INTRODUZIONE
GENERALE
6
TRASPORTATORI ABC: ATP-BINDING CASSETTE
Al giorno d’oggi le proteine ATP binding cassette (ABC transport) ancora rappresentano un’ impegnativa ricerca sul campo a causa della loro ampia distribuzione in alcuni distretti critici coinvolti nella protezione dei tessuti specialmente nel sistema nervoso centrale. La superfamiglia di proteine transmembranali ATP-dipendenti, detta ATP-binding cassette (ABC), rappresenta la più ampia famiglia di trasportatori presenti a livello umano. Attualmente sono noti 49 geni per i trasportatori ABC, raggruppati in 7 sottofamiglie: ABCA, ABCB, ABCC, ABCD, ABCE, ABCF, ABCG che differiscono per l’organizzazione dei domini aminoacidici. Sono proteine transmembrana capaci di regolare il passaggio di diverse tipologie di sostanze attraverso numerose barriere fisiologiche :
Barriera Emato-Encefalica (BEE)
Placenta
Intestino
Stomaco
Fegato
Tali proteine attivano il trasporto di substrati chimicamente diversi attraverso i doppi strati lipidici delle membrane cellulari. Esempi clinicamente rilevanti sono le proteine umane MDR1( nota anche come ABCB1 o P-glicoproteina) e MRP1( o ABCC1), che contribuiscono all’instaurarsi della resistenza multipla ai farmaci nelle cellule tumorali andando, quindi, a catalizzare l’estrusione di composti citotossici utilizzati nella terapia del cancro.
7
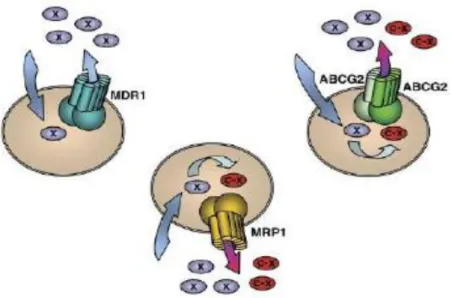
Figura 1. Funzione del trasportatore multidrug/xenobiotico ABC. Il trasportatore ABC risiede nella membrana plasmatica e estrude vari prodotti
xenobiotici e metabolici idrofobici e/o anfipatici. MDR1/Pgp trasporta composti idrofobi (X), mentre MRP1 e ABCG2 può estrudere entrambi i
farmaci idrofobi e metaboliti intracellularmente formate, ad esempio, glutatione o coniugati glucuronide (CX).
Questi trasportatori sono sovraespressi nelle cellule tumorali e sono responsabili dell'efflusso del farmaco fuori dalla cellula. Inoltre possiedono la capacità di interagire con una vasta gamma di substrati, strutturalmente e funzionalmente, molto diversi fra loro (per esempio, metaboliti, lipidi, steroli, farmaci).
8
P-GLICOPROTEINA (P-gp)
Espressione e attività della P-gp
Tra le numerose proteine trasportatrici ABC, la ABCB1, nota anche come P-Glicoproteina (P-gp), ha suscitato un grande interesse a causa del suo ruolo in alcune patologie neurodegenerative quali Parkinson, Alzheimer e in molti processi tumorali.
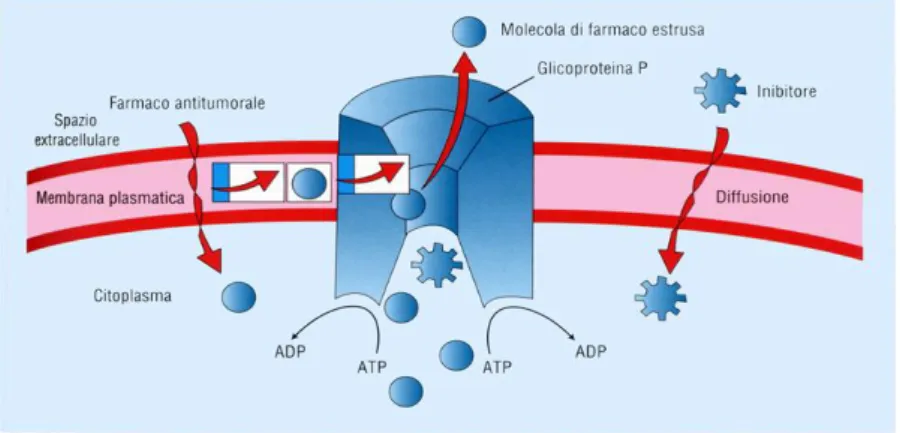
Figura 2. Rappresentazione schematica dell’attività della P-Glicoproteina per l’estrusione dei chemioterapici.
