1
CAPITOLO 1
INTRODUZIONE
1.1 Reazioni di Nitroso Diels-Alder (NDA) con Acil- e Aril-nitroso derivati
Le reazioni di cicloaddizione di Diels –Alder permettono di sintetizzare molecole mono- o policicliche di natura anche complessa in maniera selettiva e stereocontrollata a partire da precursori semplici. In particolare, le reazioni di etero Diels-Alder con nitroso-derivati come dienofili 1 permettono l’introduzione simultanea e stereospecifica di legami carbonio-azoto e carbonio-ossigeno in un solo passaggio sintetico dando l’accesso diretto a 3,6-diidro-1,2-ossazine 2 (Schema 1.1).
Schema 1.1. Reazione di nitroso Diels-Alder
Gli aril-nitroso derivati 1a sono stati i primi ad essere scoperti e sono reagenti stabili che reagiscono lentamente con dieni in reazioni di cicloaddizione [4+2]. Gruppi elettron-attrattori sull’anello aromatico accelerano la reazione. Effetti simili sono stati osservati per i composti alchilnitroso α-sostituiti come specie cloronitroso e acetossinitroso 1b. I dienofili più reattivi risultano essere gli acilnitroso derivati 1c e gli esteri del C-nitrosoformiato 1d, in cui il gruppo nitroso è direttamente legato ad un sostituente elettronattrattore (Figura 1.1).
2
Dei nitroso derivati più utilizzati nelle NDA, il nitrosobenzene è l’unico ad essere reperibile in commercio come tale; per i composti tipo 1c e 1d, altamente instabili e reattivi, è necessario che siano sintetizzati in situ a partire dai corrispondenti acidi idrossammici. A questo scopo sono stati messi a punto numerosi metodi che prevedono l’utilizzo di una serie di agenti ossidanti: Sali organici ed inorganici di periodato, ipoclorito, PCC, NMO, o ancora l’ossidazione di Swern e con Dess-Martin periodinano.
Negli ultimi anni l’attenzione si è spostata su un approccio catalitico dell’ossidazione degli acidi idrossammici ai corrispondenti nitroso-derivati. L’interesse nasce dalla possibilità di ottenere l’addotto di NDA in tempi di reazioni brevi e in condizioni blande, oltre al fatto di poter evitare l’eccesso di agente ossidante e quindi avere un consumo di tutti i reagenti presenti nell’ambiente di reazione. A questo proposito è opportuno ricordare il lavoro di Adamo e coll.del 2006, che utilizza H2O2 come ossidante, metalli di uso commerciale e a basso costo Cu(I), Cu(II), Ni(II) e Fe(III)
come catalizzatori (5 mol%), la cui attività è incrementata dall’aggiunta di liganti come ammino-alcool o trietilammina (15 mol%), in THF o DCM come solventi. Questo permette di utilizzare 1 eq di BocNHOH e 1 eq del diene. La reazione avviene a temperatura ambiente, in tempi brevi (20-30 minuti) e con buone rese (Schema 1.2)1.
Schema 1.2. Ossidazione metallo-catalizzata di acidi idrossammici
Nell’ottica di sviluppare nuovi metodi di natura “green” che prevedano condizioni blande di reazione per l’ossidazione di acidi idrossammici, Whiting ha utilizzato CuCl2 (10 mol%) in
presenza di 2-etil-2-ossazolina (20 mol%) in aria a temperatura ambiente in MeOH, ottenendo la conversione quantitativa di acido idrossammico in cicloaddotto con buone rese isolate (Schema 1.3).2
3
Schema 1.3. Ossidazione rame-catalizzata in condizioni aerobiche.
Le NDA procedono con meccanismo concertato dello stato di transizione. È stato dimostrato in alcuni studi computazionali che lo stato di transizione endo è preferito rispetto a quello eso: quest’ultimo, infatti, presenta una forte repulsione tra il lone pair sull’atomo dell’azoto e gli elettroni π del diene. Vari studi hanno dimostrato che molti dieni non simmetrici si addizionano a nitroso composti regioselettivamente. La regioselettività dipende dalla natura del sostituente per cui gruppi fortemente elettron-attrattori o fortemente elettron-donatori portano alla formazione di cicloaddotti con maggiore regioselettività.
