofecoxib
I coxib, inibendo solo la Cox-2 diminuiscono l’infiammazione provocando minori effetti collaterali a livello gastrointestinale; inoltre essi non diminuiscono il trombossano e non inibiscono l’aggregazione piastrinica né allungano il tempo di sanguinamento. Contemporaneamente all’immissione in commercio dei primi coxib, erano in corso due studi clinici randomizzati per provare la loro sicurezza a livello gastrointestinale: lo studio CLASS ( celecoxib long-term arthritis safety study) e lo studio VIGOR (vioxx gastrointestinal outcomes research) (14). Tali studi hanno dimostrato in seguito la superiorità del profilo gastrointestinale rispetto ai classici FANS anche se con qualche controversia, infatti i ridotti effetti gastrointestinali del celecoxib osservati nello studio CLASS erano evidenti per i primi sei mesi ma diminuivano
aumentare il rischio di eventi cardiovascolari avversi indotti dall’inibizione della Cox-2. Nonostante lo studio VIGOR avesse suscitato controversie riguardo alla sicurezza cardiovascolare, nessuno pensò di programmare studi clinici controllati per verificare la tossicità cardiovascolare al contrario furono pianificati molti studi per mostrare l’efficacia dei coxib in altre indicazioni quali la prevenzione della poliposi adenomatosa ricorrente, il dolore post-operatorio ed il rallentamento dell’ Alzheimer. E’ in uno di questi studi l’APPROVe (adenomatous polyp prevention on vioxx) terminato anzitempo che è stato osservato un aumento del rischio cardiovascolare nel gruppo di pazienti trattati con il rofecoxib. Allo stato attuale il celecoxib (Celebrex) è registrato in Italia per il trattamento dell’artrosi e dell’artrite reumatoide. Il rofecoxib (Vioxx) è stato ritirato nel 2004 (FDA, 2005) dal commercio a livello mondiale a causa di aumento del rischio di eventi cardiovascolari e trombotici (tra cui infarto del miocardio e ictus). Poco dopo, nel corso del meeting annuale dell'American Heart Association (AHA), sono stati presentati dati relativi ad un aumento del rischio cardiovascolare da parte di un altro inibitore Cox-2, il valdecoxib (Bextra) (15).
L’EMEA ha inoltre approvato la commercializzazione di lumiracoxib (Prexige), un nuovo inibitore Cox-2 selettivo che però non è ancora commercializzato in Italia.
Lumiracoxib
Sembra quindi evidente che il rischio cardiovascolare sia un effetto di classe dei farmaci antinfiammatori ad azione selettiva Cox-2. Nonostante il meccanismo responsabile di tale effetto non sia stato ancora completamente chiarito, nel 1998 diventò evidente che le due isoforme delle Cox incidevano differentemente sull’equilibrio dei prostanoidi vasoattivi (16). Diversi studi suggerirono che il trombossano A2 (TXA2), vasocostrittore e promotore
dell’aggregazione piastrinica, derivava principalmente dalla Cox-1 mentre la sintesi del vasodilatatore e potente inibitore dell’aggregazione piastrinica, la prostaciclina PGI2, era dovuta
della Cox-2, da cui ne consegue che i coxib potrebbero spostare l’omeostasi vascolare verso uno stadio vascolare protrombotico e ipertensivo (18).
1.3 Meccanismi Cox-2 indipendenti
Gli effetti antiproliferativi e proapoptotici Cox-2 indipendenti sono stati dimostrati in linee cellulari tumorali che non esprimono la Cox-2.
In particolare è stato evidenziato che il sulindac e i suoi metaboliti, inibitori non selettivi degli enzimi Cox-1 e Cox-2, esercitano effetti antiproliferativi senza modificare i livelli di PGE2. Quindi, in
cellule Cox-2 negative, gli effetti dei Cox-2 inibitori possono essere mediati da meccanismi Cox e PGE2 indipendenti.
In cellule tumorali di colon, il sulindac solfuro induce apoptosi sia in linee che esprimono la Cox-2, quali le HT-29, sia in quelle che non la esprimono, le HCT-15. Il trattamento di HCT-15 con varie prostaglandine (PGs) infatti non esercita un effetto protettivo nei confronti dell’apoptosi.