Sia l’espressione che l’attività della P-gp sono influenzate da numerosissimi segnali, oltre che fisiologici anche patologici, che possono o aumentare o ridurre la sua attività di trasporto. Per la P-gp, è noto come la sua espressione sia influenzata dall’azione di ben 49 geni, fra i quali, il più importante è sicuramente l’MDR1, il cui analogo umano è l’hMDR1: essi agiscono da promotori1. La regolazione avvienegrazie all’azione di molte altre proteine che regolano il suo processo trascrizionale.Finora, sono stati caratterizzati diversi elementi della zona del promotore come laGC-box, la Y-box, l’elemento p53, l’elemento X del recettore del Pregnano (PXR)1 . Questi sono siti di legame per diversi fattori di trascrizione i quali rispondono a diversi stimoli ambientali come: l’ipossia, gli xenobiotici, l’infiammazione, lo stressossidativo. Inoltre, anche meccanismi epigenetici,
9
quali l’acetilazione degli istoni o la metilazione del DNA, influiscono pesantemente sulla espressione dell’hMDR11
. È, poi, possibile un controllo post-traduzionale, grazie all’azione di numerosi segnali intracellulari, che, senza influenzare direttamente l’espressione del gene, possono darluogo a numerosi meccanismi di modulazione1. Ad esempio, la fosfo-/defosforilazione della proteina, la degradazione, la sua associazione con altre proteine di membrana.A monte dell’estremità 5’dell’hMDR1, si trova un complesso regolatore, formato dadiversi siti di legame per il recettore nucleare ligando-dipendente : il PXR o recettore “X” del pregnano1. Questo è in grado di mediare l’induzione della trascrizione su MDR11
. Il PXR, fa parte della superfamiglia dei fattori di trascrizione ligando-dipendenti. È attivato, oltre che dagli steroidi naturali (es., pregnenolone, progesterone) e sintetici (antiglicocorticoidi e glucicocorticoidi), anche da molti altri xenobiotici (farmaci, sostanze tossiche). I suoi substrati variano a seconda della sua localizzazione nei vari distretti dell’organismo. Per quanto riguarda la sua relazione con la P-gp, vari studi hanno evidenziato un’azione diretta del PXR sia sull’espressione che sull’attività di trasporto della proteina1. È stato evidenziato che l’espressione della P-gp, nell’epilessia e nell’ictus, aumenti, ma non è ancora chiaro se tale aumento sia la diretta conseguenza dell’attivazione del PXR o di altri recettori nucleari da parte di ligandi endogeni o farmaci1.
Struttura della P-gp
La P-gp è dotata di una specificità notevolmente ampia per numerosi composti, come tutti i transporters ABC esistono sottoforma di monomeri, la cui unità funzionale di base è formata da 4 domini fondamentali suddivisibili in :
2 domini Trans-membrana (TMD), che costituiscono l’estremità N-terminale
2 domini di Legame Nucleotidici (NBD), che costituiscono l’estremità C- terminale
10
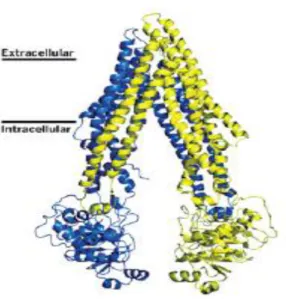
Figura 3. Struttura tridimensionale della P-gp
Sono, inoltre, presenti un linker e una cavità transmembranaria2. I TMD della glicoproteina sono costituiti da 12 α-eliche transmembrana che attraversano il doppio strato lipidico sei volte3 e sono responsabili della formazione del canale che permette il passaggio di un substrato, a seguito di un cambiamento conformazionale. La conformazione dei segmenti TMD ( TMD1 segmenti da 1 a 6, TMD2 segmenti da 7 a 12) e del sito di legame è variabile perché le sequenze amminoacidiche, nella porzione intramembranaria, sono poco conservate, ed è questa caratteristica che consente il legame di substrati strutturalmente diversi. I domini NBD sono localizzati nella parte citoplasmatica della membrana, hanno sequenze amminoacidiche altamente conservate e presentano due siti di legame per l’ATP.
Meccanismo d’azione
La P-gp è sovra-espressa in diverse linee cellulari tumorali ed è responsabile dell’efflusso dei farmaci al di fuori della cellula4. Utilizza l’energia
dell’idrolisi dell’ATP per estrudere i composti grazie ad un complesso processo di trasporto5.
11
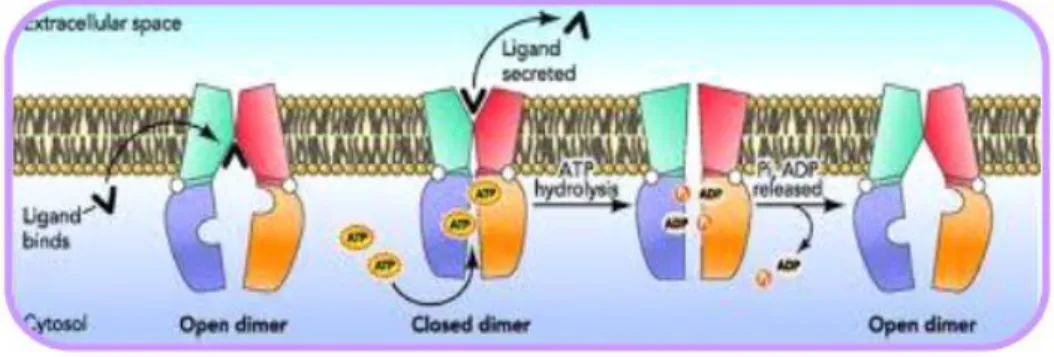
Figura 4. Meccanismo d’azione dei trasportatori ABC1
La fase iniziale del processo di trasporto coinvolge il sito di legame ad alta affinità per i farmaci e contemporaneamente il legame con ATP nei NBD. Il legame con il farmaco e l’ATP sono accoppiati; l’accoppiamento dei legami al substrato ed al nucleotide causa cambiamenti conformazionali del sito di legame, e si verifica il rilascio del farmaco. Per descrivere il meccanismo di trasporto sono stati suggeriti 3 modelli:
Modello a poro: i farmaci che legano la P-gp nel citosol vengono trasportati fuori attraverso una proteina canale6,7.