Lo sviluppo di NDA asimmetriche permette di ottenere importanti building blocks, come le 3,6-diidro-1,2-ossazine, con stereocentri aventi una ben definita configurazione.
Essenzialmente due sono gli approcci alle NDA asimmetriche: il primo è l’utilizzo di uno o entrambi i reagenti, diene e nitroso-derivato, otticamente attivi; il secondo, su cui si è focalizzata l’attenzione negli ultimi anni (ed è un campo ancora in via di sviluppo), si basa sulla catalisi asimmetrica, mediante l’aggiunta di un ligante chirale. Nel 2004 Yamamoto ed il suo gruppo di ricerca ha realizzato una NDA asimmetrica utilizzando il sistema Cu(I)-Segphos come catalizzatore e come dienofilo la 6-metil-2-nitrosopiridina 2, con l’ottenimento dell’ossazina 3 enantiomericamente pura. Il successo di questa reazione risiede nella chelazione esercitata dall’azoto piridinico che organizza il complesso catalitico(Schema 1.4). 3
4
Una reazione simile è stata analizzata anche da Studer in un lavoro del 2009 in cui utilizza come dienofilo la 2-nitrosopiridina. In questo caso si ottengono ottimi risultati utilizzando come ligante il Walphos-CF3, mentre non sono altrettanto buoni quelli ottenuti con Segphos4. Questo è
probabilmente dovuto alla mancanza dell’ingombro sterico del gruppo metilico in posizione 6, che rende più difficile l’accesso per la cicloaddizione (Schema 1.5, cfr es. b) e c)).
Schema 1.5. Nitroso Diels-Alder catalitica asimmetrica
L’importanza delle reazioni di acilnitroso Diels-Alder risiede nella ricca chimica dei risultanti prodotti di cicloaddizione, che possono essere elaborati attraverso quattro principali trasformazioni: cleavage del legame N-O, elaborazione del doppio legame, rimozione del gruppo protettore dall’azoto, apertura con differenti nucleofili o organometalli a carico del legame C-O.
Per quanto riguarda l’attinenza al lavoro di questa tesi, approfondirò esclusivamente il cleavage del legame N-O.
5
In letteratura sono riportate numerose procedure per la rottura del legame N-O, ad esempio Ni-Raney, litio alluminio idruro, Zn in acido acetico e l’idrogenolisi Pd catalizzata. Queste metodiche necessitano condizioni riduttive drastiche, spesso incompatibili con eventuali funzioni presenti nel substrato. Si è venuta a creare quindi la necessità di ricercare metodi più blandi di cleavage, che siano orientati esclusivamente sul legame N-O. Uno dei metodi più blandi per questo scopo è il molibdeno esacarbonile [Mo(CO)6], descritto per la prima volta nella sua funzione da Brandi e
coll.5 Un altro metallo di transizione, che permette condizioni di reazione blande, è il Ti(III), la cui fonte classica è il TiCl3 in HCl , che però ha molte limitazioni, sia per l’ambiente acido che si viene
a creare sia per la formazione di piccole quantità di Ti(IV) che complicano il work-up. La soluzione è stata recentemente trovata da Miller generando in situ titanocene cloruro dal dicloruro in presenza di Zn o Mg.6
In particolare il tandem NDA-cleavage riduttivo del legame N-O permette di ottenere i corrispondenti 1,4-cis-amminoalcoli, che rappresentano intermedi utili nella sintesi chimica, potendo essere ulteriormente elaborati per dare molecole dotate di attività biologica.
