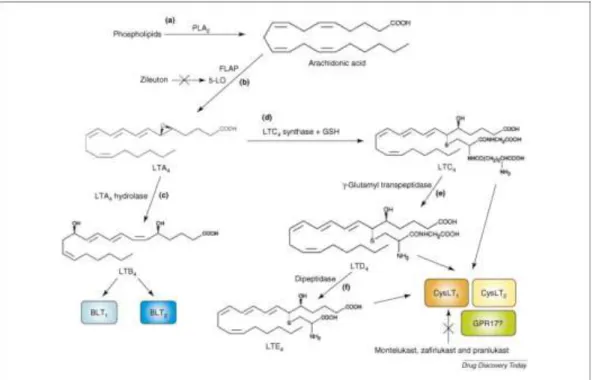
1.3 Inibitori dei cisteinil-leucotrieni
I cisteinil-leucotrieni (CysLTs), caratterizzati dalla presenza di un
residuo di cisteina nella catena laterale, appartengono alla
superfamiglia degli eicosanoidi (acidi grassi polinsaturi a 20 atomi di
carbonio) e alla famiglia dei leucotrieni, molecole con 3 doppi legami
coniugati. Questi mediatori lipidici sono prodotti dall’azione di
differenti PLA2 sull’acido arachidonico esterificato nella membrana
cellulare. La successiva attivazione della 5-lipossigenasi (5-LO) da
parte della 5-LO-activating protein (FLAP) determina la produzione di
LTA4, metabolizzato a LTB4 o LTC4. L’enzima LTC4-sintetasi o glutatione-S-transferasi, proteina della famiglia delle MAPEG
(proteine di membrana che metabolizzano gli eicosanoidi e il
glutatione), produce LTC4, il primo dei tre cisteinil-leucotrieni (Figura
Figura 1. Cascata dei leucotrieni e siti bersaglio della terapia antiasmatica
La 5-LO è particolarmente espressa in granulociti, macrofagi,
mastociti e linfociti B (Dahlén, 2006), ma eosinofili e mastociti
possono produrre gran parte di LTC4 grazie alla presenza di un pool
endogeno di acido arachidonico. Da alcuni studi in vitro risulta che
anche i fibroblasti bronchiali possiedono gli enzimi della cascata degli
eicosanoidi e producono spontaneamente cisteinil-leucotrieni (James
et al., 2006). Inoltre, anche altre cellule come piastrine, eritrociti,
possono produrre CysLTs ed LTB4 metabolizzando il LTA4 prodotto
dai neutrofili attivati (Folco et al. 2006).
I CysLTs hanno numerosi effetti immunologici e patobiologici e
nell’asma sono responsabili della maggior parte delle manifestazioni e dei fenomeni coinvolti nel rimodellamento delle vie aeree. Il
ritrovamento di LTE4, maggior metabolita dei CysLTs, nelle urine è
un parametro significativo per monitorare l’asma e molte altre patologie infiammatorie (Taniguchi et al., 2008). La ricerca si è
pertanto orientata verso lo studio di molecole capaci di impedire
l’attività di questi mediatori.
L’osservazione che gli antagonisti dei CysLT1 e gli inibitori di 5-LO hanno la medesima efficacia negli studi su modelli di
infiammazione ha portato alla conclusione che la maggior parte degli
effetti anti-asmatici degli anti-leucotrieni sia dovuta all’antagonismo
1.3.1 Cisteinil-leucotrieni e caratterizzazione dei
recettori
In base agli studi finora effettuati, i cisteinil-leucotrieni agiscono
attraverso due recettori, CysLT1 e CysLT2, appartenenti alla famiglia
dei recettori accoppiati alle proteine G. In modelli sperimentali in
vitro è stato evidenziato che i recettori dei CysLT sembrano poter
legare indipendentemente le proteine Gq/11 o le proteine Gi/o in base al
tessuto e al tipo cellulare utilizzati (Rovati e Capra, 2007).
La caratterizzazione funzionale di questi due recettori è stata
possibile grazie a studi farmacologici che hanno dimostrato una
diversa sensibilità ai più potenti antagonisti del recettore CysLT1, tra
cui il montelukast, un antagonista specifico (Labat et al., 1992). Al
contrario, gli antagonisti non selettivi risultano essere meno potenti,
soprattutto nell’uomo (Tudhope et al., 1994).