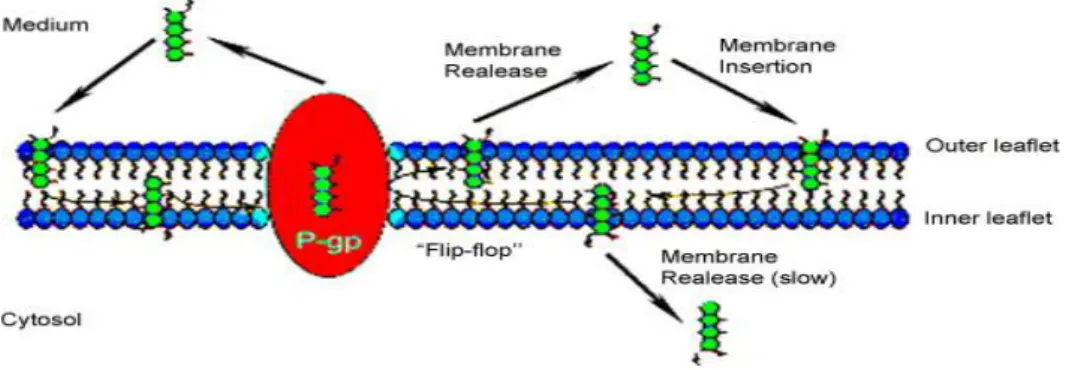
Modello flippasi: P-gp fa si che i farmaci sono trasportati dal compartimento citoplasmatico interno a quello esterno della membrana plasmatica, contro gradiente di concentrazione. L’idrolisi dell’ATP fornisce l’energia per far muovere le molecole del foglietto citoplasmatico della membrana a quello esterno. Il substrato si disperde sul lato citoplasmatico della membrana e si lega alla glicoproteina; in seguito all’idrolisi dell’ATP la molecola viene “flippata” sullo strato esterno e diviene libera di rompere il legame dalla proteina.
Modello “hydrophobic vacuum cleaner”: le molecole riconosciute dalla P-gp nel doppio strato lipidico entrano nella proteina in un sito membranoso ed escono attraverso la cavità centrale, costituita dal ripiegamento di una o più subunità.
12
Figura 5. Rappresentazione schematica del modello “ flippase”.
Localizzazione della P-Glicoproteina
La sua funzione principale in tutti questi siti è di protezione dei tessuti nei confronti di sostanze tossiche. La P-gp viene espressa sulla superficie luminale degli organi adibiti all’assorbimento, alla distribuzione, al metabolismo e all’eliminazione dei farmaci. L’attività della P-gp viene espressa tramite tre vie:
limitazione dell’assorbimento del farmaco: per trasferimento del substrato dagli enterociti al lume intestinale e la sua successiva eliminazione con le feci attraverso l’interazione con il Citocromo P450;
eliminazione attiva: per trasferimento del substrato dalle cellule del tubulo prossimale al lume tubulare e la sua successiva eliminazione con le urine o per trasferimento del substrato dagli epatociti alla bile; limitazione della distribuzione del farmaco ai tessuti: per
trasferimento del substrato dall’endotelio celebrale e testicolare ai capillari sanguigni, o per trasferimento del substrato dai capillari fetali al lato materno della placenta e da qui al sangue materno8. La P-gp risulta essere espressa in numerose barriere, come la barriera emato-encefalica, la barriera sangue-fluido cerebrospinale, e la barriera sangue-testicolo. Anche attraverso queste barriere modula l’assorbimento e l’escrezione di xenobiotici9
13
a livello di compartimenti intracellulari; in particolare sembra agire a livello di vescicole citoplasmatiche, trasportando e concentrando all’interno di queste i farmaci-substrati, impedendo loro il raggiungimento del bersaglio intracellulare.
Coinvolgimento della P-gp nelle diverse membrane fisiologiche P-gp e attività del CYP
La penetrazione del farmaco attraverso l'intestino tenue è influenzata dalla presenza della pompa di efflusso P-gp e l'enzima CYP3A4 , che modifica la biodisponibilità e la metabolizzazione dei farmaci. P-gp e l’enzima CYP3A4 agiscono sinergicamente per modulare il metabolismo pre-sistemico dei farmaci10. Il metabolismo intestinale e trasporto attivo potrebbero quindi influenzare il tasso di trasporto transepiteliale di farmaco, come le interazioni farmaco-farmaco e farmaco-cibo con conseguente diminuzione dell'efficienza del trasporto, o degli effetti indesiderati11. Il metabolismo dipende da molti fattori fisiologici, come la velocità di trasporto dei farmaci nelle cellule e l’integrità delle cellule12. Nell’uomo, la
sottofamiglia del CYP3A è composta da almeno 4 geni ed il CYP3A4 sembra essere il più importante, è espresso sia nel fegato che nell’intestino. I CYP3A rappresentano circa il 30% di tutti gli isoenzimi del CYP450 presenti a livello epatico ed essendo caratterizzati da una ampia specificità di substrato, contribuiscono al metabolismo di circa il 50% dei farmaci utilizzati. Il CYP3A4 è l’enzima maggiormente rappresentato a livello del tratto gastrointestinale, dove può essere responsabile del metabolismo di molti farmaci.