Il sulindac solfuro e il sulindac solfone inducono apoptosi, indipendentemente dall’espressione della Cox-2. Il sulindac solfone, è un metabolita inattivo del sulindac in quanto è inattivo sulla Cox-1 e sulla Cox-2; tale composto agisce senza variare i livelli di PGS.
La presenza di meccanismi antiproliferativi Cox-2 indipendenti nei FANS crea una condizione favorevole in termini di efficacia antitumorale, in quanto l’azione della molecola non dipende necessariamente dall’espressione dell’enzima Cox-2 nelle cellule neoplastiche.
1.3.1 Bersagli coinvolti nei meccanismi
Cox-2 indipendenti
L’attività Cox-2 indipendentemente dei FANS è in parte responsabile dei loro effetti sulla proliferazione cellulare e sulla regolazione dell’apoptosi e del ciclo cellulare.
Tali effetti si realizzano mediante l’interazione con numerosi bersagli molecolari quali: citocromo c, NAG-1, PDE2/5, NF-kB, PDK1, Akt, ERK1/2, RSK2/MAPKs, PPARδ, 1, 15-LOX-2, Wnt, acido carbonico, ciclina D, p21.
• NF-kB
Il fattore di trascrizione NF-kB ha un ruolo centrale nella regolazione dell’espressione di vari geni, implicati nel controllo
- Geni con azione a feedback negativo - Geni coinvolti nell’immunità
- Geni antiapoptotici
- Geni della proliferazione cellulare
Normalmente NF-kB, è sequestrato nel citoplasma dalla proteina inibitrice IKB. In seguito all’ubiquitinazione, IKB si stacca dal complesso NF-kB che può dunque traslocare nel nucleo. Tale processo è mediato dalla IKB chinasi (IKK).
Dopo la traslocazione nel nucleo NF-kB regola la trascrizione di geni quali la Cox-2, il c-myc e la ciclina D1.
È stato dimostrato che alcuni membri della famiglia di NF-kB e IKB sono coinvolti nello sviluppo del cancro. In diversi tumori solidi, tra cui quelli al pancreas e al colon, NF-kB è costitutivamente attivo; il sulindac inibisce l’attivazione di NF-Kb(19).
• Fosfodiesterasi (PDs)
Le fosfodiesterasi sono enzimi che regolano i livelli di cAMP. La loro importanza nelle malattie tumorali deriva dal ruolo svolto sulla regolazione della crescita cellulare, sull’apoptosi e sulla migrazione delle cellule tumorali.
Ad esempio, il sulindac solfone induce apoptosi in cellule tumorali umane inibendo la PDE2 e la PDE5. Il conseguente aumento della
concentrazione cellulare di cGMP induce, infatti, l’attivazione di proteine chinasi cGMP-dipendenti che si traduce nell’attivazione del meccanismo apoptotico e nel blocco della crescita cellulare. Nel tumore al colon, il sulindac solfone attiva anche JNK1 (c-Jun N- terminal kinase), una chinasi che può fosforilare ed inattivare le proteine antiapoptotiche Bcl-2 e Bcl-XL. JNK-1 può anche aumentare l’espressione di proteine proapoptotiche attraverso l’attivazione del fattore di trascrizione AP-1 (20).
• PDK-1/Akt (phosphoinositide-dependent kinase-1)
L’apoptosi è indotta anche dall’inibizione di PDK-1. PDK-1 (serine/threonine kinase) viene attivata da P13K (phosphatidylinositol 3-kinase) a sua volta attivata da Akt, una proteina coinvolta nella proliferazione cellulare. Ad esempio, in cellule tumorali di colon HT-29, il celecoxib induce apoptosi attraverso il blocco dell’attivazione di Akt (21).
• RSK-2 (ribosomal S6 kinase-2) MAPKs
Questa serina/treonina chinasi è attivata dalla MAPK. I FANS inibiscono l’attività di RSK-2 sopprimendo la fosforilazione del suo substrato cAMP.
(b) JNKs (c-Jun N-terminal kinase); (c) p38 MAPKs e JNK.
Alcuni FANS tra cui l’acido salicilico e acetilsalicilico inibiscono l’attività di ERKs, riducendo la crescita cellulare e inducono p38 MAPK e JNK, promuovendo l’apoptosi (22).
• PPAR (recettori attivati da proliferatori perossisomiali)
Questi recettori regolano la trascrizione di geni coinvolti nell’apoptosi, nella differenziazione cellulare e nell’infiammazione. Nel tumore al colon si osservano livelli elevati di PPARδ. I PPARδ vengono attivati dalla PGI.