Recenti studi suggeriscono, però, l’esistenza di altri sottotipi recettoriali, come il recettore orfano GPR17, che risulta essere attivato
sia dai CysLTs che da alcuni uracil-nucleotidi (ATP, UDP). Poiché i
nucleotidi sono coinvolti in numerosi processi biologici, disfunzioni
e i CysLT nelle risposte infiammatorie (Rovati et al., 1997). E’ stato dimostrato, infatti, che gli antagonisti dei CysLT1 come il montelukast
e il pranlukast inibiscono anche gli effetti di alcuni nucleotidi sui
recettori P2Y (Mamedova et al., 2005).
Alcuni esperimenti indicano la possibilità che i CysLTs possano
formare omodimeri o eterodimeri (Mellor et al., 2003). Tale evidenza
ha consentito di ipotizzare la presenza di un eterodimero
CysLT1/CysLT2 insensibile all’antagonismo selettivo del CysLT1
(Mellor et al., 2003), poi confermata dall’identificazione dell’eterodimero sulla membrana nucleare di mastociti umani (Rovati e Capra, 2007).
La scoperta del recettore CysLT1 ha destato particolare interesse per
l’attività anti-infiammatoria e anti-brococostrittiva esercitata dagli antagonisti selettivi. Gli effetti di maggior rilievo nella patologia
asmatica sono infatti mediati dai recettori CysLT1, che risultano avere
una distribuzione più ampia nelle cellule infiammatorie e strutturali
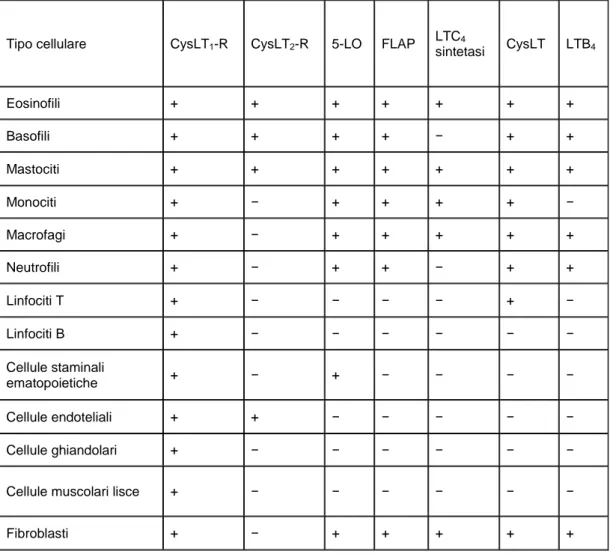
I recettori CysLT1 sono espressi in monociti, macrofagi, eosinofili,
basofili, mastociti, neutrofili, linfociti T e B, cellule staminali
ematopoietiche, cellule interstiziali della mucosa gastrica, muscolatura
liscia delle vie aeree, fibroblasti bronchiali e cellule dell’endotelio
vascolare (Tabella 1) (Montuschi et al., 2007).
Tabella 1. Produzione dei CysLT e distribuzione degli enzimi della biosintesi e dei sottotipi recettoriali
Tipo cellulare CysLT1-R CysLT2-R 5-LO FLAP LTC4
sintetasi CysLT LTB4 Eosinofili + + + + + + + Basofili + + + + − + + Mastociti + + + + + + + Monociti + − + + + + − Macrofagi + − + + + + + Neutrofili + − + + − + + Linfociti T + − − − − + − Linfociti B + − − − − − − Cellule staminali ematopoietiche + − + − − − − Cellule endoteliali + + − − − − − Cellule ghiandolari + − − − − − −
Cellule muscolari lisce + − − − − − −
I recettori CysLT2, invece, sono particolarmente espressi a livello
cardiaco e neuronale, ma sono stati individuati anche a livello di
leucociti circolanti, linfonodi e macrofagi interstiziali dei polmoni,
mentre nella muscolatura liscia delle vie aeree risultano assenti
(Capra, 2004).
