PART I
Introducció
La primera part d’aquest treball fa referència al context històric en el qual es va situar l’inici d’aquesta tesi doctoral. Es pot considerar un exemple de l’importància i dels esforços invertits per la comunitat científica per a desxifrar el contingut gènic humà. A continuació es recullen els resultats obtinguts pel nostre laboratori com a membres del Consorci EuroImage. L’estratègia utilitzada i els resultats obtinguts han contribuït a avançar en el coneixement dels gens continguts en els genomes seqüenciats durant les últimes dècades.
I. Heretabilitat, trets genètics i genoma humà
La consciència de l’existència d’heretabilitat de trets físics i del comportament va sorgir en l’antiguitat. Aquest fet queda reflectit en l’àmbit de la ramaderia (cria i millora genètica dels animals domèstics) o en l’àmbit agrícola (obtenció de cultius més productius, més resistents…). L’herència de certes característiques fenotípiques era reconeguda com a familiar. Principalment l’estudi de l’herència d’anomalies fenotípiques i malalties va establir els fonaments per a una nova visió de la biologia i la medicina on s’incorporava un nou factor: l’herència i els antecedents familiars. En un primer moment, el coneixement i la definició de diferents modes d’herència es va descriure sense conèixer la base física d’aquesta herència. El coneixement de l’existència dels gens o del material genètic va esclatar quan es van recuperar els estudis de Mendel amb les plantes del pèsol. Aquests estudis i el descobriment de la naturalesa física i molecular de l’agent responsable d’aquesta heretabilitat, l’àcid desoxiribonucleic, DNA, va suposar l’inici d’una revolució en els camps de la medicina i la biologia. Va sorgir un especial interès pel coneixement de les bases moleculars de malalties d’herència mendeliana com per exemple la malaltia de Huntington o l’anèmia de Fanconi. És per això que actualment la majoria de les malalties o alteracions hereditàries estudiades i més conegudes són aquelles causades per un sol gen. En aquest cas un sol gen quan és anòmal és responsable de donar lloc a la malaltia (mutacions puntuals, repeticions…). Aquest tipus de trets són els anomenats monogènics. Actualment, un dels objectius principals de la biomedicina és aconseguir determinar el paper dels gens en caràcters amb patrons d’herència complexos o no mendelians. És el cas d’alteracions com la diabetis, els càncers o moltes malalties mentals. S’hi inclouen també trets fenotípics complexos com l’alçada,
el comportament, l’inteligència o la pigmentació de la pell. En aquests casos és probable que més d’un gen i més d’una mutació siguin presents per a que la malaltia o tret fenotípic es manifesti. És evident a més a més, que moltes d’aquestes variacions genètiques actuen com a factors de susceptibilitat no determinants i que l’influència de l’entorn és també un factor a tenir en consideració. Aconseguir discernir entre tots aquests agents i el seu paper en diferents alteracions fenotípiques ha esdevingut essencial per a la biologia i medicina actuals. Una de les eines principals per tal d’aconseguir identificar aquests factors i determinar la seva funció és l’obtenció de la seqüència genòmica humana i d’organismes model completa, acurada i disponible sense restriccions per a la comunitat científica. Amb aquest objectiu es va engegar el Projecte Genoma Humà, objecte de la següent secció, i en el context del qual es desenvolupa la recerca duta a terme en aquesta tesi.
II. Context històric: Projecte Genoma Humà
La primavera de l’any 2003 és considerada la data oficial de l’obtenció de la seqüència completa del genoma humà. Des del seu inici va ser evident que es tractava d’un esdeveniment que revolucionaria la biologia i obriria nous camps d’aplicació a nivell de la medicina molecular, la biotecnologia, el control ambiental, els recursos energètics o l’avaluació de riscs.
La primera publicació que menciona la possibilitat i importància d’obtenir la seqüència completa del genoma humà data de mitjans dels anys 80 (Dulbecco, 1986). La creixent innovació i l’optimització experimental (el clonatge amb cromosomes artificials de llevat (YAC), els mapes genètics d’alta resolució, la seqüenciació automàtica fluorescent, l’algoritme de BLAST per alinear seqüències) va permetre que l’any 1990 el Departament d’Energia (DOE) i els National Institutes of Health (NIH) dels Estats Units iniciessin oficialment el Projecte Genoma Humà (HGP, Human Genome Project) amb l’objectiu final d’obtenir la seqüència nucleotídica completa del genoma humà. La posterior incorporació del Wellcome Trust britànic optimitzant l’ús de cromosomes artificials de bacteris (BAC) per a mapatge i seqüenciació va permetre a finals de l’any 1992 l’obtenció dels mapes físics complets del cromosoma Y (Foote et al., 1992; Vollrath et al., 1992) i del cromosoma 21 (Chumakov et al., 1992). A partir d’aquest moment i durant els anys següents es van aconseguir generar
mapes genètics de baixa resolució humans i murins i es va obtenir la primera seqüència completa d’un organisme viu de vida lliure, el genoma del bacteri Haemophilus influenzae (Fleischmann et al., 1995). Al mateix temps es produia un augment significatiu en l’obtenció de dades de seqüència i expressió gràcies a projectes com l’iniciat per l’IMAGE Consortium (Lennon et al., 1996), el RIKEN (Wada, 1994) o el projecte genoma del llevat Saccharomyces cerevisiae (Dujon, 1996).
La competència declarada per part del sector privat (Celera Genomics) per a obtenir la seqüència completa del genoma humà va accelerar dràsticament la producció i alliberació de dades de seqüència i mapatge a la comunitat científica. El cromosoma 22 es considera el primer cromosoma humà en ser totalment seqüenciat (Dunham et al., 1999). A mesura que s’anaven obtenint, les seqüències genòmiques provisionals parcials (draft) es posaven a l’abast de la comunitat científica a les bases de dades públiques. Aquesta informació va resultar, i encara ho és avui dia, molt útil per a nombrosos projectes, entre els quals s’inclou aquesta tesi. És important tenir en compte la naturalesa provisional i canviant d’aquesta informació, especialment durant la fase més productiva i competitiva del projecte. En aquest context es situen la major part dels resultats obtinguts en les dues primeres parts d’aquest treball.
L’any 2001 es va publicar la seqüència provisional completa del genoma humà representant aproximadament el 90% de la seqüència eucromàtica, és a dir, la corresponent a DNA no repetitiu i, en principi, amb capacitat de transcriure’s (Lander et al., 2001; Venter et al., 2001). Es tractava de seqüència que no arribava a complir els criteris de qualitat establerts per a la seqüència final acabada i per tant, va ser necessari utilitzar les dades amb precaució. Tot i el seu grau de provisionalitat, aquestes dades han resultat molt útils tant a nivell d’estudis genòmics globals com a nivell d’anàlisi de gens particulars.
Des d’aquest moment i fins l’any 2003 es va anar obtenint el que s’ha considerat seqüència definitiva acabada. Aquestes dades cobreixen el 99% del DNA eucromàtic i es considera que actualment existeixen menys de 400 discontinuïtats o gaps. L’1% restant no seqüenciat correspón a DNA centromèric i repetitiu, la seqüenciació del qual no és tecnològicament factible actualment.
De forma paral.lela i complementària a l’obtenció de la seqüència crua del DNA humà, nombrosos avenços tecnològics s’han produït pel que fa a les eines per a la seqüenciació, aconseguint reduïr-ne el cost econòmic i augmentar el volum i la velocitat de producció de dades.
La disponibilitat de la seqüència genòmica humana ha permès començar a avançar en el coneixement de la variabilitat nucleotídica entre individus i en l’aplicació d’eines de genètica comparativa usant els genomes d’altres organismes model seqüenciats (ratolí, rata, Drosophila melanogaster o Caenorhabditis elegans). Paral.lelament, avenços significatius s’han produït en el camp de la bioinformàtica i biologia computacional, com és el desenvolupament d’eines per a la generació, captura i anotació de dades, el desenvolupament de programes per a la representació i anàlisi de similaritat i variació de seqüència, i les millores en el contingut i usabilitat de les bases de dades.
Algunes dades concretes obtingudes a partir de la seqüència publicada del genoma humà i algunes incògnites que encara queden per esbrinar es recopilen a les Taules 1 i 2.
Taula 1. Algunes dades obtingudes a partir de la seqüència completa del genoma humà.
Conté 3 mil milions de parells de bases nucleotídiques. La mida mitjana d’un gen és de 3000 bases.
La distrofina és dels gens coneguts el més gran, 2’4 megabases. Més del 50% dels gens identificats tenen funció desconeguda. El 99’9% de la seqüència genòmica és idèntica entre individus.
Aproximadament el 2% del genoma conté informació per a codificar proteïna. Com a mínim el 50% del genoma és seqüència repetitiva no codificant.
La seqüència repetitiva té un paper en l’estructura i dinàmica dels cromosomes. Són responsables de generar reorganitzacions genòmiques donant lloc a gens completament nous o a noves seqüències gèniques.
El genoma humà conté una proporció de seqüència repetitiva major que altres organismes seqüenciats (Caenorhabditis elegans, Drosophila melanogaster o Mus musculus).
Més del 40% de proteïnes predites comparteixen similaritat de seqüència amb proteïnes de Caenorhabditis elegans o Drosophila melanogaster.