14
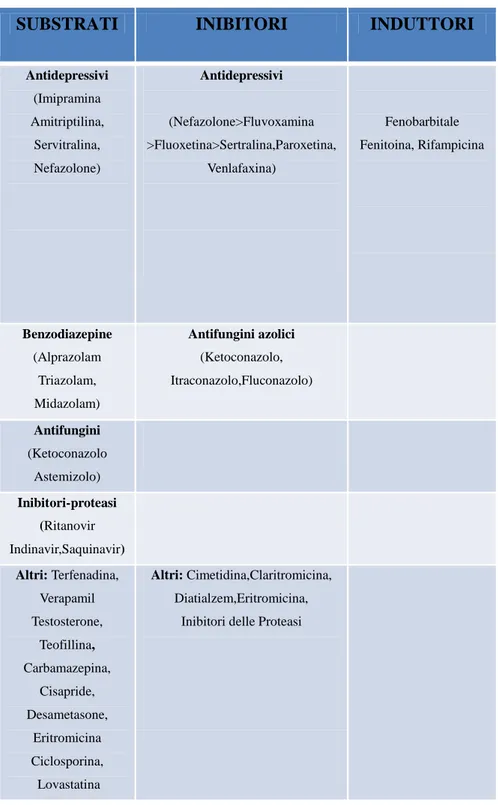
Figura 6. Tabella che indica alcuni dei substrati, inibitori ed induttori dell’enzima CYP450
SUBSTRATI INIBITORI INDUTTORI
Antidepressivi (Imipramina Amitriptilina, Servitralina, Nefazolone) Antidepressivi (Nefazolone>Fluvoxamina >Fluoxetina>Sertralina,Paroxetina, Venlafaxina) Fenobarbitale Fenitoina, Rifampicina Benzodiazepine (Alprazolam Triazolam, Midazolam) Antifungini azolici (Ketoconazolo, Itraconazolo,Fluconazolo) Antifungini (Ketoconazolo Astemizolo) Inibitori-proteasi (Ritanovir Indinavir,Saquinavir) Altri: Terfenadina, Verapamil Testosterone, Teofillina, Carbamazepina, Cisapride, Desametasone, Eritromicina Ciclosporina, Lovastatina Altri: Cimetidina,Claritromicina, Diatialzem,Eritromicina, Inibitori delle Proteasi
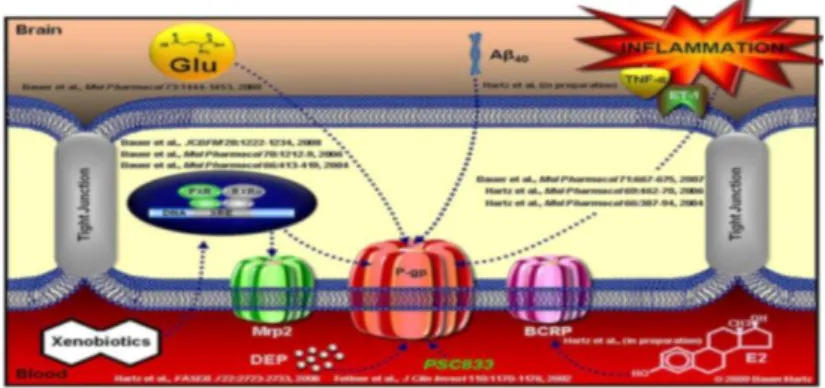
15 P-gp e la barriera emato-encefalica (BEE)
L’integrità della barriera viene controllata da astrociti, periciti e componenti della matrice extracellulare13. La barriera ematoencefalica(BEE) è una struttura anatomica costituita dalle cellule endoteliali dei capillari del Sistema Nervoso Centrale la cui funzione è quella di proteggere i tessuti cerebrali da elementi nocivi presenti nel sangue. La BEE ha il ruolo di proteggere il cervello da xenobiotici tossici mantenendo separato il cervello dalla circolazione cerebrale. Solamente composti xenobiotici e farmaci lipofili possono attraversare la barriera ed entrare nel cervello tramite diffusione semplice. Alcuni studi hanno dimostrato che esiste una relazione fra la lipofilicità di un farmaco e la sua permeabilità cerebrale14,15. Nonostante la lipofilicità sia un parametro importante per determinare la penetrazione di un farmaco nel cervello, molti composti lipofili mostrano avere una penetrazione cerebrale ridotta, ciò è dovuto all’azione della P-gp che riconoscendo lo xenobiotico, ne catalizza la immediata espulsione dal distretto cerebrale. Ne consegue che l’accumulo dei farmaci sia estremamente difficoltoso e ridotto. La P-gp sembra avere azione immuno-modulatoria poiché regola la secrezione di citochine da parte dei linfociti T e la migrazione delle cellule dendritiche verso i nodi linfatici, dai quali dipende l’immunità cellulo-mediata16. Grazie all’utilizzo di anticorpi
monoclonali è stato dimostrato che la P-gp è altamente espressa sulla superficie delle cellule endoteliali dei capillari cerebrali. Attualmente si ritiene che la ridotta penetrazione dei farmaci sia dovuta al trasporto mediato dalla P-gp17.
16 P-gp e la Placenta
Un’altra importante barriera sangue-tessuto è la membrana delle cellule trofoblastiche. Come altri organi, anche la placenta presenta espressioni di trasportatori di farmaci, inclusa la P-gp. Si ritiene ancora che questa glicoproteina contribuisca a proteggere il feto da involontarie esposizioni della madre a xenobiotici e ad eventuali terapie necessarie alla madre durante il periodo della gravidanza18,19. La proteina è presente per tutto il periodo della gravidanza, anche se sembra che la sua attività decresca progressivamente durante il corso della stessa.
P-gp e cellule neoplastiche : Fenomeno della Multidrug resistance Il trattamento del cancro è estremamente difficile a causa della grande varietà di mutazioni che possono innescarlo e, contemporaneamente, dei numerosi meccanismi che le cellule neoplastiche possono utilizzare per resistere all’azione dei farmaci. Questi meccanismi sono la diretta conseguenza di alcuni fenomeni quali :
Alterazione delle vie di segnalazione apoptotiche Amplificazione genica
Attivazione della riparazione del DNA
Iperespressione dei trasportatori che facilitano l’eliminazione dei farmaci (esempio P-gp)
Il principale ostacolo nel trattamento del cancro è rappresentato dalla “multidrug resistance”(MDR), ovvero resistenza a molteplici agenti antitumorali non correlati tra loro per struttura chimica e bersaglio molecolare. L’MDR può manifestarsi nelle cellule sane, nelle cellule alterate, in seguito ad una patologia, o nelle cellule neoplastiche; esplica la propria azione attraverso differenti meccanismi:
estrusione del farmaco dalla cellula (P-gp e altre proteine); riduzione della permeabilità;
17 alterazione dei siti di legame del farmaco; vie metaboliche alternative.