Il sulindac riduce la capacità dei PPAR nel riconoscere il proprio sito d’azione. Anche l’acido 13-S-idrossioctadecadienoico (13-S-HODE), prodotto per azione della 15-lipossigenasi 1 (15-LOX-1), regola i PPARδ nelle cellule tumorali di colon inducendo apoptosi(23).
• NAG-1 (gene attivato dai FANS)
Il gene attivato dai FANS, NAG-1, è un membro della famiglia del fattore di crescita tumorale β (TGFβ). Questo gene ha un ruolo nella crescita cellulare, nell’infiammazione e anche nell’angiogenesi.
NAG-1 ha un’azione antitumorale e proapoptotica nel tumore intestinale. In particolar modo, il sulindac solfuro regola l’espressione di NAG-1 aumentando i livelli di mRNA di circa 4-6
volte rispetto ai controlli. Tale meccanismo d’azione è stato riscontrato anche in altre forme tumorali (24).
• Inibizione della progressione del ciclo cellulare
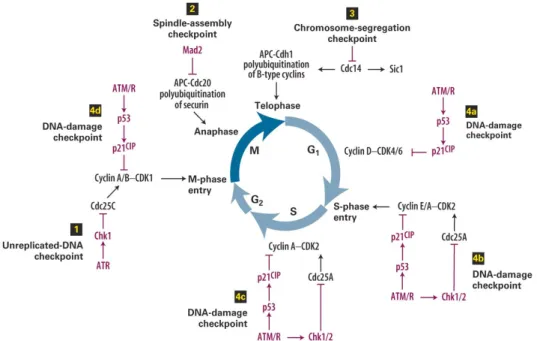
La progressione ordinata delle varie fasi del ciclo cellulare è realizzata da varie cicline, cicline-chinasi dipendenti (CDKs) e dai loro inibitori.
Le CDKs sono espresse costitutivamente durante il ciclo cellulare ma in forma inattiva; sono attivate tramite fosforilazione dopo il legame con le cicline che sono, invece, sintetizzate solo in specifiche fasi. La sintesi delle cicline D, che legano CDK4 e CDK6, è stimolata all’inizio della fase G1; successivamente viene indotta la sintesi della ciclina E che si lega al CDK2.
I complessi ciclina D/CDK4, ciclina D/CDK6 e ciclina E/CDK2 fosforilano la proteina del retinoblastoma (Rb). Quest’ultima è una proteina codificata da un gene oncosoppressore che, nelle cellule quiescenti, è presente in forma attiva ipofosforilata e blocca la progressione del ciclo cellulare da G1 a S sequestrando il fattore E2F.
In seguito a fosforilazione, la proteina Rb rilascia E2F che può, in ultima analisi, attivare la trascrizione di diversi geni target; dopo la mitosi una fosfatasi defosforila la proteina Rb e la cellula torna in
complesso con CDK1 e che permette alle cellule di passare dallo stato G2 alla fase M, fosforilando una serie di proteine richieste per la mitosi (Fig.10).
Figura 10: Ciclo cellulare
L’attività delle CDKs è regolata da due famiglie di inibitori: la prima è composta dalle tre proteine p21, p27 e p57, che hanno effetto su tutte le CDKs; la seconda famiglia, ha un effetto selettivo sui complessi ciclina D/CDK4 e ciclina D/CDK6 ed è composta da 4 membri, p14, p16, p18 e p19, chiamati anche INK4.
Il trattamento di varie linee cellulari tumorali con FANS induce l’arresto del ciclo cellulare in fase G1, riduzione dell’espressione delle cicline A, B e D, aumento dell’espressione di proteine
inibitrici del ciclo cellulare, quali p21waf1 e p27kip1, e dalla perdita di attività di CDK.
Il sulindac, il sulindac solfuro e solfone, agiscono proprio mediante tale meccanismo, che porta ad accumulare cellule in fase G0 /G1 e a ridurre i livelli di attività delle cicline-chinasi dipendenti (25).
• Citocromo c
Vari segnali apoptotici, tra cui la procaspasi 9 e i fattori che inducono apoptosi (AIF), possono indurre il rilascio di citocromo c, dal mitocondrio. Sono state identificate due famiglie di proteine che regolano l’apoptosi: Bcl-2 e Bcl-xL inibiscono l’apoptosi, mentre, Bax, Bad, Bid la promuovono.