Es considera que l’elevat nombre de tipus de proteïnes humanes en comparació amb altres espècies es deu principalment a mecanismes de splicing alternatiu i de modificació post-traduccional.
La majoria de famílies de proteïnes són comuns entre humans, Caenorhabditis elegans o Drosophila melanogaster. La principal diferència recau en el número de membres, molt més elevat en humans.
Els gens es troben concentrats en regions genòmiques a l’atzar separades per grans extensions de DNA no codificant.
A les regions riques en gens predominen les bases G-C.
Les regions pobres en gens presenten un percentatge més elevat de bases A-T. El cromosoma 1 és el més gran i conté el major nombre de gens.
El cromosoma Y és el de tamany i contigut gènic menor.
S’han identificat més de 3 milions de posicions nucleotídiques amb variabilitat entre humans.
Taula 2. Algunes incògnites i dades encara desconegudes sobre el genoma humà.
El número exacte de gens, la seva posició i la seva funció. Els mecanismes de regulació gènica.
L’organització i estructura dels cromosomes.
Els tipus de DNA no codificant, la seva distribució i funció.
La coordinació de l’expressió gènica, síntesi proteica i mecanismes post-traduccionals. El proteoma dels organismes: el conjunt de proteïnes i les seves funcions respectives. La conservació proteica entre organismes.
Les correlacions entre les variacions nucleotídiques dels individus i trets fenotípics/malalties. Les variacions en la seqüència dels gens com a factor de susceptibilitat.
Els gens implicats en l’herència de caràcters complexos i multigènics.
Els beneficis i les aplicacions de les dades de seqüència generades a partir del projecte Genoma Humà afecten àrees tan àmplies i diverses com la medicina molecular, la microbiologia, els estudis evolutius i antropològics, la medicina forènsica o l’agricultura. Algunes de les possibles aplicacions en cadascún d’aquests camps s’enumeren a la Taula 3.
Taula 3. Aplicacions futures de les dades fruit del Projecte Genoma Humà.
Medicina molecular
Aplicacions futures inclouen millores en el diagnòstic de malalties, en la detecció precoç de possibles predisposicions genètiques, en el disseny de fàrmacs, en la teràpia gènica i sistemes de control de l’acció de fàrmacs o en la síntesi de fàrmacs individuals (farmacogenòmica).
Microbiologia genòmica
S’usen els recursos i eines del projecte genoma humà per a iniciar la seqüenciació massiva de genomes de microorganismes. S’espera obtenir informació referent a noves fonts d’energia, eliminació segura de residus tòxics, detecció de productes contaminants ambientals i comprensió de les vulnerabilitats enfront malalties infeccioses.
Avaluació de risc
El coneixement de la variabilitat genètica i el seu paper en referència a la susceptibilitat està permetent avaluar el risc individual enfront agents tòxics com radiacions i substàncies mutagèniques o cancerígenes. De la mateixa manera permetrà reduïr la transmissió vertical de mutacions.
Evolució i antropologia
La comparació de genomes ha de permetre l’estudi de l’evolució dels organismes vius, establir els corrents migratoris al llarg de la història i determinar edats i dates concretes d’esdeveniments històrics.
Genòmica forènsica
Conèixer la seqüència genòmica permetrà l’identificació d’individus concrets en un context criminal, en esdeveniments catastròfics o en l’establiment de relacions familiars. A nivell ambiental permetrà l’identificació d’espècies protegides i la detecció d’agents contaminants. A nivell mèdic permetrà l’identificació de compatibilitats tissulars per a transplantaments d’òrgans.
Agricultura La genòmica de plantes i animals impulsarà la creació de varietats resistents, més productives, més nutritives o amb incorporació de vacunes.
III. Anàlisi transcripcional del genoma humà
Un cop obtinguda la seqüència completa d’un determinat genoma el repte principal resideix en l’identificació de tots els gens presents. Especialment en el cas del genoma humà la dificultat principal per a assolir aquest objectiu es troba en l’extrema complexitat del nostre genoma. La major part dels gens humans acostumen a consistir de diversos exons petits separats per seqüències no codificants de longitud variable (introns) i que fins i tot poden arribar a contenir altres gens. És aquesta una característica que compromet greument la precisió amb la que les eines informàtiques de predicció de gens són capaces d’identificar seqüències gèniques (predicció de novo). L’ús d’eines informàtiques per a l’anàlisi de similaritat de seqüència i per a l’identificació de
seqüències específiques de gens (transicions exó-intró, seqüències promotores, dianes de poliadenilació o pautes de lectura oberta) es coneix amb el terme de clonatge in silico. El creixement exponencial de les bases de dades públiques de seqüència evidencia els nombrosos avantatges d’aquesta estratègia però cal tenir en ment que un dels desavantatges principals d’aquestes eines de predicció es troba en l’imprecisió i probabilitat d’error si se’n fa un ús exclusiu o independent. Indicacions i evidències sobre potencials seqüències gèniques poden també obtenir-se mitjançant genòmica comparativa. En aquest cas s’utilitza la comparació d’una seqüència genòmica, l’humana per exemple, amb altres genomes més petits, de menor complexitat i amb menys seqüència no codificant (fugu, llevat, ratolí) amb la finalitat d’identificar regions de conservació i els gens que hi estàn continguts.
Taula 4. Comparació i aplicacions de l’mRNA i del cDNA.
mRNA cDNA
Cadena senzilla Doble cadena Poca estabilitat Estable
Manipulació complexa Fàcil manipulació
Traducció a proteïna Transcripció a RNA i traducció a proteïna
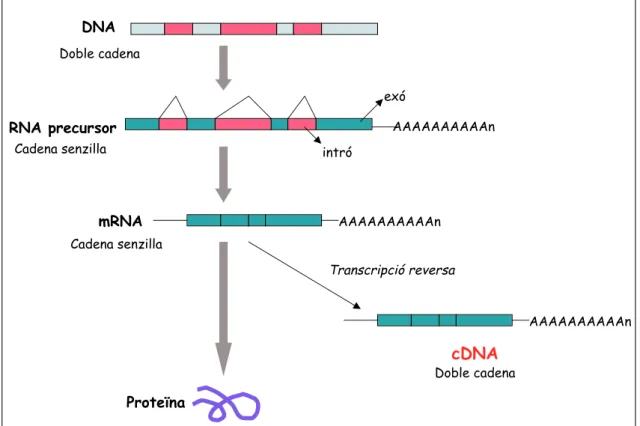
Tot i els avantatges de les eines informàtiques per a l’anàlisi de la seqüència genòmica, és imprescindible obtenir evidència experimental directa sobre la naturalesa gènica d’una determinada seqüència. L’estratègia transcripcional per a l’identificació de seqüències gèniques utilitza com a punt de partida el producte en forma d’RNA missatger (mRNA) o de proteïna, dels gens expressats en una determinada cèl.lula, teixit, organisme o estadi de desenvolupament i permet demostrar empíricament que una determinada seqüència nucleotídica correspón a un gen (Figura 1). Es parteix de la producció de còpies en DNA de l’RNA missatger (cDNA) d’una cèl.lula, teixit o organisme (Taula 4). Els clons de cDNA poden ser aleshores amplificats i seqüenciats (Figura 2). Els avantatges principals de l’identificació de gens seguint aquesta estratègia resideixen en la seva rapidesa relativa per a identificar seqüències transcrites. A més a més, cada clon de cDNA aporta
informació adicional sobre aquell gen, com l’identificació de seqüència codificadora de proteïna (ja que no contenen introns), informació sobre el patró d’expressió (el teixit o cèl.lules o estat de desenvolupament d’on prové el cDNA analitzat dóna informació valuosa d’on s’expressa el gen corresponent), l’identificació de fenòmens de transcripció alternativa (clons de cDNA
DNA mRNA Proteïna Cadena senzilla Doble cadena RNA precursor exó intró AAAAAAAAAAn AAAAAAAAAAn Cadena senzilla AAAAAAAAAAn Doble cadena Transcripció reversa cDNA
Figura 1. Generació de còpies de DNA (cDNA) a partir del RNA missatger (mRNA) generat per la transcripció de gens en organismes eucariotes.
mRNA AAAAAAAAAAn
TTTTTTTT
Síntesi de la primera cadena de DNA
Tractament amb RNAsa H i síntesi de la segona cadena de DNA (DNA polimerasa I)
TTTTTTTT Lligació en vector
adient (amb extrems roms o adaptadors)
Clon de cDNA
procedents d’un mateix gen que difereixen parcialment en la seva seqüència), i l’identificació de gens homòlegs en altres espècies (ja que només contenen la part del gen més conservada –codificadora- no tenen introns).
IV. Consorci IMAGE. Projecte EUROIMAGE
L’importància d’aconseguir identificar empíricament seqüències gèniques ha esdevingut cada vegada més evident a mesura que s’han anat obtenint dades de seqüència genòmica crua. El Consorci IMAGE (Integrated Molecular Analysis of Genomes and their Expression) es va iniciar l’any 1993 amb l’intenció de compartir recursos amb l’objectiu d’optimitzar la comprensió del genoma humà partint d’una estratègia d’anàlisi transcripcional (Lennon et al., 1996). Per assolir tal objectiu es va generar una col.lecció de clons de cDNA a partir de llibreries normalitzades pre-existents. Es van establir conjunts o arrays de clons representatius i es van caracteritzar parcialment a nivell de seqüència (ESTs, expressed sequence tags, seqüències de 500 nucleòtids aproximadament, corresponents als extrems dels clons) i mapatge de baixa resolució. L’anàlisi d’aquestes dades de seqüència va permetre agrupar els clons en funció del transcrit del qual provenien. Aquesta informació, els clons i les llibreries usades han estat a l’abast de tota la comunitat científica a través de les bases de dades públiques i dels centres distribuidors de clons. El consorci IMAGE ha contribuït molt notablement a l’identificació de nombrosos gens humans, així com a l’ensamblatge i l’anotació final de la seqüència del genoma humà.