Kouklovsky sintetizza pirroli polisostituiti a partire dal prodotto di cleavage, attraverso una reazione di ossidazione per MnO2 a cui segue una reazione di ciclizzazione spontanea. Le
3.6-diidro-1,2-ossazine 4 e 5 sono sottoposte al cleavage del legame N-O mediato da Mo(CO)6 in
acetonitrile/acqua, con l’ottenimento dei corrispondenti (Z)-idrossicarbammati 6 e 7 come miscela di regioisomeri. Questi, ossidati con MnO2 in diclorometano, vanno incontro a ciclizzazione
6
Schema 1.6. Sintesi di pirroli polisostituiti da cicloaddotti di nitroso Diels-Alder
Streith utilizza in diversi lavori il tandem Nitroso-Diels-Alder/cleavage del legame N-O con lo scopo di ottenere gli intermedi 1,4-cis-amminoalcooli e da questi, con una serie di manipolazioni, derivati piperanosidici (o azazuccheri)8. L’interesse verso questi composti risiede nella loro alta capacità di inibire enzimi quali glicosidasi e glicosil-transferasi, trovando applicazione nella terapia del diabete mellito di tipo 2. Questi composti sono infatti analoghi strutturali degli zuccheri in cui l’ossigeno endociclico è sostituito da un atomo di azoto. La reazione di Nitroso-Diels-Alder sulla diidropiridina 9 come diene, condotta usando il nitrosobenzene come dienfilo porta selettivamente alla formazione del biciclo di tipo “diretto” 10, mentre nel caso del fenil-acetilnitroso si ottiene il biciclo di tipo “inverso” 13 (Schema 1.7). La reazione di cis-idrossilazione su i bicicli 10 e 13 con quantità catalitiche di OsO4 in presenza di un eccesso di NMO in una soluzione di acqua/acetone,
procede in maniera stereospecifica attraverso l’approccio anti dell’OsO4 rispetto al ponte
N-metilenico. La successiva idrogenolisi con H2-Pd/C porta alla formazione del derivato
piperidinosico ricercato. L’unico inconveniente è che le condizioni drastiche in cui viene condotta la reazione di idrogenolisi comporta, oltre alla ricercata rottura del legame N-O, anche la deprotezione dell’azoto esociclico 12 e 15.
7
Schema 1.7. Sintesi di derivati di azazuccheri a partire da addotti di nitroso Diels-Alder con
8
1. 2 Metodi di sintesi di 1,2-diidropiridine.
Semplici 1,2-diidropiridine (DHP) sono dieni elettronricchi in cui il sistema π è attivato dall’atomo di azoto eterociclico. Questi composti sono noti subire le reazioni caratteristiche delle enammine, nonché comportarsi come partners reattivi nelle reazioni di cicloaddizione.
A causa di questa loro versatilità potrebbero avere considerevoli potenzialità nella sintesi chimica. Tuttavia, le 1,2-DHP che non contengono sostituenti elettrondonatori sull’anello sono relativamente instabili rispetto a reazioni di dimerizzazione, polimerizzazione e ossidazione. Le 1,2-DHP si ottengono per addizione nucleofila all’anello piridinico; quest’ultimo, però, non è sufficientemente reattivo per ottenere il prodotto desiderato in buone rese, se non in presenza di sostituenti che attivino l’anello stesso.
Questo ha portato all’uso di piridine funzionalizzate sull’atomo di azoto in modo da generare sali di piridinio che risultano più elettrofili del corrispondente eterociclo non attivato. Inoltre, l’acidità del legame Cα-H è incrementata in queste specie, consentendo una più facile deprotonazione.
Quindi le reazioni vengono eseguite attraverso l’addizione di un reagente attivante alla miscela di piridina e oganometallo, in modo da ottenere il sale di piridinio in situ, fornendo uno dei metodi più lineari per la sintesi di diidropiridine.