La P-gp, è di particolare rilevanza clinica in quanto questo trasportatore possiede un substrato esterno aspecifico da cui deriva il termine “multidrug transporter”, capace di legare una varietà di farmaci aventi struttura e meccanismo d’azione diversi20
. La P-gp, essendo espressa dai tessuti con funzioni escretorie (intestino, fegato e rene) e dalle barriere sangue-tessuto (barriera emato-encefalica, barriera sangue-testicolo e placenta), è in grado di limitare l’entrata dei farmaci all’interno del corpo dopo somministrazione orale e di promuoverne l’eliminazione attraverso la bile e le urine21
. Questo fenomeno di resistenza, quindi, risulta essere il maggior ostacolo quando vengono effettuate terapie indirizzate verso il sistema nervoso centrale, inclusa la chemioterapia per il cancro. La MDR mediata dalla P-gp gioca un ruolo clinico importante in molti tumori umani, ed è per questo che l’iperespressione di questa glicoproteina nelle cellule tumorali appresenta un bersaglio terapeutico importante al fine del superamento della resistenza.
18
LE MALATTIE NEURODEGENERATIVE
Molte malattie neurologiche hanno un carattere multieziologico e i correnti approcci farmacologici, che usano farmaci orientati verso un singolo obiettivo, sono spesso inefficaci. Quindi il progetto razionale e lo sviluppo di nuove strategie terapeutiche che s’indirizza simultaneamente verso diversi obiettivi coinvolti in questa patologie può offrire migliori benefici. Recentemente è stato dimostrato17,22 che la funzione del P-gp a livello cerebro-vascolare diminuisce negli stati avanzati di malattie neurodegenerative come Parkinson e Alzheimer. La disfunzione del P-gp sembrerebbe, quindi, contribuire al danno neuronale indotto dall’incremento di accumulo di tossine come succede nel Parkinson (PD) o dalla diminuita abilità del cervello di espellere le proteine accumulate lì come nell’Alzheimer (AD). Inizialmente in entrambe queste patologie neurodegenerative si verifica un’ up-regulation di espressione e funzione di P-gp seguito dal danno neuronale. Nel processo neuroinfiammatorio c’è un’attivazione estensiva di microglia che esprimono P-gp e anche altri fattori coinvolti in processi infiammatori come: tumori, necrosi, fattore alfa (TNF-α), interleuchine 6 (IL 6) , e ossidonitrico che possono aumentare l’attività del P-gp23,24
19 Alzheimer (AD) e P-gp
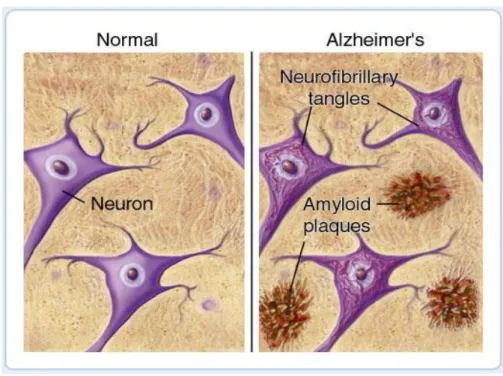
L’alzheimer (AD) è una malattia neurodegenerativa multifattoriale e eterogenea caratterizzata da una progressiva perdita delle funzioni cognitive, fattore primario che porta alla demenza e talvolta anche alla morte. Le principali peculiarità sono:
Accumulo di β-amiloide peptide (A-β) aggregati nel cervello che formano delle placche amiloidi indissolubili;
Grovigli di neurofibrille (NFTS) che consistono in coppie di
filamenti elicali di abnormi proteine τ- fosforitate.
Figura 8. Sezione di un cervello sano (a destra) e sezione di un cervello affetto da Alzheimer ( a sinistra)
Oggi sappiamo che la secrezione di β-amiloide è l’evento scatenante nella patogenesi dell’alzheimer, mentre l’aggregazione delle τ-proteine può essere considerato un importante fattore secondario implicato nella neurodegenerazione25,26. Le placche amiloidi sono principalmente composte dal peptide β-amiloide (A-β) che si forma dalla scissione proteolica della proteina precursore β-amiloide (APP). La prima scissione di APP è effettuata dall’enzima β-secretasi anche conosciuto come BACE. Successivamente la scissione del frammento c-terminale di APP dal γ
-20
secretasi genera Aβ peptidi di lunghezza di 40 e 42 amminoacidi, Aβ40 e Aβ42 rispettivamente. Questi peptidi si aggregano e si depositano formando delle placche senili e insolubili. Ad oggi l’inibizione di BACE rimane un obiettivo terapeutico attraente per il trattamento e la prevenzione dell’Alzheimer. Questo tipo di approccio rimane piuttosto infruttuoso perché troppo spesso gli inibitori BACE sembrano essere anche substrati della P-gp27,28. Recenti studi hanno dimostrato che la quantità di Aβ depositata nel cervello è inversamente connessa all’espressione di P-gp cerebrovascolare suggerendo così un ruolo chiave della glicoproteina-p nella sua eliminazione22,29,30. I peptidi Aβ possono essere eliminati dopo la degradazione proteolica, o attraverso il flusso passivo o tramite trasporto attivo attraverso la BEE. Con il progredire della malattia si verifica una diminuizione dell’ espressione di P-gp a livello della BEE, e ciò comporta una riduzione del rilascio di Aβ nel sangue inducendo così il suo accumulo all’interno del cervello. Anche se la possibilità di usare induttori di P-gp può essere un valido approccio per ridurre il livello di Aβ nel cervello negli stadi iniziali, durante gli stadi avanzati della patologia, la sovra-espressione di P-gp rappresenta il principale limite per l’efficacia delle terapie correnti farmacologiche.
21
Recenti studi hanno mostrato che molti inibitori di BACE1 che possono essere usati per abbassare la formazione di Aβ, sono efficaci solo ad alte dosi perché sono anche substrati di Pg-p. La co-somministrazione di inibitori di BACE1 assieme a inibitori di Pg-p induce una forte riduzione di Aβ nel cervello27
.