Amb l’objectiu de consolidar el treball portat a terme i els recursos generats pel consorci IMAGE, l’any 1997 es va impulsar un nou subprojecte, l’EuroImage (European Integrated Analysis of Genes and their Expression), finançat pel programa BIOMED2 de la Comunitat Europea (Biomed BMH4-CT97-2284). Entre els objectius proposats en el projecte destacaven els següents:
• Generació d’una col.lecció mínima no redundant de clons de cDNA corresponents a la majoria dels transcrits humans.
• Creació d’una col.lecció ‘master’ de clons de cDNA complets utilitzant els recursos establerts pel consorci IMAGE (llibreries i clons).
• Caracterització de la col.lecció ‘master’ de clons de cDNA mitjançant seqüenciació de qualitat, una fiabilitat mínima del 99.99% per a cada cadena i un mínim de tres lectures per cada base nucleotídica.
• Mapatge d’alta resolució dels gens identificats en humans i organismes model.
• Obtenció dels perfils d’expressió en humans i organismes model per tal d’aprofundir en el coneixement de transcrits específics d’especial interès pel grup de recerca responsable.
• Integració de l’informació generada pel projecte en bases de dades públiques disponibles a tota la comunitat científica.
Per tal d’assolir els objectius proposats pel Consorci EuroImage es va establir una col.laboració entre vuit grups de recerca europeus complementant recursos, eines i resultats. Els laboratoris participants en el projecte són els llistats a continuació:
• Centre National de la Recherche Scientifique, CNRS (Dr Auffray, França) • Max-Planck Institut fur Molekulare Genetik, MPI (Dr Lehrach, Alemanya) • Deutsches Krebsforschungszentrum Stiftung des offentlichen Rechts, DKFZ
(Dr Poutska, Alemanya)
• Kungl Tekniska Hogskolan, KTH (Dr Uhlen, Suècia)
• European Molecular Biology Laboratory, EMBL (Dr Ansorge, Alemanya) • Human Genome Mapping Project Resource Centre, HGMP (Dr Gibson,
Gran Bretanya)
• Telethon Institute of Genetics and Medicine, TIGEM (Dr Ballabio, Itàlia) • Medical and Molecular Genetics Department, Institut de Recerca
Oncològica, IRO (Dr Estivill, Espanya)
Enmarcats en el context del projecte EuroImage i com a membres del consorci, el nostre laboratori al Departament de Genètica Mèdica i Molecular de l’Institut de Recerca Oncològica es va centrar en l’anàlisi del contingut gènic de regions cromosòmiques considerades d’especial relevància biomèdica per al departament. Entre aquestes regions d’interès es troba la zona cromosòmica objecte d’anàlisi en la segona part d’aquest treball, la regió q24-q26 del cromosoma 15 humà.
V. Aïllament de gens in silico
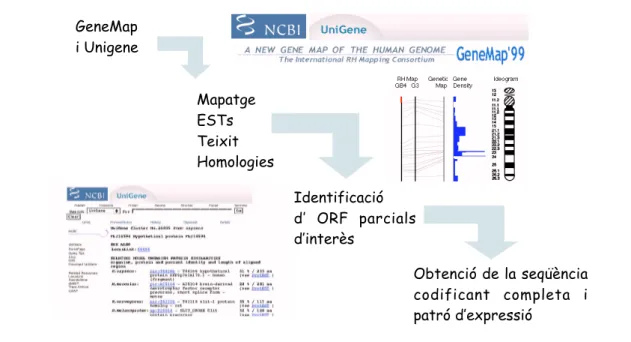
L’obtenció de la seqüència completa crua del genoma humà i altres organismes ha comportat el desenvolupament de noves estratègies per a l’identificació de gens. En el cas de l’aïllament de gens in silico, s’obtè benefici de les eines i dades presents a les bases de dades públiques generades de forma sistemàtica, se n’obté el màxim d’informació, es comprova aquesta informació empíricament i es completa posteriorment en funció dels objectius de l’estudi corresponent (Figura 3).
S’acostuma a partir de les bases de dades d’ESTs, seqüències dels extrems dels clons de cDNA, per a identificar potencials seqüències expressades. A partir d’aquest punt els passos seguits per a l’identificació i aïllament del gen o gens corresponents s’esquematitzen tot seguit.
1/ Predicció de gens a partir d’ESTs
S’utilitzen dades de mapatge presents a les bases de dades públiques (Genemap, NCBI) per a definir grups d’ESTs representatius de gens no coneguts. S’usen programes bioinformàtics d’aliniament per homologia de
Identificació d’ ORF parcials d’interès GeneMap i Unigene Obtenció de la seqüència codificant completa i patró d’expressió Mapatge ESTs Teixit Homologies
Figura 3. Etapes principals de l’estratègia de clonatge i identificació de seqüències transcrites usada en aquest treball. ORF, pauta de lectura oberta; EST, expressed sequence tag.
seqüència (CAP Assembly, Sequencher, BLAST) i la comparació posterior amb seqüències de les bases de dades públiques (dbEST, GenBank, Unigene). L’objectiu final d’aquest apartat és identificar clons corresponents a un únic gen, no quimèrics i del màxim número de parells de bases possible per tal que continguin el màxim de seqüència codificant del gen corresponent.
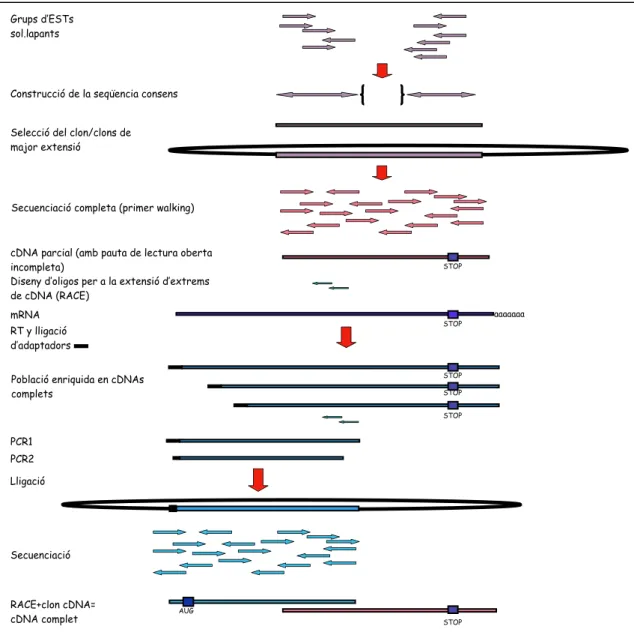
2/ Seqüenciació dels clons seleccionats
A partir dels clons sel.leccionats en el punt anterior se n’obté la seqüència completa mitjançant l’estratègia de primer walking. Això comporta dissenyar nous oligonucleòtids a partir de cada seqüència parcial obtinguda (500-700 nucleòtids). Es realitza aleshores la següent reacció de seqüència, es repeteix el procediment fins que es completa la seqüència de l’insert i s’obté seqüència
STOP aaaaaaa STOP STOP STOP STOP Grups d’ESTs sol.lapants
Construcció de la seqüencia consens
Selecció del clon/clons de major extensió
Secuenciació completa (primer walking)
cDNA parcial (amb pauta de lectura oberta incompleta)
Diseny d’oligos per a la extensió d’extrems de cDNA (RACE)
mRNA RT y lligació d’adaptadors
Població enriquida en cDNAs complets PCR1 PCR2 Lligació Secuenciació RACE+clon cDNA=
cDNA complet STOP
AUG
Figura 4. Gràfic mostrant el procés de sel.lecció, seqüenciació i obtenció de clons de cDNA complets.
de vector, indicant així, que l’insert ha estat cobert d’extrem a extrem. Les dades de seqüència obtingudes per a cada reacció són processades i editades amb eines bioinformàtiques específiques per a l’ensamblatge i edició de seqüències nucleotídiques com Sequencher (GeneCodes). Per al disseny de seqüències oligonucleotídiques adients pel seu ús en reaccions de seqüenciació s’empren programes com OLIGO o GCG PRIMER (Figura 4).
3/ Detecció d’homologies
Les seqüències completes dels inserts dels clons de cDNA seleccionats poden ser analitzades per a detectar seqüències codificadores de proteïna (pautes de lectura oberta) completes o parcials (Sequencher). Tant la seqüència nucleotídica com la seqüència aminoacídica predita permeten cercar homologies a les bases de dades (FASTA, BLAST). En funció del grau d’homologia de seqüència els gens o proteïnes detectades a les bases de dades poden suggerir l’inclusió de la nova seqüència en una determinada classe de gens o proteïnes, així com la seva implicació en determinats processos cel.lulars i l’identificació de gens ortòlegs en altres espècies.