CysLT1
Il gene del recettore CysLT1 è stato localizzato sul cromosoma X e
consiste di 3 esoni ed un singolo promoter con numerosi siti di inizio
trascrizione (Woszczek et al., 2005). Lo splicing da origine a 5
differenti trascritti, diversi in base al tessuto e al tipo cellulare, che
codificano per proteine identiche (Zhang et al., 2006).
Il recettore CysLT1 ha una maggiore omologia (32%) con il
recettore purinico P2Y1 e il recettore del PAF, ma una più bassa
omologia (28%) con un’altra classe di recettori leucotrienici, i BLT dei LTB4.
Piastrine − − − − + + +
Eritrociti − − − − + + +
Inoltre, possiede 5 potenziali siti di fosforilazione da parte di PKA e
PKC, alcuni dei quali all’estremità carbossi-terminale (Lynch et al., 1999). E’ stato infatti dimostrato che il CysLT1 può andare incontro ad internalizzazione dovuta all’attivazione diretta della PCK, ma anche all’azione delle arrestine 2 e 3 e della GRK2 (Naik et al., 2005; Deshpande et al., 2007). Invece, la desensitizzazione provocata da
nucleotidi tramite attivazione della PKA induce una internalizzazione
molto breve ed un rapido ripristino della responsività, rispetto alla
desensitizzazione omologa provocata dagli agonisti (Capra et al.,
2005).
Uno studio che ha caratterizzato la risposta agli agonisti ha
dimostrato che LTD4 è il più potente tra i CysLTs e che in ordine di
potenza LTD4>LTC4, mentre LTE4 è qualificato come agonista
parziale, con minore affinità (Lynch et al., 1999).
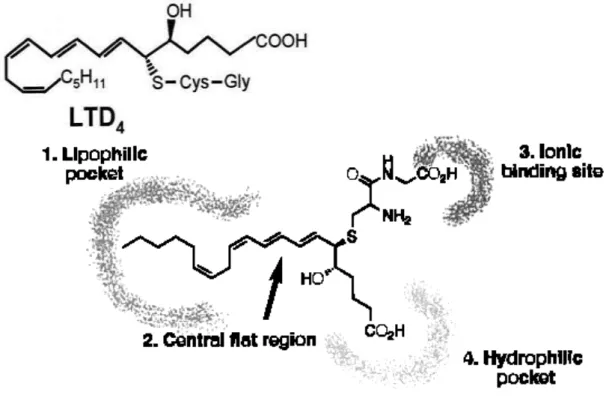
Lo sviluppo degli antagonisti dei CysLT1 si è basato sul modello
dell’agonista naturale LTD4, infatti piccoli cambiamenti strutturali degli agonisti dei CysLT1 risultano in composti con attività
antagonista (Figura 8). La somiglianza nelle relazioni struttura-attività
agonisti naturali ha pertanto suggerito che possa esserci una simile
interazione con il recettore CysLT1 (Young, 1989) .
Figura 2. Struttura base degli antagonisti del LTD4
Nel valutare la potenza degli antagonisti dei CysLT1 nei confronti
di LTD4, uno studio di binding ha dimostrato che zafirlukast e
montelukast sono più potenti degli altri antagonisti attualmente in
commercio, mentre nei confronti di LTC4 zafirlukast è l’unico
antagonista incapace di competere con il sito di legame (Ravasi et al.,
distinti per LTD4 e LTC4 sul recettore CysLT1 o che il sito ad alta
affinità per LTC4 possa essere un altro recettore, come il CysLT2
(Capra et al., 1998; Ravasi et al., 2000).
CysLT2
Il gene del recettore CysLT2 è localizzato sul cromosoma 13
(Takasaki et al., 2000), ma l’organizzazione genomica non è stata ancora completamente caratterizzata.
L’omologia con CysLT1 è ridotta (38%), ma in comune con l’altro sottotipo possiede 5 siti che possono essere fosforilati. Pertanto,
CysLT2 può essere soggetto a desensibilizzazione omologa ed
eterologa mediata da PKC o PKA (Heise et al., 2000).
Esperimenti di binding hanno provato che LTD4 e LTC4 sono
agonisti di ugual potenza, mentre LTE4 risulta un agonista parziale
(Capra, 2004).