Morbo di Parkinson (PD) e P-gp
Analogamente ad AD , anche PD è una malattia neurodegenerativa attualmente incurabile. Si manifesta con la perdita di cellule dopaminergiche nella substantia nigra del mesencefalo e dal deterioramento dei neuroni catecolaminergici31 .
Figura 10. Morbo di Parkinson
I sintomi caratteristici della malattia sono: tremori, rigidità, lentezza dei movimenti e, negli stadi più avanzati, demenza e discinesia. Quest’ultimo sintomo, è caratterizzato da contrazioni involontarie della muscolatura volontaria che impediscono, sempre più, al soggetto di controllare i propri movimenti. Come nell’ AD, si osserva la presenza di aggregati proteici noti come corpi di Lewy (LBs). La patogenesi della PD è ancora sconosciuta32, tuttavia, un ruolo chiave sembra essere giocato dalla formazione di LBs composti principalmente da α-sinucleina, una proteina solubile neuronale
22
che in condizioni patologiche è convertita in oligomeri insolubili. Sebbene non sia ancora noto se l’ α-sinucleina sia un substrato della P-gp, è stato ampiamente dimostrato che la P-gp è in grado di espellere alcuni pesticidi33 e tossine considerate responsabili dell’inizio della malattia di Parkinson34
. MPP+, una neurotossina coinvolta nella sindrome, rappresenta un substrato della P-gp35; inoltre, pesticidi e metalli hanno recentemente dimostrato di interagire con α-sinucleina, promuovendo così la sua fibrillazione in vitro36
, e quindi inducendo sporadico PD37. Lo studio condotto in vivo indica che la funzione del P-gp al livello BEE diminuisce principalmente in regioni della materia bianca e nelle regioni orbito-frontale. Bartels ha dimostrato che la riduzione della P-gp dipende dall'età, e si verifica anche a livello di bulbi olfattivi, suggerendo una stretta relazione tra invecchiamento e la suscettibilità alla comparsa di PD38. Questi risultati evidenziano che la concentrazione celebrale e la conseguente tossicità di molti medicinali antiparkinson potrebbe essere dovuta al P-gp. Quindi, la co-somministrazione di un modulatore o un inibitore di questo tipo di trasportatore può essere un utile mezzo, o per provare l'efficacia della terapia antiparkinson, o per ridurre la tossicità molto spesso associata all'uso di queste medicine.
23 Epilessia e P-gp
L’ epilessia è una malattia neurologica che colpisce approssimativamente l’ 1-2% della popolazione mondiale ed è caratterizzata da un insieme di cronici e comuni disordini neurologici che, nella maggior parte dei casi, provocano convulsioni. Le convulsioni epilettiche, sono dovute ad una anormale ed eccessiva attività elettrica dei neuroni che fanno parte della sostanza grigia dell’encefalo. L’aggregato di neuroni dai quali parte la scarica epilettica è definito focolaio epilettogeno. I farmaci antiepilettici (AEDs) agiscono attraverso differenti meccanismi, accomunati da una comune causa scatenante che induce all’epilessia-farmaco resistente39
. Uno dei meccanismi che causano la resistenza AED è la limitazione dell’accesso di questi farmaci alla regione epileptogenica risultante da una sovra-espressione di trasportatori di efflusso come la P-gp a livello della BEE40. Recenti studi sono stati effettuati per valutare gli effetti della co-somministrazione di AEDs e inibitori della P-gp40,41,42, con lo scopo di valutare il coinvolgimento della glicoproteina-P nell’inizio dell’epilessia farmaco-resistente. Tutti questi risultati mostrano che la co-somministrazione di inibitori P-gp insieme a Fenobarbital o Fenitoina può migliorare le proprietà antiepilettiche di farmaci anticonvulsivi aumentando la locale concentrazione di AEDs attraverso l’interferenze con P-gp ,con un conseguente miglioramento del controllo degli spasmi in pazienti con epilessia refrattaria43.
Fenitoina Carbamazepina
24
In aggiunta alla sovra-espressione di P-gp in regioni del cervello epileptogenica e di pazienti con epilessia AED resistente44, recentemente è stato suggerito anche che diversi farmaci antiepilettici di prima linea sembrano essere substrati della glicoproteina-P45,41,46,47.
Figura 12. Legenda: (VPA: valproato di sodio, CBZ: carbamazepina, LTG: lamotrigina, PB: fenobaribital)
Terapia utilizzata all'esordio
VPA CBZ LTG PB25
Malattia di Huntington e malattia di Creutzfeldt-Jakob
La malattia di Huntington (HD) così come la malattia di Creutzfeldt- Jakob (CJD) condividono alcune caratteristiche con le malattie neurodegenerative precedentemente discusse. HD è una condizione neurodegenerativa caratterizzata dal disordine del movimento, declino cognitivo e disturbi psichiatrici.
CJD è la più comune forma trasmittibile all’uomo della encefalopatia spongiforme bovina che porta alla morte.
Figura 13. Morbo di Creuzfeldt.Jakob
La sindrome clinica è caratterizzata da perdita di memoria, cambiamenti di personalità, allucinazioni, disartria, mioclono, rigidità posturale e convulsioni. La principale caratteristica di entrambe le neuropatie è l’accumulo di proteine mal ripiegate all’interno del cervello. Il meccanismo con il quale le proteine huntigtine (HTT) inducono questo tipo di cambiamento a livello cellulare non è stato ancora chiarito48. È stato suggerito che piccoli aggregati di HTT mutate possono essere responsabile degli effetti tossici49 che portano all’inizio dell’HD. Al giorno d’oggi il coinvolgimento della P-gp in questa patologia non è stato ancora investigato.
26
L’espressione del P-gp indotto dalle simvastatine che si è osservato in AD rimane ancora una possibile ipotesi che spiega almeno in parte la neuroprotezione contro HD50.