4/ Obtenció de la seqüència codificadora completa de cada gen
La gran majoria de clons de les llibreries contenen inserts corresponents a cDNAs parcials que no representen el total de la seqüència codificadora de proteïna del gen. Una vegada cercades les bases de dades amb la seqüència obtinguda en el pas anterior amb l’objectiu de detectar seqüències solapants, cal adoptar procediments moleculars per extendre els clons parcials fins a obtenir el cDNA complet amb una pauta de lectura oberta (ORF) sencera. Alguns d’aquests procediments estàn basats en l’amplificació mitjançant PCR, com és el cas dels experiments de RACE (extensió ràpida d’extrems de cDNA) o la transcripció reversa (RT-PCR), i altres es basen en la hibridació de llibreries de cDNA de teixits o d’òrgans específics.
5/ Predicció de l’estructura i dominis de proteïnes
Un cop predita la seqüència codificadora de proteïna a partir de la seqüència completa del cDNA poden identificar-se dominis proteics coneguts i conservats amb altres proteïnes de les bases de dades. S’usen programes de lliure accés com PROSITE (Sigrist et al., 2002), PFAM (http://pfam.wustl.edu/index.html),
SMART (Letunic et al., 2004; Schultz et al., 1998) o MOTIF (http://motif.genome.jp) per a identificar dominis proteics. Es pot predir l’estructura secundària de la proteïna putativa amb programes com GENEQUIZ
( A n d r a d e e t a l ., 1 9 9 9 ) , P R E D I C T P R O T E I N
(http://cubic.bioc.columbia.edu/predictprotein) o PSIPRED (McGuffin et al., 2000), així com obtenir indicacions sobre la seva funció potencial.
6/ Predicció de l’estructura genòmica
La seqüència nucleotídica corresponent a un cDNA no conté seqüència intrònica. Gràcies a aquest fet, un aliniament amb la corresponent seqüència genòmica permet deduir l’estructura exònica-intrònica del gen. La presència de la seqüència genòmica humana completa a les bases de dades públiques facilita enormement aquest procés.
7/ Mapatge
De la mateixa manera que en el punt anterior la disponibilitat de tot el genoma humà a les bases de dades públiques permet determinar fàcilment la localització cromosòmica del gen seqüenciat. Un cop coneguda la seva situació cromosòmica és possible analitzar la presència en la regió d’interès de loci responsables de malalties per les quals el gen/gens responsables encara romanen desconeguts. D’aquesta manera és possible identificar gens candidats per a determinades característiques o patologies (McKusick). En el cas de no obtenir correspondència (per exemple per errors de mapatge o per discontinuïtats de seqüència) es pot confirmar experimentalment el mapatge mitjançant diversos mètodes (FISH (hibridació in situ fluorescent) o híbrids de radiació per exemple)
8/ Anàlisi de seqüències adjacents
L’anàlisi de les regions genòmiques flanquejants pot permetre identificar potencials seqüències reguladores i promotores. Es poden utilitzar eines de
lliure accés com GENSCAN (Burge & Karlin, 1997) o NIX
9/ Eines i enllaços útils per l’anàlisi in silico (adreces web de setembre 2004) GENERALS NCBI http://www.ncbi.nlm.nih.gov/ EBI http://www.ebi.ac.uk/ DDBJ http://www.ddbj.nig.ac.jp/ ExPASy http://www.expasy.ch/ PROJECTES GENOMA
Entrez Genome http://www.ncbi.nlm.nih.gov/genomes/static/euk_g .html
The Institute for Genome Research (TIGR) Microbial Database http://www.tigr.org/tdb/mdb/mdb.html Integrated Genomics Inc. http://www.genomesonline.org/ NHGRI List of
Genetic and Genomic Resources
http://www.nhgri.nih.gov/Data The Sanger Centre http://www.sanger.ac.uk
Washington University-St.Louis http://genome.wustl.edu Ohlahoma University http://www.genome.ou.edu/ Microbial Genome Database http://mbgd.genome.ad.jp ANÀLISI DE GENOMES MAGPIE http://genomes.rockefeller.edu/magpie GeneQuiz http://jura.ebi.ac.uk:8765/ext-genequiz/ http://www.cmbi.kun.nl/swift/genequiz/info_entry.ht ml PEDANT http://pedant.gsf.de/ Clusters of Orthologous Groups of Proteins (COGs) http://www.ncbi.nlm.nih.gov/COG Kyoto Encyclopedia of Genes and Genomes (KEGG) http://www.genome.jp/kegg/ What Is There (WIT) http://wit.integratedgenomics.com/IGwit
ANÀLISI D’ESTS dbEST home page http://www.ncbi.nlm.nih.gov/dbEST/ EST Projects at Washington University http://genome.wustl.edu/est/ The I.M.A.G.E. Consortium http://image.llnl.gov/ UniGene http://www.ncbi.nlm.nih.gov/UniGene/ The UniGene build procedure http://www.ncbi.nlm.nih.gov/UniGene/build.html UniGene query engine http://www.ncbi.nlm.nih.gov/UniGene/query.cgi HomoloGene http://www.ncbi.nlm.nih.gov/HomoloGene/ STACK http://www.sanbi.ac.za/Dbases.html TIGR Gene Indices http://www.tigr.org/tdb/tgi.html TIGR Orthologous Gene Alignment database http://www.tigr.org/tdb/tgi/ego/ GeneMap http://www.ncbi.nlm.nih.gov/genemap/ dbSNP http://www.ncbi.nlm.nih.gov/SNP/ Cancer Genome Anatomy Project (CGAP) http://www.ncbi.nlm.nih.gov/ncicgap/ CGAP Digital Differential Display (DDD) http://www.ncbi.nlm.nih.gov/UniGene/ddd.cgi?ORG=Hs CGAP xProfiler http://cgap.nci.nih.gov/Tissues/xProfiler ALINIAMENT DE SEQÜÈNCIES BLAST http://ncbi.nlm.nih.gov/BLAST/ CLUSTAL W http://www.ebi.ac.uk/clustalw/ dotter ftp://ftp.sanger.ac.uk/pub/dotter/ FASTA lalign http://www.ebi.ac.uk/fasta/
hmmer http://hmmer.wustl.edu/
RepeatMasker http://ftp.genome.washington.edu/RM/RepeatMasker.h tml
seg ftp://ncbi.nlm.nih.gov/pub/seg/ sim4 http://globin.cse.psu.edu
PREDICCIONS A PARTIR DE SEQÜÈNCIES DE DNA
Banbury Cross http://igs-server.cnrs-mrs.fr/igs/banbury
FGENEH http://www.softberry.com/berry.phtml?topic=fgenesh &group=programs&subgroup=gfind GeneID http://www1.imim.es/geneid.html GeneMachine http://genome.nhgri.nih.gov/genemachine GeneParser http://beagle.colorado.edu/~eesnyder/GeneParser.ht l GENSCAN http://genes.mit.edu/GENSCAN.html Genotator http://www.fruitfly.org/~nomi/genotator/ GRAIL http://compbio.ornl.gov/tools/index.shtml GRAIL-EXP http://compbio.ornl.gov/grailexp/ HMMgene http://www.cbs.dtu.dk/services/HMMgene/ MZEF http://www.cshl.org/genefinder PROCRUSTES http://www-hto.usc.edu/software/procrustes RepeatMasker http://ftp.genome.washington.edu/RM/RepeatMasker.h tml Sputnik http://rast.abajian.com/sputnik/
BASES DE DADES I EINES PER ANALITZAR DOMINIS PROTEICS
ProDom http://protein.toulouse.inra.fr/prodom.html Pfam http://pfam.wustl.edu http://www.sanger.ac.uk/Software/Pfam/ SMART http://smart.embl-heidelberg.de CDD search http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi BLOCKS http://blocks.fhcrc.org PRINTS http://www.bioinf.man.ac.uk/dbbrowser/PRINTS/PRINTS. html ProfileScan http://www.isrec.isb-sib.ch/software/PFSCAN
PREDICCIÓ DE PROPIETATS A PARTIR DE SEQÜÈNCIES D’AMINOÀCIDS
Compute pI/MW http://www.expasy.ch/tools/pi tool.html MOWSE http://srs.hgmp.mrc.ac.uk/cgi-bin/mowse PeptideMass http://www.expasy.ch/tools/peptide-mass.html TGREASE ftp://ftp.virginia.edu/pub/fasta/
SAPS http://www.isrec.isb-sib.ch/software/SAPS form.html AACompIdent http://www.expasy.ch/tools/aacomp/
AACompSim http://www.expasy.ch/tools/aacsim/ PROPSEARCH http://www.embl-heidelberg.de/prs.html
PREDICCIÓ ESTRUCTURA PROTEICA Nnpredict http://www.cmpharm.ucsf.edu/~nomi/nnpredict.html PredictProtein http://www.embl-heidelberg.de/predictprotein/ SOPMA http://pbil.ibcp.fr/ Jpred http://www.compbio.dundee.ac.uk/~www-jpred/ PSIPRED http://bioinf.cs.ucl.ac.uk/psipred/psiform.html PREDATOR http://menu.hgmp.mrc.ac.uk/menu-bin/run?option=predator COILS http://www.york.ac.uk/depts/biol/units/coils/coi lcoil.html MacStripe http://www.york.ac.uk/depts/biol/units/coils/coi lcoil.html PHDtopology http://www.embl-heidelberg.de/predictprotein SignalP http://www.cbs.dtu.dk/services/SignalP/ TMpred http://www.ch.embnet.org/software/TMPRED_form.ht ml DALI http://www2.ebi.ac.uk/dali/ FSSP http://www2.ebi.ac.uk/dali/fssp/ SWISS-MODEL http://www.expasy.ch/swissmod/SWISS-MODEL.html TOPITS http://www.embl-heidelberg.de/predictprotein/
RECUPERACIÓ D’INFORMACIÓ A PARTIR DE LES BASES DE DADES
Entrez http://www.ncbi.nlm.nih.gov/Entrez/ FlyBase http://flybase.bio.indiana.edu GDB http://www.gdb.org/ GeneCards http://bioinfo.weizmann.ac.il/cards/ HomoloGene http://www.ncbi.nlm.nih.gov/HomoloGene/ Kinemage http://www.umass.edu/microbio/rasmol/mage.htm LocusLink http://www.ncbi.nlm.nih.gov/LocusLink/ MIPS http://www.mips.biochem.mpg.de/ MMDB http://www.ncbi.nlm.nih.gov/Structure/MMDB/mmdb.sht ml OMIM http://www.ncbi.nlm.nih.gov/Omim PDB http://www.rcsb.org/pdb/ Sacch3D http://www-genome.stanford.edu/Sacch3D/ SGD http://www.yeastgenome.org/ VAST http://www.ncbi.nlm.nih.gov/Structure/VAST/vast.sht ml YPD http://www.proteome.com/databases/index.html
PART I: Objectius
• Identificació de nous gens humans en el marc del Projecte Genoma Humà i del Consorci EuroImage a partir de llibreries de cDNA establertes i de les bases de dades i eines bioinformàtiques públiques
• Caracterització dels nous gens humans identificats a nivell de la seva seqüència nucleotídica, patró d’expressió, predicció de la proteïna codificada i determinació d’homologies amb altres espècies
PART I
Resultats
Resultats
La participació del nostre grup de recerca en el projecte europeu EuroImage destinat a la seqüenciació de clons de cDNA humans ha permès l’identificació, l’obtenció de la seqüència completa i la caracterització de l’expressió de diversos gens humans desconeguts fins aleshores. Tots els casos presentats a continuació corresponen a gens novells no identificats anteriorment en humans i configuren un recull dels gens identificats i publicats com a resultat de la participació directa del nostre laboratori en el projecte EuroImage. Cadascún d’ells constitueix un exemple de l’estratègia usada per a l’aïllament, anàlisi de l’expressió i identificació d’homologies utilitzada per a la caracterització sistemàtica de gens nous a petita escala en el marc d’un projecte amb l’ambiciós objectiu d’identificar i caracteritzar tots els gens del genoma humà.