Studi recenti hanno dimostrato l’esistenza di polimorfismi nella
regione codificante il recettore CysLT2 associati allo sviluppo di asma
1.3.2. Meccanismo di trasduzione dei recettori dei
CysLTs
Il meccanismo di trasduzione dei recettori dei CysLTs si basa
sull’attivazione delle proteine Gq/11 e Gi/o; pertanto, sono capaci di modulare non solo i canali ionici, ma anche la fosfolipasi C (PLC)
(Rovati e Capra, 2007).
LTD4, tramite i recettori CysLT1 e CysLT2, induce un aumento del
Ca2+ intracellulare e del metabolismo del fosfatidil-inositolo (PI)
(Winkler et al., 1988) e nelle cellule muscolari umane provoca
l’attivazione dei canali del K/Ca2+
-dipendenti a larga conduttanza, con
conseguente depolarizzazione (Snetkov et al., 2001). Questa evidenza
ha suggerito che LTD4 causi l’ingresso di Ca 2+
soprattutto attraverso i
canali voltaggio-indipendenti, ma in parte anche attraverso le
fosfolipasi C specifiche per la fosfatidilcolina (PC-PLC). Inoltre, la
contrazione può essere dovuta all’incremento di isoforme di PKC sia
Ca2+-dipendenti che indipendenti (Accomazzo et al., 2001).
Studi sul CysLT1 hanno dimostrato che il legame con la proteina
Gi/o determina la fosforilazione delle proteine chinasi attivate da
immunitario tramite attivazione della PKCα e della Raf-1 (Hoshino et al., 1998). Studi successivi hanno invece dimostrato che, nella risposta
chemiotattica, l’attivazione della fosfoinositolo 3-chinasi (PI3K) risulta avere un ruolo predominante rispetto alla Rho-chinasi e alla
proteina p38 (Parameswaran et al.,2002). Probabilmente, con questi
meccanismi i CysLTs promuovono l’interazione dei leucociti con l’endotelio vascolare e aumentano l’espressione della p-selettina (Kanwar et al., 1995) e della proteina Mac-1 (Fregonese et al., 2002).
Inoltre, studi in vitro hanno anche dimostrato che LTD4, attraverso il
recettore CysLT1, provoca la fosforilazione di ERK e l’attivazione di STAT-1 e di MEK, che inducono l’espressione delle ICAM-1
sull’epitelio (Profita et al., 2008).
LTD4 risulta responsabile della secrezione mucosa caratteristica
dell’infiammazione delle vie aeree, tramite induzione della trascrizione del gene MUC2, mediata dal recettore CysLT1 attraverso
l’attivazione della proteina G e conseguentemente di PKC, MEK, ERK-2 ed NF-kB (Suzuki et al., 2008).
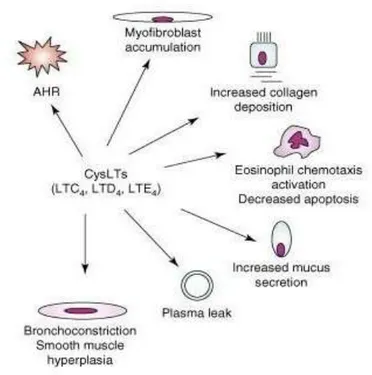
Nel processo di rimodellamento delle vie aeree, CysLT1 provoca la
proteina Gi/o, della Rho-GTPasi e della tirosina-chinasi, sia in presenza
che in assenza di elevati livelli di Ca2+ intracellulari (Saegusa et al.,
2001). In concentrazioni nanomolari, infatti, LTC4 induce
l’espressione della collagenasi in linee di fibroblasti umani polmonari, il che contribuisce significativamente al rimodellamento della matrice
extracellulare nei processi infiammatori cronici (Holgate et al., 2000).