Oltre a non richiedere uno stadio di preattivazione, i sali N-acil-piridinio sono molto più elettrofili rispetto agli N-alchil-derivati, ciò è dovuto ad una maggior attivazione del sistema π. Inoltre, la protezione del gruppo acilico sull’atomo di azoto aumenta la stabilità dei prodotti diidropiridinici. Fraenkel e coll. riportano che l’anello piridinico possa essere rapidamente attaccato da un reagente di Grignard in presenza di etil-clorofomiato ad ottenere 1-etossi-1,2-diidroiridina 2-sostituita.9 Lyle e coll. dimostrarono che anche i cloruri acilici, come acetilcloruro e benzoilcloruro, sono efficaci nell’attivare l’anello piridinico e renderlo sufficientemente elettrofilo per subire l’attacco del Grignard. In presenza di cloruri acilici o cloroformiati, la piridina esiste in equilibrio con il sale N-acil-piridinio che presenta siti elettrofili reattivi alle posizioni 2,4 e 6 dell’anello piridinico, così come al carbonio carbonilico (Schema 1.8).10
9
Dopo l’aggiunta di un nucleofilo organometallico si ottengono spesso miscele di diidropiridine 1,2- e 1,4-sostituite, mentre l’addizione al carbonio 1-acilico è comune in reazioni di catalisi nucleofila. Nonostante l’equilibrio possa essere spesso spostato verso la piridina non complessata, a seconda della natura del nucleofilo e delle condizioni di reazioni, la piridina reagisce solo lentamente con nucleofili organometallici, ed è proprio la presenza di sali di piridinio ad essere responsabile della formazione del prodotto desiderato.
Il modello HSAB (hard/soft acid/base) può essere utilizzato per razionalizzare la regioselettività dell’addizione di reagenti organometallici, e la scelta giudiziosa del nucleofilo, del cloruro acilico opportuno e delle condizioni di reazione, può fornire addizioni selettive alle posizioni C2 o C4: gli
organometalli soft, come gli organocuprati, generalmente si addizionano alla posizione C4; i
nucleofili hard, come i Grignard, alla posizione C2; i nucleofili very hard, come i derivati di
alchillitio (es. BuLi) si addizionano sul carbonio acilico.
Uno studio di Wanner e coll., ha dimostrato che, cambiando la natura del controione dal cloruro ad uno meno nucleofilo come il triflato (in particolare lo studio si è concentrato sull’utilizzo di quantità catalitiche di trimetilsilitriflato), si ottiene un significativo incremento della specie piridinio in soluzione all’equilibrio, ed una maggiore resa11
.
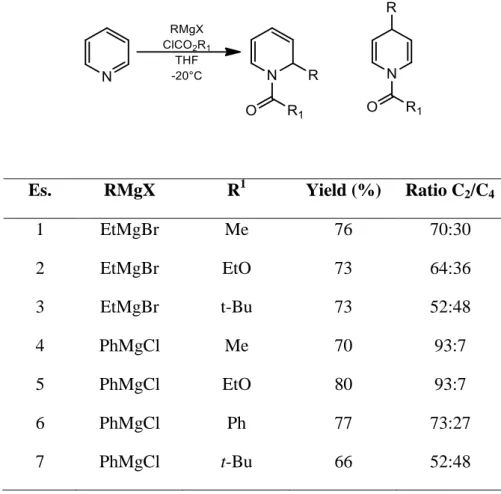
Un lavoro di Comins e Abdullah sull’addizione di reagenti di Grignard (alchilici e arilici) a ioni piridinio N-protetti con una serie di gruppi attivanti ha dimostrato che gli alchil-Grignard sono poco regioselettivi, al contrario dei corrispondenti aril-Grignard, che prediligono un attacco in posizione C2 (Tabella 1.1, cfr. es 1 e 4)12.