Figura 14. Morbo di Huntington
A differenza della malattia di Huntington il coinvolgimento di P-gp in CJD è stato provato da Vogelgesang51. Questa malattia neurodegenerativa è caratterizzata dall’accumulo di un abnorme isoforma PrPsc
della proteina prione PrP che forma aggregati responsabili del progresso patologico di questa malattia. Come AD, l’aumento nella concentrazione di proteine prioni nel cervello è associato a un diminuire nell’espressione di P-gp vascolare51. Un’ efficace terapia per curare o prevenire questo tipo di malattia neurodegenerativa potrebbe essere rappresentato dall’uso di induttori dell’ espressione della P-gp o da appropriati modulatori abili a aumentare la loro funzione a livello del cervello. Anche se non esiste nessuna prova che PrP è un substrato di P-gp due percorsi sono stati ipotizzati nei quali la riduzione di P-gp può essere collegata alla formazione di aggregati tossici di PrPsc. Un meccanismo è basato sull’ipotesi che la perdita di funzioni P-gp migliora l’aggregazione di PrPsc 29,51,52, mentre un’altra teoria concerne il danneggiamento tra l’accumulazione di PrPsc
e la sua degradazione indotta dalla perdita dipendente dall’età della funzione di P-gp53.
27 Sclerosi amiotrofica laterale
La sclerosi laterale amiotrofica (SLA) è una malattia neurodegenerativa caratterizzata da un’insorgenza tardiva, una progressiva perdita di neuroni motori (motoneuroni) nel cervello e nel midollo spinale che porta alla paralisi e alla morte54.
Figura 15. Morbo di Lou Gehrig (dal nome del primo paziente alla quale venne diagnosticata) o Malattia di Charcot (dal nome del medico che per
primo la individuò), è una terribile malattia degenerativa e progressiva, solitamente mortale, che colpisce i motoneuroni (quelli preposti al
movimento e al controllo dei muscoli).
Sebbene le cause molecolari dell’SLA sembrano ancora poco chiare, anche in questa malattia neurodegenerativa si ha un mal ripiegamento e la conseguente aggregazione di proteine. Attualmente il Riluzone è l’unico agente terapeutico comprovato in prove cliniche in pazienti con SLA55.
28
Riluzone
Il Riluzone è stato studiato in un modello animale di SLA56 in combinazione con Minocicline una tetracicline con un ampio spettro antimicrobica. Questa combinazione ritarda l’insorgenza della SLA riduce i deficit motori, prolungando, inoltre, la sopravvivenza del paziente. E’ evidente che la combinazione di questi farmaci aumenta la concentrazione di riluzone nel cervello a causa dell’inibizione della P-gp dovuto all’utilizzo di monocicline.
Il ruolo del P-gp nel trattamento dell’ SLA è stato studiato anche da Kirkinezos57 usando ciclosporina A (CsA) un famoso substrato P-gp. La neuroprotezione indotta da CsA è chiaramente visibile solo in situazioni in cui la BEE é compromessa; questo fatto suggerisce che l’efficacia della terapia sulla SLA potrebbe essere appropriatamente aumentata attraverso la co-somministrazione di un inibitore P-gp.
29
FARMACI E P-GP
Visto il ruolo ricoperto della P-gp nei tumori, nelle malattie neurodegenerative e nella multidrug resistance MDR, la ricerca si è concentrata sullo sviluppo di farmaci in grado di modularne l’attività e/o l’espressione. Le varie indagini hanno evidenziato non solo le caratteristiche chimico-strutturali che la sostanza deve possedere per poter interagire con la P-gp, ma anche le caratteristiche dei suoi substrati.
Substrati e modulatori della P-gp
La P-gp può trasportare un gran numero di composti molto diversi tra loro per funzione e struttura, ma accomunati da alcune caratteristiche quali alto grado di idrofobicità, un basso peso molecolare (250-900 Da), una carica positiva a pH neutro e la capacità di attraversare la membrana citoplasmatica per diffusione passiva.
Fra i substrati della P-gp troviamo farmaci appartenenti a molte categorie terapeutiche, come antineoplastici quali gli alcaloidi della vinca (vinblastina, vincristina), le antracicline (doxorubicina, daunorubicina, epirubicina), i taxani (paclitaxel, docetaxel) ed anche antiaritmici, antistaminici, immunosoppressori, antivirali e farmaci utilizzati nella terapia del Parkinson, dell’Alzheimer, e di altre malattie neurodegenerative.
30
Figura 16. Tabella che indica alcuni agenti terapeutici substrati della Glicoproteina-P
SUBSTRATI INIBITORI
Bloccanti del canale delCa++: Diltiazem, Mibefradil Antibiotici: Eritromicina, Rifampicina, Levofloxacina, Tetraciclina Verapamile Valspodar Chinidina Ciclosporina Ketoconazolo Antitumorali: Paclitaxel Docetaxel Vinblastina Vincristina Doxorubicina Daunorubicina Epirubicina ß- antagonisti: Carvediolo Talinolo Reserpina Antiaritmici: Digossina Digitossina Steroidi: Desametasone Metilprednisolone Aldosterone Progesterone Idrocortisone Cortisolo Corticosterone Antistaminici: Fexofenadina Oppioidi: Loperamide Domperidone Morfina Metadone Fentanile Antiacidi: Cimetidina Ranitidina
Queste molecole, dopo essere penetrate nella cellula, vengono riportate nella regione extracellulare dalla proteina mediante trasporto attivo, e quindi contro gradiente di concentrazione58. I modulatori, attraverso un’interazione allosterica negativa, riducono la capacità della proteina di formare legami con i substrati.
E’stato dimostrato, attraverso l’utilizzo di radioligandi, che queste molecole alterano il legame della P-gp con il substrato in maniera non competitiva; in questo modo viene ridotta la massima densità recettoriale (Bmax) per il legame con il substrato, ma non cambia la costante di equilibrio di associazione (Kd), perché interagiscono con un sito diverso della P-gp.