I cisteinil-leucotrieni risultano essere potenti broncocostrittori in
vitro (Yang et al., 2004) e, in vivo, aumentano la responsività tissutale
alla metacolina e all’istamina (O’Hickey et al., 1991). Recenti studi dimostrano che LTD4 incrementa la produzione di mRNA del TGFβ
in maniera proporzionale al tempo di esposizione e alla
concentrazione, causando iperplasia ed iperresponsività (Bossè et al.,
2008). Recentemente, è stato anche rivelato che l’unico effetto fisiologico del LTE4 in vivo è rappresentato dalla fosforilazione di
ERK e CREB e dalla up-regulation delle COX-2. La conseguente
produzione di PGD2, che potenzia l’iperresponsività, è suscettibile sia agli antagonisti del CysLT1 che agli antagonisti dei PPARγ (Figura 9)
Figura 3. Effetti dei cisteinil-leucotrieni sullo sviluppo della patologia asmatica
Numerosi studi hanno dimostrato che il CysLT1 induce la
proliferazione di cellule emopoietiche maligne, cellule epiteliali,
cellule muscolari delle vie aeree, progenitori degli eosinofili e
fibroblasti con diversi meccanismi (Rovati e Capra, 2007):
la cooperazione con il fattore di crescita insulino-simile (IGF), attraverso l’induzione da parte di LTD4 della MMP-1, capace di bloccare l’inibitore della IGF-binding protein (Saegusa et al., 2001).
la fosforilazione di ASK1, una chinasi a monte di c-Jun e p38 MAPK, a loro volta capaci di regolare il fattore di trascrizione AP-1
responsabile della proliferazione e differenziazione cellulare
(Kumasawa et al., 2005).
la transattivazione del fattore di crescita dell’epidermide (EGF) tramite produzione di specie reattive dell’ossigeno (ROS) e fosforilazione di ERK1/2 (Ravasi et al., 2006).
la produzione di TGF-β1 per attivazione della p38 MAPK (Perng et al., 2006)
l’aumento di RasGTP-asi, PLC e tirosin-chinasi Ca2+
-dipendenti
in seguito ad attivazione di ERK1/2 (Capra et al., 2004)
l’attivazione di ERK e p38 mediata da PI3K e PKC (McMahon
et al., 2000)
la transattivazione del fattore di crescita delle piastrine (PDGF), associata all’attivazione di c-Src (Gq/11-mediata) e di Ras (McMahon et al., 2002).
L’induzione trascrizionale dei geni pro-infiammatori sembra essere dovuta all’attivazione di NF-kB ed AP-1 (Thompson et al., 2006), che potrebbe essere mediata direttamente dal recettore CysLT1. Infatti,
studi recenti hanno rivelato la presenza del CysLT1 all’esterno della
all’interno del nucleo dopo prolungata esposizione agli agonisti nelle cellule epiteliali intestinali (Nielsen et al., 2005). A questo scopo, la
sequenza localizzata all’estremità carbossi-terminale risulta essenziale per la traslocazione ed è tipica di altre GPRS e proteine normalmente
localizzate nel nucleo (Rovati e Capra, 2007).
Alcuni studi hanno riportato che l’espressione di questo recettore
può essere aumentata dai linfociti Th2, che stimolano la produzione di
IL-5 da parte degli eosinofili (Thivierge et al., 2000) e di IL-4 e IL-13
da parte dei macrofagi e dei monociti (Thivierge et al., 2001).
Tuttavia, altri autori non rilevano una up-regulation né del recettore né
del trascritto dei CysLT1, bensì la comparsa di un altro recettore
attivato dal ligando pirimidinico UDP, escludendo la possibilità che si
tratti del recettore CysLT2 (Mellor et al., 2001). Pertanto resta ancora
da chiarire se l’up-regulation riguardi una forma inducibile o normalmente espressa.
Inoltre, è stato dimostrato che l’attivazione della PKC da parte del CysLT1 modula l’attività dei recettori β2, provocando la
desensitizzazione e la riduzione della risposta ai β2 agonisti in vitro.
desensitizzazione possa essere mediata anche dalla regolazione di
cAMP e PKA (Rovati et al., 2006).
Questo fenomeno contribuisce al fallimento terapeutico dei β2
agonisti e suggerisce una nuova possibilità di utilizzo degli antagonisti
dei CysLT1 (Coreno et al., 2005).
L’aumento del Ca2+
intracellulare nelle cellule muscolari da parte
del CysLT2 è mediato, come per il CysLT1, dalla proteina Gi/o (Mellor
et al., 2003). Nelle HUVECs, dove il CysLT2 è espresso in maniera
esclusiva, provoca l’induzione trascrizionale di 37 geni, tra cui quelli di EGR, vari fattori di trascrizione nucleari, IL-8, ADAMTS1, fattore
tissutale (TF) e COX-2 (Uzonyi et al., 2006). L’effetto sull’induzione
trascrizionale è causato dalla fosforilazione della p38 MAPK, che
media anche la produzione di IL-8 (Mellor et al., 2001).