La famiglia di Bcl-2 regola l’attivazione di enzimi proteolitici responsabili della morte cellulare, le caspasi. La formazione di pori sulla membrana mitocondriale favorisce la fuoriuscita del citocromo c ed è regolata dalle proteine della famiglia Bcl-2 e dall’attivazione della caspasi-9 e caspasi-3.
I FANS agiscono mediante una riduzione dell’espressione delle proteine antiapoptotiche Bcl-2, Bcl-xL e Mcl-1, favorendo un aumento delle proteine proapoptotiche Bad, il rilascio di citocromo c, l’attivazione di Apaf-1 e delle caspasi 3, 8 e 9 (26).
• Inibizione dell’anidrasi carbonica
L’anidrasi carbonica è un enzima che catalizza la conversione di anidride carbonica e acqua in acido carbonico, che a sua volta si dissocia in ioni H+ e ioni bicarbonato. È caratterizzata dalla presenza dello ione zinco nel sito attivo.
Tale enzima è stato studiato in particolare nel tumore intestinale e pancreatico, dal momento che riveste un ruolo nella carcinogenesi legato ad una incontrollata proliferazione cellulare e all’invasione tessutale da parte di cellule maligne.
Le prostaglandine PGE1, PGE2 e PGI2 inibiscono l’anidrasi
carbonica sia in vitro che in vivo, mentre PGF2α, TXA2 e i
leucotrieni LTB4 e LTC4, aumentano l’attività dell’enzima.
L’inibizione dell’anidrasi carbonica ha un ruolo nell’attività antitumorale dei FANS (27).
1.4 Angiogenesi
L’angiogenesi rappresenta un insieme di processi funzionali finalizzati alla formazione di nuovi vasi sanguigni a partire da quelli preesistenti. I vasi sanguigni sono costituiti da cellule endoteliali, a diretto contatto con il sangue, periciti subendoteliali, cellule della muscolatura liscia, fibroblasti, membrana basale e matrice extracellulare.
La formazione di un vaso passa attraverso stadi ben definiti, caratterizzati da modificazioni dell’endotelio e della matrice
extracellulare. Si suddividono quattro tappe nel processo angiogenico:
1. Destabilizzazione dei vasi preesistenti in seguito ad un aumento della permeabilità vasale e ad una perdita delle connessioni tra cellule endoteliali.
2. Migrazione e proliferazione delle cellule endoteliali in una zona del tessuto dove è necessaria la formazione di nuovi vasi. In questa fase occorre la liberazione di enzimi proteolitici che modificano la matrice extracellulare, facilitando la migrazione delle cellule endoteliali.
3. Differenziazione delle cellule endoteliali caratterizzata da un arresto della proliferazione cellulare e dalla formazione di capillari primitivi.
4. Richiamo di cellule subendoteliali di supporto quali i periciti e le cellule della muscolatura liscia. Avviene così una riorganizzazione delle interazioni cellulari.
L’angiogenesi è di fondamentale importanza in molti processi sia fisiologici che patologici, come il tumore. Nelle prime fasi della crescita il tumore utilizza sia per il rifornimento di ossigeno e nutrienti, sia per l’eliminazione di sostanze nocive, il letto di capillari proprio del tessuto di origine. Tale struttura non è più sufficiente quando il tumore supera i 2-3 mm.
1. VEGF (vascular endothelial growth factor) e Angiopoietina
2. Chemochine e citochine
3. TNFα (tumor necrosis factor) e TGFβ (trasforming growth factor)
Vari fattori inducono l’espressione di VEGF: – condizioni di ipossia
– il fattore di trascrizione HIF-1, il TNFα , il TGFβ e l’EGF (epidemal growth factor)
– i mediatori della risposta infiammatoria quali IL e PGE2
– IGF-1 (insulin-like growth factor)
– l’attivazione delle proteine chinasi C ed A – oncogene RAS
– le proteine regolatrici del ciclo cellulare quali p53 e VHL (soppressore del gene di Von Hippel-Lindau).
VEGF si lega a due classi di recettori della famiglia delle tirosin chinasi: FLT-1, recettore con maggiore affinità espresso nelle cellule endoteliali vascolari, e FLK-1, recettore con minore affinità identificato su cellule endoteliali tumorali e monociti.