31 Inibitori della P-gp
L’attività degli inibitori ha perfezionato la comprensione del meccanismo di trasporto della P-gp. L’ampio numero dei composti che interagiscono con la glicoproteina ha portato ad ipotizzare la presenza di diversi siti di legame, sulla P-gp58,59. Attualmente sono stati individuati 4 distinti siti di interazione sulla proteina: tre sono stati riconosciuti essere quelli adibiti al trasporto in quanto interagiscono sia con i substrati che con i modulatori. Il quarto sito è di regolazione, sul quale alcune molecole come elacridar e nicardipina sembrano interagire come modulatori.
Struttura della Nicardipina
La formazione di un legame in uno di questi siti provoca sugli altri un cambiamento conformazionale58. Attraverso studi SAR si è dimostrato come gli aspetti strutturali sono strettamente correlati con la capacità inibitoria delle molecole. Ad oggi si riconoscono diverse classi di inibitori: prima, seconda e terza generazione.
Inibitori di prima generazione
Gli inibitori di prima generazione annoverano composti come il verapamile, un Ca++ antagonista, in grado di determinare la regressione dei fenomeni di MDR. Tale farmaco sensibilizza le cellule leucemiche all’azione della vincristina e vinblastina, annullando, così, i fenomeni di MDR60. Il verapamile è capace di modulare in modo non specifico diverse vie secretive e di alterare le proteine di trasporto Ca++ dipendenti grazie alla capacità di intercalarsi nel doppio strato fosfolipidico. E’ stato inoltre dimostrato che la ristabilita citotossicità degli alcaloidi della vinca, è effettivamente dovuta ad un’inibizione della P-gp da parte del verapamil. Studi SAR effettuati utilizzando diversi farmaci ad attività vasodilatatoria ed in grado di modulare l’attività dei canali al Ca++
32
nicardipina, hanno stabilito che un composto, per essere considerato un inibitore, deve esplicare uno o più dei seguenti effetti:
Aumentare la potenza del farmaco contro cellule resistenti Aumentare la sua concentrazione all’interno di tali cellule
Dimostrare interferenza con la P-gp durante le analisi di “photoaffinity labeling”(tecnica utilizzata per evidenziare i siti di legame delle proteine)
Quindi attraverso questo tipo di screening, furono scoperte numerose sostanze, anche strutturalmente molto diverse fra loro, in grado di modulare l’attività della P-gp. Alcuni esempi di inibitori sono rappresentati dagli antagonisti della calmodulina, ciclosporina A, analoghi della chinina, tamossifene (antiestrogeno). Gli ormoni steroidei rappresentarono, invece, la prima generazione di modulatori della P-gp poiché sono in grado di invertire i fenomeni di MDR61. La co-somministrazione di questi inibitori con farmaci antitumorali non determina alcun miglioramento nel trattamento delle patologie, questo perché si ha un:
Mancato raggiungimento di una concentrazione plasmatica sufficiente per inibire la P-gp
Profilo di cardiotossicità troppo elevato alle dosi necessarie a modulare l’attività della P-gp.
33 Inibitori di seconda generazione
La seconda generazione di inibitori include analoghi del verapamile (dexverapamil e dexiniguldipine), il valspodar derivato della ciclosporina ed il biricodar derivato del tacrolimus62. Questi inibitori sono più potenti di quelli di prima generazione sebbene continuino a presentare interazioni farmacocinetiche critiche dovute all’inibizione del citocromo P450 e riduzione dell’escrezione biliare con conseguente aumento della tossicità63,64.
Dexverapamil Biricodar
Inibitori di terza generazione
L’ultima generazione di inibitori della P-gp è stata sviluppata attraverso studi di SAR e di chimica combinatoriale per superare le limitazioni dei modulatori di seconda generazione. Farmaci come acridonecarboxamide, antranilammide, ciclopropildibenzosuberano, l’inibitore della farnesil transferasi e diarilimidazolo, sono alcuni inibitori di terza generazione65. L’elacridar (Acridonecarboxammide) è uno dei più potenti modulatori MDR conosciuti. Studi clinici hanno dimostrato che questo composto presenta un’elevata affinità per la P-gp e un’attività inibitoria sul trasportatore BCRP, accompagnata ad una modesta interazione con i sistemi enzimatici del citocromo P450, ad esempio il 3A466.
34
Elacridar
Il tariquidar (antranilammide) è un’altro inibitore della P-gp molto potente, selettivo ed efficace, inoltre è molto ben tollerato ed opera un’inibizione di tipo non competitivo con farmaci antitumorali come la vinblastina ed il paclitaxel. Ha attività inibitoria sulla glicoproteina a basse concentrazioni ed è caratterizzato da una lunga durata d’azione67
. Questo composto agisce inibendo l’attività ATP-asica basale della pompa, per cui impedisce il legame del substrato e l’azione idrolitica sull’ATP68.
Tariquidar
Lo zosuquidar è un composto ad elevata specificità per la P-gp ed è dotato di scarsa affinità per il citocromo P450 per cui l’interazione con altri farmaci risulta minima66.
35
Il valspodar, PSC833, è un inibitore, analogo della ciclosporina A, ma non ha attività immunosoppressiva, per cui può essere somministrato anche a dosi elevate69. L’utilizzo del valspodar in terapia, però, è limitato dalla sua attività inibitoria nei confronti del citocromo P450 3A470. Dato che molti agenti antitumorali, come la doxorubicina, sono metabolizzati da questo sistema enzimatico, la loro co-somministrazione con il valspodar determina un aumento degli effetti tossici dei chemioterapici stessi e costringe ad una conseguente riduzione dei dosaggi del P-gp inibitore69.