10
Tabella 1.1. Regioselettività dell’addizione di Grignard su N-acil-piridina attivata con gruppi aventi
diverso ingombro sterico
Es. RMgX R1 Yield (%) Ratio C2/C4
1 EtMgBr Me 76 70:30 2 EtMgBr EtO 73 64:36 3 EtMgBr t-Bu 73 52:48 4 PhMgCl Me 70 93:7 5 PhMgCl EtO 80 93:7 6 PhMgCl Ph 77 73:27 7 PhMgCl t-Bu 66 52:48
Inoltre, l’ingombro sterico dell’agente attivante legato all’atomo di azoto ha un ruolo chiave nella determinazione della regioselettività C2 vs C4. Infatti un reagente attivante di dimensioni maggiori
porta ad un aumento dell’ addizione 1,4 rispetto alla 1,2 (Tabella 1.1, cfr. es. 1-3 e 1-7).
Yamaguchi e coll., hanno esaminato sistematicamente la regioselettività dell’addizione di diversi nucleofili sull’ N-metossicarbonilpiridinio cloruro, dimostrando che:
a) Con alchil Grignard si ottengono miscele di 1,2- e 1,4-DHP in rapporti variabili in dipendenza del gruppo alchilico;
b) Con alchenil- e alchinil- Grignard, più forti degli alchil-derivati, la reazione procede in maniera altamente selettiva, portando esclusivamente alla formazione della 1,DHP 2-sostituita (Tabella 1.2., es. 7-9).
c) Reagenti più hard, come organo-litio derivati, hanno prodotto esclusivamente anidride pentanoica, probabilmente attraverso l’attacco al gruppo carbonilico del sale di piridinio (Tabella 1.2, es. 6).13,14
11
Come si può vedere dalla tabella 1.2, con MeMgI, che è un nucleofilo più forte rispetto al BuMgBr, attacca prevalentemente in posizione C2 (Tabella 1.2, cfr. es. 1 e 2).
Tabella 1.2. Reazioni dell’ 1-Metossicarbonilpiridinio cloruro con alchil-Grignard e altri reagenti organometalli
Es. R M Yield (%) Ratio C2/C4
1 Me MgI 0 54 92:8 2 Bu MgBr 0 41 78:22 3 Bu MgBr -78 99 67:33 4 i-Pr MgBr -78 99 19:81 5 Bu ZnCl 0 99 19:81 6 Bu Li -78 n.c. 7 CH2=CH- MgBr -78 81 >99:1 8 PhCC- MgBr -78 85 >99:1 9 TMSCC- MgBr -78 99 >99:1 10 CH2=CHCH2- SnBu3 0 87 94:6 11 CH2=CHCH2- MgBr 57 79:21
Fowler ha descritto un metodo fondamentale per la sintesi di diidropiridine non sostituite, in particolare della N-carbometossi-1,2- e 1,4-diidropiridina.15 Tale metodo prevede il trattamento di una miscela di piridina e sodio boro idruro (NaBH4) con metilcloroformiato. Sebbene questa
reazione possa essere condotta in vari solventi (etere, THF, metanolo, acqua) il tetraidrofurano, THF, risulta essere il solvente di scelta. Eseguendo la reazione in THF e mantenendo una temperatura al di sotto dei 10 °C, Fowler ottiene una miscela delle due DHP contenente il 35-40% dell’isomero 1,4 (Schema 1.9 a) Il sostituente carbossimetil- stabilizza la struttura diidropiridinica, rendendola più resistente all’ossidazione da parte dell’aria rispetto alle N-alchil-diidropiridine. Per l’interazione del lone pair sull’azoto con il gruppo carbonilico, il doppio legame carbonio-carbonio di queste diidropiridine ha poco carattere enamminico. Il sostituente carbometossi è anche un versatile gruppo funzionale in sintesi organica. Esso può sia essere convertito ad un derivato
H-12
sostituito, sia essere ridotto a metile, che è comune in prodotti naturali. Fowler ha, infatti, osservato che il trattamento di queste diidropiridine con LiAlH4 porta alla formazione dei poco stabili
N-metil-derivati (Schema 1.9 b). 15
Schema 1.9. Sintesi di Fowler delle 1,2- e 1,4-DHP.