I. Identificació i caracterització del gen humà PDCD9
La publicació següent exposa de quina manera l’anàlisi de transcrits desconeguts permet l’identificació de PDCD9 (programmed cell death 9), un nou gen humà homòleg a la proteïna pro-apoptòtica p52 de Gallus. El nostre estudi va permetre refinar la seva localització cromosòmica a 5q11. Es van obtenir dades sobre el patró d’expressió en teixits humans i es va determinar el grau de conservació a nivell de seqüència proteica entre diferents espècies. La baixa conservació amb altres famílies de proteïnes va indicar que possiblement es tracta d’una nova família de proteïnes amb una funció potencial en processos apoptòtics. Aquestes dades constitueixen el cos principal de l’article publicat l’any 1999 pel nostre grup.
Cytogenet Cell Genet 87:85–88 (1999)
Cloning, expression, and mapping of PDCD9,
the human homolog of
Gallus gallus
pro-apoptotic protein p52
L. Carim, L. Sumoy, M. Nadal, X. Estivill, and M. Escarceller
Centre de Genètica Mèdica i Molecular, Institut de Recerca Oncològica, Hospital Duran i Reynals, Barcelona (Spain)
Supported by EU Biomed Project No. BMH4-CT97-2284 to X.E. M.E. is funded by the Spanish Ministry of Education (CIDYT contract FPI-070-97) and L.S. by the Catalan autonomous government (RED contract 1998-64).
Received 5 May 1999; revision accepted 30 July 1999.
Request reprints from Dr. Mònica Escarceller, Centre de Genètica Mèdica i Molecular, Institut de Recerca Oncològica, Hospital Duran i Reynals, Autovia de Castelldefels km 2,7, L’Hospitalet de Llobregat,
08907 Barcelona (Spain); telephone: 34-93-260-7775; fax: 34-93-260-7776; e-mail: [email protected].
Abstract. We report the sequence, tissue distribution, and chromosome location of a novel gene, PDCD9 (programmed cell death 9). PDCD9 is the mammalian counterpart of the
Gal-lus galGal-lus pro-apoptotic protein p52. The human cDNA has an
open reading frame of 1,314 nucleotides and was predicted to encode a protein of 438 amino acids with a calculated mass of
50 kDa. The protein sequences of chicken, mouse, and human PDCD9 are remarkably conserved. PDCD9 mRNA is ex-pressed ubiquitously in adult tissues, displaying a stronger sig-nal in heart, skeletal muscle, kidney, and liver. PDCD9 was mapped to chromosome 5q11.
Copyright © 2000 S. Karger AG, Basel
To date, just a fraction of the entire human gene set has been identified. Our laboratory, a member of the EUROIMAGE Consortium, is engaged in the isolation and mapping of novel human genes. The EUROIMAGE Consortium was constituted in 1997 with the objective of completing the cDNA sequence and identifying genes involved in human biology and inherited diseases by correlating precise map locations and gene expres-sions with phenotypic data (Adams et al., 1991; Lennon et al., 1996; Schuler, 1997; Deloukas et al., 1998).
We now report the cloning, tissue distribution, and chromo-some location of PDCD9 (programmed cell death 9), the human homolog of Gallus gallus pro-apoptotic protein p52. Sun et al. (1998) reported the purification of protein p52 from chicken embryos and the cloning of its corresponding cDNA. It was also shown that p52 expression in mouse fibroblasts caused apoptotic cell death, upregulation of the c-Jun transcription factor, and activation of the c-Jun N-terminal kinase (Jnk1),
presenting p52 as a new cell-death protein. Our results show a striking degree of similarity between PDCD9 and p52, which, together with the lack of homology to other related protein fam-ilies, suggests the possibility of PDCD9 being one of the pro-teins associated with apoptotic pathways that have yet to be identified.
Apoptosis is a morphologically distinct form of program-med cell death (for a review, see Steller, 1995). It is an active process that plays a major role during cellular differentiation, development, tissue homeostasis, and metamorphosis, as well as in many diseases, including cancer, acquired immunodefi-ciency syndrome, and neurodegenerative disorders (Thomp-son, 1995; Vaux et al., 1999). The basic machinery appears to be present in essentially all mammalian cells at all times, but the activation of the cellular suicide program is regulated by many different extracellular and intracellular signals. Numer-ous genes involved in apoptosis have been cloned, and many of them are highly conserved among different species (Jacobson et al. 1997).
Materials and methods
Cluster assembly and sequence analysis
EST clusters were assembled using the EST CAP assembly program (http://www.tigem.it) and Sequencher software (GeneCodes) for the Macin-tosh computer. Clones were obtained from the EUROIMAGE distribution centers (DHGP and HGMP). Sequences were determined by primer walking
86 Cytogenet Cell Genet 87:85–88 (1999)
with custom-synthesized primers (LifeTech), using Perkin-Elmer BigDye reagents, following the manufacturer’s instructions, on an ABI 377 auto-mated fluorescence sequence analyzer. For each clone, both strands were sequenced with at least three independent reads per base. PDCD9 nucleotide sequence is available from GenBank under accession number AF146192. Sequence comparisons were performed using ClustalW 1.7 (http://dot. imgen.bcm.tmc.edu:9331/multi-align/multi-align.html). Boxed multiple se-quence alignments were obtained with the BOXSHADE 3.21 program (http: //www.isrec.isb-sib.ch/software/BOX_form.html). The protein pattern and domain databases Prosite, SMART, and Pfam (http://www.hgmp.mrc.ac.uk/ GenomeWeb/prot-domain.html) were searched for known motifs or func-tional domains.
Northern blot analysis
Multiple-tissue Northern blots (MTN-12 blot, Clontech) were hybridized with a 1-kb PCR product corresponding to the 3) region of the human IMAGE clone 1368574 for detection of PDCD9 and with a commercial (Clontech) 2-kb ß-actin cDNA as a control for quantification. Probes were labeled using a random primer DNA labeling kit (BioRad). Blots were hybridized overnight at 65 ° C in ExpressHyb hybridization solution (Clon-tech) and washed at 68° C in 0.2 × SSC, 0.5 % SDS.