La formazione di nuovi vasi sanguigni e capillari è essenziale per la crescita e la formazione di metastasi nei tumori solidi. Gli effetti sulle cellule endoteliali sono mediati da VEGF e da bFGF (basic fibroblastic growth factor).
Sia la Cox-1 che la Cox-2 sono importanti per l’angiogenesi, infatti, i FANS hanno dimostrato di possedere un’azione antiangiogenica. Il meccanismo d’azione sembra essere dovuto ad un effetto diretto di blocco dello sviluppo delle cellule endoteliali, mediante l’inibizione della proteina chinasi ERK2.
L’interferenza nella traslocazione a livello nucleare di ERK, è indipendente dalla proteina chinasi C, ed ha una componente prostaglandina-dipendente e una prostaglandina-indipendente. È stato inoltre evidenziato un meccanismo Cox-indipendente, tipico dell’aspirina, che consiste nella riduzione dei livelli di VEGF che porta all’inibizione della crescita delle cellule endoteliali, all’aumento dell’apoptosi e alla riduzione dell’angiogenesi.
L’effetto anti-angiogenetico del sulindac è stato dimostrato in vitro su cellule endoteliali umane (HUVEC). Il sulindac agisce mediante l’inibizione di VEGF e Ang-1 riducendo in questo modo la fosforilazione di residui critici per l’attivazione di ERK1/2, p38 e Akt.
Anche il blocco di sGC (guanylyl cyclase), e la conseguente diminuzione della produzione di cGMP comportano una riduzione dei livelli di VEGF. Tale effetto determina una riduzione della migrazione delle cellule della membrana basale e della proliferazione.
Ang-1 però determina un segnale di trasduzione e di regolazione della maturazione vasale, mentre Ang-2, è un antagonista naturale di Ang-1.
L’azione del VEGF nel promuovere la differenziazione e la proliferazione di cellule endoteliali e la formazione di vasi maturi, si integra quindi con quella prodotta dal complesso Ang-1/Tie2 il quale induce il rimodellamento e la stabilizzazione dei vasi neoformati (28).
1.5 Effetto chemiopreventivo dei
FANS
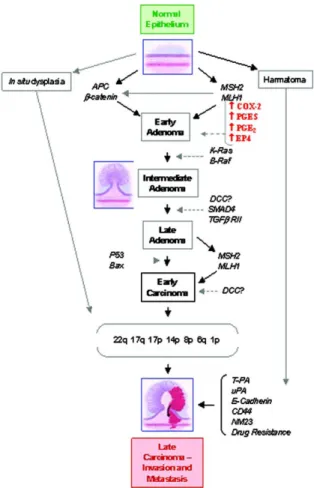
I FANS hanno dimostrato di esercitare un effetto chemiopreventivo nel cancro colorettale. Il sulindac e i suoi metaboliti, ad esempio, inducono una regressione dei polipi intestinali, neoformazioni benigne della mucosa che possono degenerare attraverso varie modificazioni cellulari (displasie), fino a diventare lesioni maligne (Fig. 11).
Figura 11: Evoluzione del tumore
Per questo motivo il loro utilizzo è stato proposto nei casi di poliposi adenomatosa familiare, una condizione ereditaria in cui, a causa di un gene alterato, l’intestino sviluppa numerosi polipi che arrivano a coprire tutta la mucosa (29).
Un aumento della Cox-2 dell’80-90% è stato osservato nelle neoplasie intestinali; tale modificazione biochimica promuove la sopravvivenza e la crescita cellulare, la migrazione, l’invasione e l’angiogenesi. In accordo con questa osservazione la PGE2
rappresenta uno dei fattori coinvolti nello sviluppo del cancro colorettale. È stato osservato, infatti, un aumento dell’espressione di PGES e quindi di PGE2 in cellule tumorali umane. La PGE2 viene
sintetizzata a partire da un precursore PGH2, (prodotto dall’acido
arachidonico mediante l’azione di Cox-1 o Cox-2) grazie all’intervento della PGES che catalizza la trasformazione di PGH2
in PGE2 (30).
Il secondo effetto chemiopreventivo è indipendente dalla biosintesi delle prostaglandine e riguarda un’azione legata alla riduzione del potenziale cancerogeno di agenti chimici, svolto a vari livelli.