Il metodo di Arndtsen16 è stato utilizzato per la sintesi di 1,2-diidropiridine-2-sostituite con alchini terminali. Questo è un metodo recente che permette di funzionalizzare le piridine anche in modo asimmetrico attraverso una reazione rame-catalizzata. La catalisi da parte del rame fa sì che le reazioni avvengano in tempi brevi e a temperatura ambiente (Schema 1.10). Considerando l’efficienza di questo coupling catalitico e la disponibilità di ciascuno dei materiali di partenza, questo fornisce un metodo lineare per sintetizzare derivati diidropiridinici. La reazione prevede che alla miscela di piridina e cloruro acilico/cloro formiato venga addizionato l’alchino, il catalizzatore (10mol% CuI, CuOTf·C6H6, o Zn(Otf)2) ed infine il ligante, iPr2NET.
Schema 1.10: Metodo di Arndtsen per la sintesi di 1,2-diidropiridine sostituite con alchini
13
1.3 Reazioni di Nitroso Diels-Alder con diidropiridine come dieni e loro
elaborazione.
A differenza dei molti lavori riportati in letteratura per nitroso Diels-Alder coinvolgenti dieni di natura ciclopentadienica e cicloesadienica, pochi sono gli articoli che descrivono le diidropiridine in qualità di dieni in queste reazioni.
La presenza dell’azoto adiacente al diene ne perturba la simmetria elettronica, questo fa pensare che si possano ottenere bicicli regioselettivamente. In realtà, come osservato da Knaus, è esclusivamente la scelta del gruppo legante il nitroso il responsabile della distribuzione dei prodotti. Facendo reagire la diidropiridina N-acilprotetta con nitrosobenzene, si ottiene selettivamente il regioisomero 16, che lo stesso Knaus definisce di tipo “diretto”. Quando il gruppo nitroso è legato ad un acile, la regioselettività del cilcoaddotto 17 è opposta alla precedente e per questo viene definito di tipo “inverso”. Con nitroso formiati e carbamoil nitroso derivati si ottiene una miscela dei due regioisomeri 18a e 18b (Schema 1.10).17,18
Schema 1.10. Regioselettività di reazioni di nitroso Diels-Alder coinvolgenti 1,2-DHP acil protette.
Come descritto da Streith la regioselettività è razionalizzata in termini di FMO (frontier molecular orbitals): l’effetto elettronico esercitato dal gruppo legante il nitroso modula i coefficienti orbitalici dell’ossigeno e dell’azoto a tal punto che questa differenza si trasferisce sulla distribuzione dei prodotti19. Più in dettaglio:
14
nel LUMO del nitrosobenzene, i coefficienti orbitalici sono maggiori sull’atomo di azoto, che quindi dirige l’attacco diretto;
nel LUMO degli acilnitroso derivati, i coefficienti sono, al contrario, maggiori sull’atomo di ossigeno, a causa del gruppo elettron-attrattore legato, portando ad una regioselettività opposta e prediligendo quindi un attacco di tipo inverso.
Nel caso dei carbamoil nitroso derivati, come ad esempio Cbz- o Boc-nitroso, i coefficienti orbitalici degli atomi di ossigeno e azoto sono comparabili e difatti si ottengono miscele dei due regioisomeri.
Negli studi 1H NMR condotti da Knaus si osserva spesso la presenza di rotameri in questo tipo di addotti aventi legami carbammici o ammidici. Per esempio, sono visibili segnali appartenenti alla molecola che risuonano per campi diversi a causa della rotazione dell’acetile (Schema 1.11, cfr 20a e 20b). I cambiamenti a carico dei chemical shift sono associati quindi alla prossimità degli idrogeni all’ossigeno acilico e all’effetto di schermo/deschermo su questi. La possibilità di osservare i rotameri è data dalla struttura rigida del biciclo e, quindi, dall’ostacolata rotazione interna
La presenza di rotameri è confermata dal studi 1H NMR condotti a caldo, in cui si ha coalescenza dei segnali, che tornano alla situazione iniziale dopo raffreddamento.