Fluorescence in situ hybridization (FISH)
A previously described protocol (Nadal et al., 1997) was used with some modifications. Briefly, 2 Ìg of PAC clone 273D21 were labeled with bio-16-dUTP (Boehringer Mannheim) in a standard nick-translation reaction. Four hundred nanograms of the product were precipitated along with 1 Ìg of Cot-1 DNA (GIBCO BRL) and 1 Ìg of salmon sperm DNA (Sigma) and the pellet resuspended in hybridization mix containing 50 % formamide and 10 % dextran sulfate in 1.5 × SSC. Ten microliters of the hybridization mix was applied to each slide. Slides were incubated overnight in a humid cham-ber at 37 ° C. Post-hybridization washes were performed in three changes of 50 % formamide, 2 × SSC at 42 ° C, followed by three changes of 2 × SSC at 42 ° C. For signal detection, slides were incubated at 37 ° C with avidin-FITC (Vector Laboratories) for 20 min and washed in three changes of 4 × SSC, Tween 20 at 37 ° C. Slides were mounted with 40 Ìl of antifade solution (Vec-tor Labora(Vec-tories) containing 150 ng/ml of DAPI. Slides were viewed with an Olympus AH-3 (VANOX) fluorescence microscope. Images were analyzed with the Cytovision system (Applied Imaging).
Results and discussion
In our effort to identify new genes, we constructed and ana-lyzed in silico unique gene EST clusters on the basis of clone size, chromosomal localization, and tissue expression. Among all clusters studied, we isolated a partial human cDNA se-quence with a single open reading frame (ORF). The EST-derived sequence encompassed only nucleotide (nt) 570 to nt 1465 of the definitive PDCD9 sequence. To obtain the com-plete coding sequence, we selected cDNA clones by screening the Ïgt11 human cDNA library and by BLAST homology searching against dbEST (NCBI) (http://www.ncbi.nlm.nih. gov/cgi-bin/BLAST/) (Altschul et al., 1997). From the results of the search, two human IMAGE clones were chosen for sequenc-ing: 549763 (EST GenBank accession number AA101062) and 1368574 (EST GenBank AA836428). None of the Ïgt11 clones obtained completed the coding sequence, the maximum size clones spanning from nt 814 to nt 1465. Of the two IMAGE clones selected, one, 549763, also revealed a partial coding sequence. Only clone 1368574 represented the full-length cDNA.
The human cDNA had an ORF of 1,314 nucleotides (from nt 40 to nt 1354). The 5) untranslated region (UTR) contained an in-frame stop codon at nucleotide position 6, 27 bp
up-stream of the deduced initiation codon. The 3)-UTR contained a consensus polyadenylation signal ATTAAA (1,441 nt) and a poly(A) sequence at the end (1465 nt). This cDNA sequence was predicted to encode a protein of 438 amino acids with a calculated mass of 50 kDa.
BLAST homology searching against nonredundant data-bases (NCBI) gave a single significant hit with the G. gallus pro-apoptotic protein p52 (GenBank AF029071; Sun et al., 1998).
Nucleotide sequence comparison between chicken p52 and its human homolog showed 70 % identity; this gene was there-fore designated PDCD9 (programmed cell death 9) after the G.
gallus gene. In the original report (Sun et al., 1998), a fragment
of the human protein sequence was presented (amino acids 282 to 407) containing an erroneous stop codon at amino acid posi-tion 407, possibly due to the fact that a single EST sequence was considered. By determining the full-length cDNA spanning the entire ORF, we have corrected this error. The deduced human and chicken amino acid sequences are 51 % identical and 61 % similar (Fig. 1).
Sun et al. (1998) also reported the partial C-terminal mouse amino acid sequence derived from three ESTs. We extended the coding sequence by performing a BLAST homology search against mouse dbEST (NCBI), which revealed a collection of additional ESTs homologous to PDCD9. The cluster was assembled in a single contig comprising a unique sequence in mouse (UniGene Collection, Mm. 29109 (http://www.NCBI. nlm.nih.gov/UniGene/Hs.Home.html). None of the murine clones selected for sequencing were available because of yeast contamination originating at the distribution sources. Nev-ertheless, we obtained the complete putative amino acid se-quence from the consensus of all ESTs (Fig. 1). Human and mouse PDCD9 have 66 % identity and 76 % similarity at the protein level. Protein domain analysis of the PDCD9, murine Pdcd9, and chicken p52 proteins did not show any known motif or functional domain with significant probability.
Northern blot analysis of poly(A) RNA isolated from var-ious tissues (MTN Human 12-lane blot, Clontech), using a 1-kb PCR product corresponding to the 3) region of the human IMAGE clone 1368574 as a probe, revealed a ubiquitously expressed 1.5-kb mRNA species (Fig. 2). A high steady-state level of PDCD9 mRNA was observed in heart, skeletal muscle, kidney, and liver and a lower level in placenta and peripheral blood leukocytes. A barely detectable level of expression was seen in the remaining tissues tested. In addition, three minor higher molecular weight forms (7.5, 6, and 4 kb) were detected, which might have resulted from alternate promoter or polyA site usage or from alternative mRNA splicing. The pattern of PDCD9 mRNA expression differed notably from that of its homolog in chicken, which showed a wide distribution in embryonic and adult tissues. In particular, p52 mRNA was more abundant in embryonic chicken heart and liver, whereas in the adult chicken, a high hybridization signal was detected in testis, brain, heart, kidney, and lung (Sun et al., 1998).
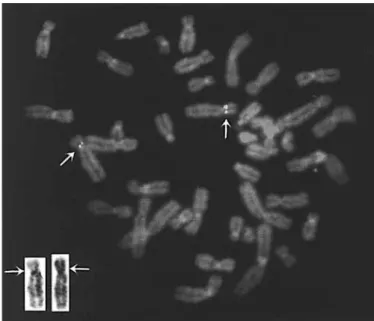
To assign the gene encoding PDCD9 to a human some, FISH analysis was performed on metaphase chromo-somes prepared from peripheral blood lymphocytes. A suitable probe was obtained by screening of the RPCI[1,3-5] human PAC library using the full-length PDCD9 cDNA. Three
posi-Cytogenet Cell Genet 87:85–88 (1999) 87
Fig. 1. Multiple sequence alignment of the human PDCD9, mouse Pdcd9, and chicken p52 polypeptides. Identical residues are printed in reverse type, and similar residues are shaded. Consensus sequence is shown at the bottom, with identical amino acids in uppercase symbols and similar amino acids in lowercase. Extents of partial amino acid sequences previously published by Sun et al. (1998) are bracketed by ! ... 1 for the human and by !! ... 11 for the mouse polypeptide.
Fig. 2. Multiple-tissue northern blot analysis of PDCD9. The 1-kb PDCD9 PCR product was used as a probe, revealing a ubiquitously expressed 1.5-kb mRNA species. PDCD9 and ß-actin transcripts are indi-cated with arrows.
Fig. 3. Localization of PDCD9 to chromosome 5q11 by FISH using the PAC clone 273D21 DNA as a biotinylated probe. The arrows point to the location of the signal on chromosome 5.
tive clones were obtained, the clones corresponding to those provided by the Resource Center of the Deutsches Humange-nomprojekt (DHGP) in Germany. Among them, PAC 273D21 was chosen and confirmed to contain the PDCD9 gene by hybridization and PCR amplification. Using PAC clone 273D21 DNA as a probe for FISH, we were able to assign the PDCD9 gene to the pericentromeric region (band 5q11) of human chromosome 5. A positive signal on both chromosome
homologs was observed in 22 (88 %) of 25 metaphase spreads (Fig. 3). This result is in agreement with the previous mapping to chromosome 5 of STS SHGC-186 in the Radiation Hybrid Stanford G3 panel. Marker SHGC-186 is contained in Uni-Gene cluster Hs. 28555, in which the selected IMAGE clones are included. Searching OMIM (http://www.ncbi.nlm.nih.gov/ htbin-post/Omim), we did not find any known hereditary con-dition mapping to 5q11 that could be associated with PDCD9.
88 Cytogenet Cell Genet 87:85–88 (1999)
The syntenic region in mice, on chromosome 13, also lacks rele-vant mutations or phenotypes related to Pdcd9.
In summary, we have cloned, mapped, and studied the expression of the human homolog of the G. gallus pro-apoptotic protein p52. We have determined that the amino acid sequence is well conserved between human, mouse, and chicken, which suggests the possibility of a similar pro-apoptotic role for PDCD9. The lack of homology with other protein families indi-cates that PDCD9s could constitute a novel class of apoptotic proteins involved in alternative cell-death pathways that have not yet been defined. However, the differences in adult tissue expression might represent the existence of distinct functions of
PDCD9 in human and chicken. Further experiments should be undertaken to elucidate the putative apoptotic role of PDCD9 in humans.
Acknowledgements
We are grateful to Michael Lynch and Mònica Gratacòs for the screening of the Ïgt11 and PAC libraries. We also want to thank A. Puig and D. Otero for their technical support with the DNA sequencing. We wish to thank the HGMP Resource Center in Hinxton, UK, and the DHGP in Berlin, Germa-ny, for supplying us with IMAGE cDNA and PAC clones.
References
Adams MD, Kelley DIM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, Kerlavage AR, McCombie WR, Venter JC: Complementary DNA sequenc-ing: expressed sequence tags and human genome project. Science 252:1651–1656 (1991). Altschul SF, Maden TL, Schaffer AA, Zhang J, Zhang
Z, Miller W, Lipman, DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search program. Nucl Acids Res 25:3389–3402 (1997).