Un aumento del trascritto dei CysLT2 sembra essere causato da IL-4
(Lotzer et al., 2003), TNFα e IL-1β (Sjostrom et al., 2003) in maniera parziale e reversibile.
Tuttavia, il ruolo dei CysLT2 risulta predominante soprattutto in
1.3.3
Ruolo
del
montelukast
nell’asma
e
nell’infiammazione
Montelukast è un inibitore selettivo dei CysLT1, con notevole
affinità per il sito recettoriale del LTC4 (Ravasi et al., 2002). Secondo
le linee guida GINA, recenti studi hanno attribuito agli antagonisti dei
leucotrieni un ruolo determinante come farmaci di base nel
trattamento dell’asma, soprattutto nell’adulto, anche in considerazione
dell’aumento del rischio di eventi avversi gravi osservati a seguito del trattamento con farmaci β2-agonisti a lunga durata d’azione in regime
di monoterapia.
Il suo uso è attualmente approvato nella terapia di combinazione
con glucocorticoidi nel trattamento dei bambini di età superiore ai 2
anni, nell’asma non controllato con glucocorticoidi inalatori, nell’asma lieve persistente (Reiss et al., 1998; Jarvis et al., 2000) e in alcuni casi di asma indotto da aspirina (Dahlén et al., 2002). Infatti,
elevati livelli di CysLTs sono associati spesso ad asma sensibile
all’aspirina (Sousa et al., 2002), nel cui trattamento, il montelukast consente un controllo superiore dei sintomi rispetto ai glucocorticoidi
Il trattamento con montelukast (10 mg) risulta utile nel prevenire la
brococostrizione indotta da esercizio fisico dopo 12 settimane di
trattamento e riduce il decremento del volume espiratorio forzato
(FEV) (Leff et al, 1998). Questo effetto è osservabile dopo appena due
ore dalla somministrazione di una singola dose orale di montelukast
(10 mg) e persiste per 24 ore (Pearlman et al., 2006). Inoltre, il
montelukast risulta più efficace del salbutamolo nel trattamento
cronico di questo tipo di asma, come dimostrato dal mantenimento del
controllo e dalla ridotta incidenza di esacerbazioni della malattia
(Villaran et al., 1999).
Sia nell’asma moderato e lieve non controllato con glucocorticoidi che nella terapia di prevenzione delle esacerbazioni, la combinazione
di un glucocorticoide con montelukast risulta meno efficace della
combinazione con un β2-agonista (Joos et al., 2008; Ducharme et al., 2006). Tuttavia, la combinazione
glucocorticoide-montelukast-salmeterolo risulta molto più efficace della combinazione del
glucocorticoide con il solo β2-agonista (Storms et al., 2004). Questo
effetto di potenziamento tra montelukast e β2-agonisti può essere attribuito alla capacità degli antagonisti dei CysLTs di antagonizzare
la desensibilizzazione eterologa dei recettori β2 dovuta alle PKA
(Coreno et al., 2005).
Un attuale studio sul trattamento dell’asma e delle riniti allergiche ha dimostrato un miglioramento del controllo in soggetti resistenti alla
terapia con glucocorticoidi inalatori e LABAs (Korn et al., 2009).
Montelukast (10 mg), somministrato per via orale in tre dosi (36 ore
e 12 ore prima e 12 ore dopo la sensibilizzazione immunologica),
inibisce la risposta immediata e tardiva all’inalazione di allergeni (Diamant et al., 1999).
Inoltre, in associazione con la budesonide aumenta il controllo
dell’asma con un’efficacia simile a quella ottenuta con una dose doppia del glucocorticoide (Price et al., 2003; Vaquerizo et al., 2003).
In pazienti affetti da concomitante rinite allergica la combinazione
montelukast/budesonide risulta anche più efficace del cortisonico ad
alte dosi (Price et al, 2006) con un migliore profilo di tollerabilità
(Tohda et al., 2002). Il potenziamento dell’effetto ottenuto con la combinazione può essere correlato al controllo di alcuni parametri
l’azione dei CysLTs sono influenzati dai corticosteroidi negli adulti e nei bambini (Gyllfors et al., 2006).