Gli agenti procancerogeni vengono infatti metabolizzati mediante un prima fase ossidativa dovuta all’azione del CYP450, che porta alla formazione di epossidi. Tali prodotti del metabolismo di fase 1, vengono detossificati da enzimi di fase 2 quali l’epossido idrolasi, che scinde il legame con l’ossigeno, portando alla formazione di un glicole etilenico.
Gli ossidrili possono poi legarsi all’acido glucuronico oppure interagire con il glutatione ridotto, formando dei complessi solubili che possono essere successivamente eliminati.
Il sulindac e i suoi metaboliti inducono un aumento della trascrizione del gene CYP1A1 e di conseguenza dei livelli di mRNA dell’enzima. La trascrizione del CYP450 è regolata da AhR,
una proteina citosolica che, quando attivata, subisce una traslocazione a livello nucleare dove lega il suo ligando (hydrocarbon nuclear traslocator). Questo eterodimero forma un fattore di trascrizione che lega una specifica sequenza nucleotidica XRE, che si trova nella regione promoter di vari geni coinvolti nel metabolismo degli xenobiotici, tra cui quello che codifica per il CYP450. Il sulindac agisce aumentando la capacità di AhR di legare XRE.
Nel metabolismo di fase 2, i FANS, intervengono inducendo l’attività di enzimi importanti per la detossificazione, mediante l’aumento dei livelli di mRNA. Gli enzimi coinvolti sono: NQO1, UGT1A1, GST-pi. L’intervento su tutte queste fasi del metabolismo, contribuisce ad una riduzione della trasformazione di procancerogeni in cancerogeni, con conseguente riduzione della probabilità di trasformazione neoplastica (31).
1.6 Principali FANS con effetto
antitumorale
Negli ultimi 30 anni sono stati compiuti numerosi studi che hanno evidenziato che i FANS possono essere utili per proteggere dallo sviluppo e progressione di diversi tipi di cancro inclusi quello alla
Parecchi studi epidemiologici hanno mostrato una importante associazione inversa tra l’assunzione di Aspirina ed altri FANS convenzionali (Piroxicam, Indometacina, Sulindac, Ibuprofene, Ketoprofene) e inibitori Cox-2 selettivi (Celecoxib) ed il rischio di cancro al colon retto.
O HO O O CH3 N CH3O COOH CH3 O Cl Aspirina Indometacina OH O O COOH CH3 Ibuprofene Ketoprofene COOH S O F N O N H N S OH O O Sulindac Piroxicam
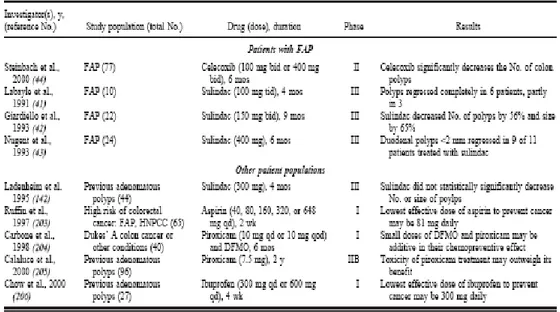
E’ stata dimostrata la loro efficacia nella riduzione di polipi in pazienti affetti da poliposi adenomatosa familiare (FAP) e nella riduzione della gravità del tumore in diversi modelli di colon retto
animale. E’ da sottolineare che la dose più alta tollerabile di questi farmaci può ridurre il numero e la grandezza dei polipi, ma non inibire del tutto la loro formazione (Fig. 12).
Altri studi basati su casi di donne in post-menopausa in età compresa tra 50-79 anni con nessuna storia di cancro alla mammella o altri tipi di tumori, sono stati eseguiti per valutare i fattori di rischio di cancro alla mammella e l’uso di FANS. I risultati mostrano che un uso regolare di FANS per 5-9 anni o superiore a 10 anni, è associato ad una riduzione del 21% e del 28% rispettivamente nell’incidenza di cancro alla mammella. In particolare lo studio mostra che, l’assunzione per un lungo periodo di ibuprofene fornisce una diminuzione maggiore rispetto all’aspirina di questa forma di cancro, mentre l’acetaminofene o basse dosi di aspirina (< 100 mg) non sono efficaci nella riduzione dell’incidenza di cancro alla mammella.
Parallelamente sono stati eseguiti studi che valutano la relazione tra l’uso di Aspirina/FANS ed il cancro all’esofago ed anche in questo caso è stato dimostrato il loro effetto chemiopreventivo legato ad una assunzione cronica.