Deloukas P, Schuler GD, Gyapay G, Beasley EM, Sod-erlund C, Rodriguez-Tome P, Hui L, Matise TC, McKusick KB, Beckmann JS, Bentolila S, Biho-reau M, Birren BB, Browne J, Butler A, Castle AB, Chiannilkulchai N, Clee C, Day PJ, Dehejia A, Dibling T, Drouot N, Duprat S, Fizames C, Fox S, Gelling S, Green L, Harrison P, Hocking R, Hollo-way E, Hunt S, Keil S, Lijnzaad P, Louis-Dit-Sully C, Ma J, Mendis A, Miller J, Morissette J, Muselet D, Nusbaum HC, Peck A, Rozen S, Simon D,
Slon-im DK, Staples R,. Stein LD, Stewart EA, Suchard MA, Thangarajah T, Vega-Czarny N, Webber C, Wu X, Hudson J, Auffray C, Nomura N, Sikela JM, Polymeropoulos MH, James MR, Lander ES, Hudson TJ, Myers RM, Cox DR, Weissenbach J, Boguski MS, Bentley DR: A physical map of 30,000 human genes. Science 282:744–746 (1998).
Jacobson MD, Weil M, Raff MD: Programmed cell death in animal development. Cell 88:347–354 (1997).
Lennon G, Auffray C, Polymeropoulos M, Soares MB: The I.M.A.G.E. Consortium: an integrated molec-ular analysis of genomes and their expression. Ge-nomics 33:151–152 (1996).
Nadal M, Moreno S, Pritchard M, Preciado MA, Esti-vill X, Ramos-Arroyo MA: Down syndrome: char-acterisation of a case with partial trisomy of chro-mosome 21 owing to a paternal balanced transloca-tion (15;21) (q26;q22.1) by FISH. J med Genet 34:50–4 (1997).
Schuler GD: Pieces of the puzzle: expressed sequence tags and the catalog of human genes. J molec Med 75:694–69 (1997).
Steller H: Mechanisms and genes of cellular suicide. Science 267:1445–1449 (1995).
Sun L, Liu Y, Fremont M, Schwarz S, Siegmann M, Matthies R, Jost JP: A novel 52 kDa protein induces apoptosis and concurrently activates c-Jun N-terminal kinase 1 (JNK1) in mouse C3H10T1/2 fibroblasts. Gene 208:157–166 (1998).
Thompson CB: Apoptosis in the pathogenesis and treatment of disease. Science 267:1456–1462 (1995).
Vaux DL, Korsmeyer SJ: Cell death in development. Cell 96:245–254 (1999).
II. Identificació, caracterització i mapatge del gen humà VPS33B
El treball següent descriu el procés d’aïllament i caracterització del nou gen humà VPS33B (vacuolar protein sorting 33B), ortòleg a l’identificat prèviament a rata. L’anàlisi de la seqüència aminoacídica va suggerir una implicació en l’organització i transport de proteïnes i vesícules a la cèl.lula. Es va estudiar el seu patró d’expressió i la seva posició en el genoma humà mitjançant mapatge per híbrids de radiació. Aquests resultats van ser publicats l’any 2000 i van refermar l’importància d’aquests treballs de caracterització preliminar de gens nous.
Cytogenet Cell Genet 89:92–95 (2000)
Cloning, mapping and expression analysis of
VPS33B, the human orthologue of rat
Vps33b
L. Carim, L. Sumoy, N. Andreu, X. Estivill and M. Escarceller
Medical and Molecular Genetics Center, Institut de Recerca Oncològica, Hospital Duran i Reynals, L’Hospitalet de Llobregat, Barcelona (Spain)
Supported by EU Biomed Project No. BMH4-CT97-2284 to X.E and by CICYT-IN95-0347. M.E. is funded by the Spanish Ministry of Education (CIDYT con-tract FPI-070-97) and L.S. by the Catalan autonomous government (CIRIT-RED contract 1998-64).
Received 1 December 1999; revision accepted 23 February 2000.
Request reprints from Mònica Escarceller, Medical and Molecular Genetics Center, Institut de Recerca Oncològica, Hospital Duran i Reynals, Autovia de Castelldefels km 2,7, L’Hospitalet de Llobregat, 08907 Barcelona (Spain); telephone: 34-93-260-7775; fax: 34-93-260-7776; e-mail: [email protected]
Abstract. We have identified VPS33B, the human ortholog of rat Vps33b. VPS33B encodes a transcript of 2482 nt with an ORF of 617 amino acids and a predicted protein size of 70.6 kDa. VPS33B contains a Sec-1 domain shared with a family of
proteins involved in protein sorting and vesicular trafficking. Enriched expression of VPS33B was observed in testis. VPS33B was positioned at chromosome 15q26.1 by radiation hybrid mapping.
Copyright © 2000 S. Karger AG, Basel
Our laboratory, a member of the EUROIMAGE Consor-tium, is engaged in the isolation and mapping of novel human genes. We sequence cDNA clones corresponding to ESTs selected on the basis of chromosome location on the long arm of chromosome 15. Using this approach we have identified VPS33B, the human orthologue of rat Vps33b, a mammalian homologue of yeast Slp1/vps33p (Pevsner et al., 1996).
The sorting of vacuolar proteins in the yeast Saccharomyces
cerevisiae has become an important genetic model system due
to the insight it has given into lysosomal biogenesis in animal cells (Conibear and Stevens, 1995; Wendland et al., 1998). To date, few human homologues of the so called vps (vacuolar pro-tein sorting) yeast genes have been described.
Protein transport to the lysosome-like vacuole in yeast is mediated by more than 40 genes in S. cerevisiae. The yeast Vps mutants are implicated in Golgi-to-lysosome trafficking (Coni-bear and Stevens, 1995; Wendland et al., 1998; Pevsner et al.,
1996) and are subdivided into six classes (A–E). Mutations in the so called fourth class C Vps, including Vps33p among oth-ers, result in the most severe vacuolar protein sorting and mor-phology defects. It is believed that Vps gene products physically and functionally interact to mediate a late step in protein trans-port to the vacuole (Rieder and Emr, 1997).
Slp1/Vps33p belongs to the Sec-1 domain family (Halachmi and Lev, 1996). Members of this family are involved in protein sorting, synaptic transmission and general secretion. Some of them are the yeast Sec1, Sly1, Slp1/Vps33p and Vps45; three nematode proteins: Unc-18, and the C. elegans homologues of Sec1 and Sly1; the Drosophila rop and the rat Munc-18/nSec1, mouse Munc18b and Munc18c, and bovine Munc18 and Sec1 (Halachmi and Lev, 1996 and references therein). The Sec1 proteins are mostly hydrophilic and lack a transmembrane domain but are membrane bound proteins. The Sec-1 proteins play a positive role in exocytosis.
Materials and methods
cDNA isolation and sequencing
Unigene cluster Hs. 26510 was built and analyzed in silico during the EUROIMAGE full-length cDNA sequencing project. It was shown to con-tain a single open reading frame (ORF) and to share homology with vacuolar protein sorting proteins. ESTs cluster Hs.26510 (http://www.NCBI.nlm. nih.gov/UniGene) was assembled using the EST CAP assembly program (http://gcg.tigem.it/cgi-bin/uniestass.pl) and Sequencher (GeneCodes) se-quence assembly software. Additional ESTs corresponding to VPS33B but
Cytogenet Cell Genet 89:92–95 (2000) 93
not included in the cluster were found by searching the dbEST database using the BLASTN program (Altschul et al., 1997). IMAGE cDNA clones whose ESTs extended most 5) and 3) in the cDNA were chosen for sequencing: 531662, 2171628, 41400 and 531649. Clones were obtained from the EUROIMAGE distribution centers. Sequence was determined by primer walking using the PerkinElmer BigDye reagents on an ABI PRISM-377 fluo-rescent automated sequencer and custom synthesized sequencing primers (LifeTech).
Full-length cDNA sequence was obtained using the rapid amplification of cDNA ends (RACE) method on Marathon-Ready cDNA from adult human heart (Clontech), according to the manufacturer’s instructions. The following primers were used: G1 (5) AGAGAGTGCTGAGAAGGTG-TAAGGC 3)), G2 (5) ATCCACATCAAGAGGCAGCAAAGAG 3)) and G3 (5) AGCCCTCAAAAGTTCTATGCGTGTG 3)) for 5) VPS33B extension. PCR extended products were subcloned into the pGEM-T-easy vector (Promega) and sequenced as above. We sought at least three independently generated fully extended clones to determine the cDNA ends.
Sequence comparisons were performed using ClustalW 1.7 (http://dot. imgen.bcm.tmc.edu:9331/multi-align/multi-align.html). Boxed multiple se-quence alignments were obtained with the BOXSHADE 3.21 program (http: //www.ch.embnet.org/software/BOX–form.html). To search for known mo-tifs or functional domains, protein pattern and domain databases consulted were Prosite, SMART and Pfam (http://www.hgmp.mrc.ac.uk/Genome-Web/prot-domain.html).
VPS33B nucleotide and protein sequences are available in GenBank under Acc. No. AF201694. The name has been approved by the Human Gene Nomenclature Committee (http://www.gene.ucl.ac.uk/nomenclature/).
Northern blot analysis
A multiple-tissue Northern blot (MTN II blot, Clontech) was hybridized to a 1.9-kb HindIII-PstI restriction product corresponding to the cDNA insert from IMAGE clone 41400; and to a 2-kb ß-actin cDNA supplied com-mercially (Clontech) as control for quantification. Probes were labeled using a random primer DNA labeling kit (Amersham Pharmacia). Blots were hybridized overnight at 65° C in ExpressHyb solution (Clontech) and
washed at 68° C in 0.2 × SSC, 0.5 % SDS.