Risentono dell’effetto di anisotropia diamagnetica esercitata dal carbonile il C1-H, il C5-H e il
metile (R1=Me) o metossicarbonile (R1=OMe) legato al pendaglio acetilico sull’N6 (Schema 1.11).17
Schema 1.11. Rotameri presenti nel cicloaddotto
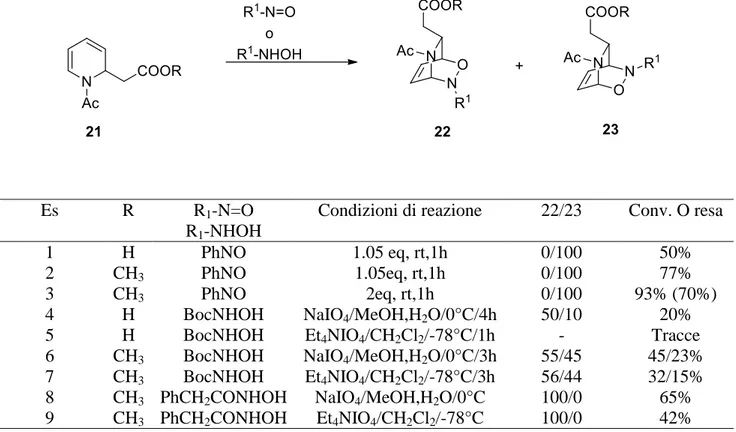
Precedenti studi compiuti nel laboratorio in cui ho svolto il presente lavoro di Tesi sperimentale condotti sulla N-acetill-2-metossicarbonilmetil-1,2-diidropiridina 21 rappresentano un’ulteriore dimostrazione della dipendenza della regio- e stereoselettività delle NDA esclusivamente dalla natura del gruppo legante il nitroso, e non dai sostituenti sull’anello diidropiridinico, confermando ulteriormente le previsioni di Streith e Knaus (Tabella 1.3).
Come si evince dalla Tabella 1.3, con nitrosobenzene è stato ottenuto in entrambi i casi esclusivamente l’addotto di tipo diretto (Tabella 1.3, es 1,2,3); con acilnitroso derivati, in questo caso PhCH2CONHOH, è stata osservata la sola formazione del biciclo di tipo inverso, con rese
15
diverse a seconda dell’ossidante utilizzato, trovando migliori le condizioni in MeOH/H2O (esempi 8
e 9); con derivati carbammici, come BocNHOH, invece, sono state ottenute miscele dei due regioisomeri ed anche in questo caso con rese migliori in condizioni MeOH/H2O (esempi 6 e 7).
Anche il CbzNHOH fornisce risultati in linea con quello già osservato, portando ad una miscela dei due regioisomeri, come ci si aspetterebbe dai dati presenti in letteratura in nostro possesso.
Tabella 1.3. Risultati della regioselettività della reazione di nitroso Diels-Alder sulla DHP 21
ottenuti precedentemente nel nostro laboratorio.
Es R R1-N=O
R1-NHOH
Condizioni di reazione 22/23 Conv. O resa
1 H PhNO 1.05 eq, rt,1h 0/100 50%
2 CH3 PhNO 1.05eq, rt,1h 0/100 77%
3 CH3 PhNO 2eq, rt,1h 0/100 93% (70%)
4 H BocNHOH NaIO4/MeOH,H2O/0°C/4h 50/10 20%
5 H BocNHOH Et4NIO4/CH2Cl2/-78°C/1h - Tracce
6 CH3 BocNHOH NaIO4/MeOH,H2O/0°C/3h 55/45 45/23%
7 CH3 BocNHOH Et4NIO4/CH2Cl2/-78°C/3h 56/44 32/15%
8 CH3 PhCH2CONHOH NaIO4/MeOH,H2O/0°C 100/0 65%
9 CH3 PhCH2CONHOH Et4NIO4/CH2Cl2/-78°C 100/0 42%
Un altro dato interessante fornitoci da Knaus riguarda l’instabilità del biciclo di tipo diretto 24, il quale risulta essere abbastanza labile da subire l’attacco dell’acqua durante il processo di work-up e purificazione con conseguente apertura del biciclo e formazione del prodotto 25 (Schema 1.12).