Una simile associazione inversa è stata descritta anche per altri tipi di cancro, inclusi quello al polmone, all’ovaia ed alla prostata (30). Da questi studi sono state ricavate tre ipotesi fondamentali:
riduzione del tumore del 30% se il trattamento viene cominciato subito dopo l’esposizione al cancerogeno ma soltanto del 12% se viene cominciato 23 settimane dopo tale esposizione.
2- Sia FANS selettivi che non selettivi inibiscono i primi stadi dello sviluppo di un tumore, ma soltanto i Cox-2 selettivi sono efficaci se il trattamento avviene nelle fasi successive. Ad esempio il celecoxib riduce l’incidenza e la molteplicità dei tumori di circa la metà, anche quando il trattamento avviene nelle fasi più tardive.
3- Il trattamento con i FANS deve essere continuativo e senza interruzione per prevenire un’ eventuale ripresa della crescita del tumore (32).
Il limite all’impiego di FANS nella chemioprevenzione è rappresentato dagli effetti collaterali legati ai loro meccanismi d’azione.
I FANS non selettivi come aspirina, piroxicam, ibuprofene, indometacina, sulindac, ketoprofene provocano effetti gastrolesivi dovuti principalmente all’inibizione della Ciclossigenasi-1. L’uso cronico di questi farmaci può portare a dispepsia, nausea, vomito, fino ad ulcerazioni e sanguinamenti gastrici e nefrotossicità. I FANS Cox-2 selettivi come i Coxib invece presentano un miglior profilo d sicurezza gastrointestinale a confronto con i FANS classici ma recenti studi hanno dimostrato per essi elevati rischi cardiovascolari.
Figura 12: Studi clinici pubblicati sull’uso dei FANS sulla poliposi adenomatosa colorettale
1.7 Sulindac
Il sulindac è un FANS derivato dell’indene, acido (Z)-5-fluoro-2-metil-1[(4-metilsulfinil) fenilmetilen]-1H-inden-3-acetico, che esplica un’azione antinfiammatoria, analgesica e antipiretica. Viene somministrato per via orale alla dose di 150-200 mg. Studi sia biochimici che farmacologici hanno evidenziato che l’attività del sulindac è dovuta al suo metabolita solfuro. Prima
un’ossidazione irreversibile a solfone che è il metabolita inattivo (Fig.13).
Figura 13: Metabolismo del sulindac
Circa il 50% della dose somministrata viene eliminata per via urinaria sotto forma di solfone, il 25% si trova nelle feci come solfuro e solfone. L’eliminazione urinaria nella forma biologicamente inattiva, riduce gli effetti collaterali a livello renale, dovuti all’azione sulla Cox-2. L’emivita (t1/2) è di 7,8 ore e di 16,4
ore rispettivamente, per il sulindac e per il suo metabolita attivo. L’attività del sulindac sulla Cox è dovuta all’azione del metabolita solfuro, il quale agisce mediante una potente inibizione di entrambi gli enzimi Cox-1 e Cox-2; il solfone, invece, manca dell’attività inibitoria sulle ciclossigenasi.
L’attività antitumorale di questi farmaci è particolarmente evidente a livello intestinale (riduzione della poliposi intestinale del 56%), ed è associata alla riduzione dei livelli di PGE2, e ad altri meccanismi
Cox-2 indipendenti. Infatti, sia il sulindac solfuro che il sulindac solfone inducono apoptosi indipendentemente dall’espressione della Cox-2 (33).
Gli studi in vitro consentono di valutare separatamente l’azione del sulindac solfone e del sulindac solfuro e quindi di indagare i diversi meccanismi con cui queste molecole esercitano la loro attività antitumorale.
Il sulindac solfuro agisce mediante meccanismi Cox-dipendenti e Cox-indipendenti; il sulindac solfone, esclusivamente con meccanismi Cox-indipendenti.
I meccanismi Cox-indipendenti su cui agisce il solfuro riguardano fondamentalmente l’inattivazione di NF-kB, mentre il solfone ha un importante ruolo pro-apoptotico dovuto all’inibizione di PDE2 e
PDE5. Questa inibizione induce un aumento dei livelli di cGMP,
che portano ad una degradazione delle β-catenine e all’attivazione di c-Jun (34).
Sia il solfuro che il solfone inducono un arresto del ciclo cellulare in fase G0/G1 .