In modelli animali di asma, montelukast previene i cambiamenti
strutturali delle vie aeree, come l’inspessimento e la fibrosi subepiteliale, che non sono modificati in modo clinicamente rilevante
dal trattamento con desametasone e altri corticosteroidi (Henderson et
al., 2006). Inoltre, montelukast, alla dose di 10 mg al giorno per otto
settimane di trattamento, riduce l’accumulo di miofibroblasti nell’asma atopico (Kelly et al., 2006).
Esiste, tuttavia, una variabilità individuale nella risposta al
montelukast e agli antileucotrieni, così come ai glucocorticoidi
(Szefler et al., 2005). Numerosi studi hanno infatti dimostrato che
polimorfismi del promoter del gene che codifica per la 5-LO sono
associati ad un’alterazione nella risposta agli antileucotrieni (In et al., 1997) e all’iperresponsività (Kim et al., 2005). Inoltre, dei cinque polimorfismi del gene che codifica il CysLT1, nessuno è stato
associato ad asma o a rinite allergica, ma un recente studio ha
individuato un aplotipo nella regione promoter che modifica la
Esclusa la possibile insorgenza della sindrome di Churg-Strauss e
considerata l’elevata tollerabilità e l’assenza di effetti secondari, anche gastrointestinali (Dahlén et al., 2006), l’utilizzo degli inibitori dei CysLT1 risulta favorevole e privo di controindicazioni nel controllo
del processo infiammatorio, specie nelle infiammazioni eosinofile
(Nagata e Saito, 2003).
Studi in vitro hanno infatti dimostrato che, sia in pazienti normali
che in soggetti asmatici, l’esposizione a CysLTs in concentrazioni nanomolari determina il reclutamento degli eosinofili circolanti (Imai
et al., 1997; Foster et al., 1991) e l’attivazione dei neutrofili. Montelukast è capace di regolare l’attività dei neutrofili (Theron et al., 2009) riducendo il Ca2+ intracellulare ed aumentando il cAMP, forse
per inibizione delle fosfodiesterasi (Anderson et al., 2009); il farmaco
riduce il numero di eosinofili nel sangue e nella saliva inducendone
l’apoptosi (Abadoglu et al., 2005; Minoguchi et al., 2002). Inoltre, è stato dimostrato che l’associazione del montelukast con il prednisolone risulta più efficace nel sopprimere l’infiltrazione degli eosinofili nelle vie aeree rispetto al solo corticosteroide (Mao et al.,
Il CysLT1 provoca l’up-regulation delle molecole di adesione delle
cellule CD34+ tramite la produzione di β2-integrine (Mohle et al.,
2003). Tuttavia, montelukast risulta meno efficace, rispetto al
prednisolone, nell’inibire la loro proliferazione e migrazione cellulare (Mao et al., 2004).
Gli effetti degli inibitori dei CysLTs sulle molecole di adesione
dovranno essere valutati meglio, ma sembra che il montelukast possa
inibire l’espressione della MMP-9 (Fregonese et al., 2002) e l’attività protesica degli eosinofili con un meccanismo indipendente
dall’antagonismo nei confronti del CysLT1 (Langlois et al., 2006). Inoltre montelukast, al contrario del salbutamolo, inibisce la
produzione di anioni superossido da parte degli eosinofili, mediata da
LTD4 (Kushiya et al., 2006).
E’ stato dimostrato che i CysLTs aumentano la produzione di numerose citochine e dei linfociti T. Montelukast riduce l’espressione
di mRNA di IL-5 (Ihaku et al., 1999), IL-6, MCP-1, TNFα e di
conseguenza l’espressione di IL-8, sopprimendo l’attività istone acetilasica di NF-kB (Maeba et al., 2005; Tahan et al., 2008).
Infine, montelukast ha un significativo effetto anti-remodelling ed
inibitorio sulle cellule strutturali (Kelly et al., 2006) contrastando
TGFβ1 e IL-13, che inducono una up-regulation del recettore CysLT1 sui miofibroblasti (Asakura et al., 2004).