16
Inoltre Knaus aveva osservato come il biciclo di tipo inverso 26 si converte lentamente nel composto 29 avente una struttura diossazinica mediante una trasposizione [3.3] sigmatropica. Questo forniva agli autori un ulteriore prova della correttezza della regiochimica assegnata visto i bicicli di tipo diretto 27 non riescono a riarrangiare a diossazine di tipo 29 (Schema 1.13). Secondo gli autori anche i regioisomeri di tipo inverso 28 non reagivano a causa della diminuzione dell’effetto elettron-attrattore del carbonile legato a N3. Questo è dovuto all’effetto mesomerico
dell’atomo di ossigeno legato al carbonile all’interno di una struttura carbammica.
Schema 1.13. Trasposizione [3.3] sigmatropica dei vari cicloaddotti.
Nel laboratorio in cui ho svolto la presente Tesi sperimentale erano state condotte prove di cleavage del legame N-O di cicloaddotti di tipo inverso impiegando per questo scopo il Mo(CO)6 (Schema
1.14). Infatti, in letteratura erano riportati molti esempi che impiegano questo reattivo per effettuare riduzioni del legame N-O su strutture analoghe ai nostri composti per fornire i corrispondenti ammino alcoli. Nel nostro caso i risultati attesi prevedevano la presenza di un doppio legame in posizione β rispetto all’azoto piperidinico come riportato nel composto 30 (Schema 1.14).
17
Schema 1.14. Prodotto atteso dalla reazione di cleavage del legame N-O.
In realtà era stato osservato che il prodotto ottenuto con il Mo(CO)6 in CH3CN/H20 aveva una
struttura completamente diversa e si trattava di una eneacetammide di tipo 31 che necessariamente doveva essere ottenuta tramite un riarrangiamento di tipo [3.3] sigmatropico seguito da un processo di idrolisi (Schema 1.15). Si poteva quindi ammettere che, diversamente da quanto riportato in letteratura,17 era possibile avere un processo di riarrangiamento [3.3] sigmatropico anche partendo da un cicloadddotto ottenuto da una specie nitroso carbammica, quale il Boc-NO. Inoltre, non sembrava possibile isolare l’ossazina intermedia (composto 32, Schema 1.15) in quanto questa si idrolizzava nell’ambiente di reazione.
Schema 1.15. Plausibile meccanismo di formazione dell’eneacetammide 31.
A questo punto poteva risultare interessante verificare se questo tipo di reazione potesse avere carattere generale e potesse essere impiegata per cicloaddotti “inversi” con sostituenti variabili. Infatti, la possibilità di ottenimento di composti di tipo 31 poteva essere interessante dal punto di vista sintetico in quanto tale composto comprende due elementi strutturali rilevanti:
a) la presenza di un doppio legame attivato dall’azoto (eneammide o enecarbammato) che riveste un ruolo importante in una varietà di trasformazioni sintetiche.20,21
18
b) La presenza di una funzionalità ossidrilica in posizione C-4 all’interno di una piperidina è di una certa importanza per la sintesi di alcaloidi dotati di attività biologica22.
Partendo da questi due presupposti di interesse sintetico per questo tipo di composti, risultava interessante una esplorazione quanto più dettagliata dei sostituenti e gruppi protettivi in grado di fornire risultati analoghi a partire da cicloaddotti di natura diversa. Scopo principale di questa tesi sperimentale è infatti quello di verificare i requisiti strutturali necessari per fornire eneacetammidi endocicliche a sei termini avente una funzionalità ossidrilica ortogonalmente protetta in posizione C4 a partire da addotti di nitroso Diels-Alder di natura “inversa”.