VPS33B radiation hybrid mapping
To precisely localize the VPS33B gene we used the Stanford TNG4 whole genome radiation hybrid panel (Stewart et al. 1997). Twopoint linkage analy-sis was performed using the RHMAP-2.0 on the RH Server at the Stanford Human Genome Center (http://www-shgc.stanford.edu/RH/index.html). We used primers F (5) CTCAGTGAGATGCAGGCATC 3)) and R (5) TATCCTGGGAGCAGGAAGTG 3)) which amplify STS14369. The PCR conditions were 1 cycle at 94° C for 3 min; 35 cycles at 94 ° C for 30 s, 61 ° C
for 30s and 72° C for 1 min; and 1 cycle at 72 ° C for 5 min.
Results and discussion
Cloning of VPS33B, the human orthologue of rat Vps33b
Within the EUROIMAGE full-length cDNA sequencing project underway in our laboratory (Lennon et al., 1998) we sequence cDNA clones corresponding to ESTs from the same cluster to identify new genes. The EST contigs that we build and analyze in silico represent unique genes and they are select-ed on the basis of clone size, chromosome location and tissue distribution. One of the partial human cDNA sequences, belonging to Unigene cluster Hs.26510, contained a single open reading frame (ORF). Additional ESTs not included in the clus-ter were found by searching the dbEST database using the BLASTN program at NCBI (http://www.ncbi.nlm.nih.gov/ cgi-bin/BLAST/) (Altschul et al., 1997). IMAGE cDNA clones corresponding to the ESTs that extended most 5) and 3) were chosen for sequencing: 531662 (EST GenBank Acc. No.
AA074549), 2171628 (EST GenBank Acc. No. AI589203), 41400 (EST GenBank Acc. No. R56540) and 531649 (EST GenBank Acc. No. AA074575).
Of the four human clones selected, 2171628 and 531649 were unable to grow in standard conditions and clone 531662 was shown to be miss-assigned and corresponded to an uniden-tified cluster of ESTs. Only clone 41400 was fully sequenced and extended the ORF obtained with the EST assembly. Since the clone did not cover the entire transcript, the full-length cDNA sequence was obtained by 5) RACE extension (see Methods). The assembly of the different clones gave as a result a total transcript length of 2482 bp (including the polyA tail), with an ORF (from nt 304–2157) encoding a 617 amino acid product with a calculated mass of 70.6 kDa. The 5) untrans-lated region (UTR) contained an in-frame stop codon at nucleotide position 217. A polyadenylation signal (AATAAA) was observed at nt 2434 and a polyA tail at the end (2458 nt).
A single hit was obtained after BLAST homology searching against non redundant nt databases (NCBI): r-vps33b, the “va-cuolar protein sorting” homolog from Rattus norvegicus (Pevsner et al., 1996). Nucleotide sequence comparison showed 90 % identity between our gene and rat Vps33b.
At the amino acid level, VPS33B showed homology to the family of proteins related to Sec1 (Halachmi and Lev, 1996): after BLAST search, the best hit was r-vps33b with a 96 % iden-tity (97 % similarity) between them (Fig. 1); the second most significant score was rat vps33a, with a 31 % identity (50 % sim-ilarity). The hits following were the “vacuolar protein sorting 33” homologues of yeast SLP1/vps33 in Drosophila,
Arabidop-sis, C. elegans and Aspergillus, as well as SLP1/vps33 itself.
Lower scores were observed for rop (Drosophila), vps45 (yeast) and unc-18 (C. elegans ).
All these close homologues to VPS33B are known members of the Sec-1 domain family and contain this motif (Halachmi and Lev, 1996). Protein pattern analysis of VPS33B revealed the presence of the domain, extending from amino acid 105 to 612, with a significant score of 3.6e-185 (SMART, http://www. hgmp.mrc.ac.uk/GenomeWeb/prot-domain.html). It has been proposed that the evolution of this gene family parallels the spe-cialization of vesicle trafficking to distinct intracellular com-partments (Pevsner et al., 1996). Indeed, loss of function in the yeast genes Sec1, Sly1, Slp1/vps33 and Vps45 results in block-ing of protein transport between distinct subcellular compart-ments, that is, Sly1 from endoplasmic reticulum to Golgi; Sec1 from Golgi to plasma membrane; Vps-45 from Golgi to preva-cuolar and Slp1/vps33 from pre-vapreva-cuolar to the vacuole (Pevsner et al., 1996; Tellam et al., 1997; and references there-in). Because of sequence identity with their yeast counterpart, it has been suggested that rat vps33a and rat vps33b might also participate in vesicular trafficking between the Golgi and the lysosome (Pevsner et al., 1996). Moreover, it is also possible that rat vps33a and rat vps33b localize to distinct intracellular compartments such as lysosomes, endosomes or peroxisomes. Here we suggest, based on the striking amino acid homology shared between human and rat vps33b, that our protein could also be involved in this step in humans. In this context, it will be of great interest to determine the subcellular location of VPS33B in future experiments.
94 Cytogenet Cell Genet 89:92–95 (2000)
Fig. 1. Multiple sequence alignments of human VPS33B (VPS33B); rat vps33b (vps33b); rat vps33a (vps33a); the yeast Slp1/vps33b homologs in
Drosophila melanogaster (D.mel), Aspergillus fumigatus (A.fum), Arabidopsis thaliana (A.thal) and Caenorhabditis elegans (C.el) and SLP1 yeast poly-peptide itself. Identical residues are printed in reverse type, and similar residues are shaded.
Fig. 2. Multiple-tissue Northern blot analysis of VPS33B. The 1.9-kb
HindIII-PstI restriction product was used as a probe revealing ubiquitous expression: (A) results after an overnight exposure and (B) after a 10 day exposure; (C) ß-actin control probe.
Expression of VPS33B
Expression studies of VPS33B with Northern blots of hu-man tissues (MTN II blot, Clontech), were carried out by hybridizing with a specific probe (see Methods). In adult tis-sues, basal expression was largely ubiquitous (Fig. 2), showing an mRNA species migrating above the 2.4-kb marker. Remark-ably, VPS33B expression was highly enriched in testis. This contrasts with the broad tissue distribution of rat Vps33b, where an equal amount of a 2.8-kb transcript was seen in all rat tissues (Pevsner et al., 1996). We suggest that, even though both orthologs display a high degree of identity, the differences in the expression pattern indicate that the gene could hold a spe-cific and distinct role in humans, perhaps in protein trafficking from Golgi to the acrosome during spermatogenesis. This spec-ificity could be mediated through the interaction with other
Cytogenet Cell Genet 89:92–95 (2000) 95 proteins, the syntaxins and syntaxin-like proteins being good
candidates. The genetic interaction between the Sec1-1 family members and genes encoding syntaxin and syntaxin homolo-gues has been shown previously (Bennett et al., 1993; Tellam et al., 1997).
Mapping of VPS33B
Chromosome location of the human VPS33B gene was determined by radiation hybrid mapping using the Stanford TNG4 panel. The gene was linked to STS SHGC-83061 with a LOD score of 7.49 at an approximate distance of 128 kb. This STS is contained in RPCI-11 BAC 51D4 which in turn contains the alpha-mannosidase II isozyme gene (D15S1173). This is in agreement with the previous mapping of STSs mp2120 and WI-22047 using the Genebridge 4 panel (between D15S202 and D15S157), which are located in 15q26.1 near the Bloom syndrome locus (Deloukas et al., 1998). These STSs are con-tained in UniGene cluster Hs. 26510, in which the selected IMAGE clones are included.
In summary, we have identified, characterized and finely mapped, within the 15q26.1 region, a new human gene, VPS33B. Comparisons with previously described genes at the protein and nucleotide level indicate that VPS33B is the human orthologue of rat Vps33b. The presence of the function-al Sec1 domain in the highly conserved amino acid sequence of human and rat vps33b suggests that VPS33B is holding a role in vesicular protein trafficking to the lysosome. Localized expres-sion of VPS33B in testis may account for a specific role of the protein in humans.
Acknowledgments
We are grateful to A. Puig and D. Otero for technical support with DNA sequencing. We wish to thank the HGMP Resource Center in Hinxton, UK, and the RZPD in Berlin, Germany, for supplying us with IMAGE cDNA clones.
References
Altschul SF, Maden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman, DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search program. Nucl Acids Res 25:3389–3402 (1997).
Bennett MK, Garcia-Arraras JE, Elferink LA, Peterson K, Fleming AM, Hazuka CD, Scheller RH: The syntaxin family of vesicular transport receptors Cell 74:863–873 (1993).
Conibear E, Stevens TH: Vacuolar biogenesis in yeast: sorting out the sorting proteins. Review. Cell 83:513–516 (1995).
Deloukas P, Schuler GD, Gyapay G, Beasley EM, Sod-erlund C, Rodriguez-Tome P, Hui L, Matise TC, McKusick KB, Beckmann JS, Bentolila S, Bihor-eau M, Birren BB, Browne J, Butler A, Castle AB, Chiannilkulchai N, Clee C, Day PJ, Dehejia A, Dibling T, Drouot N, Duprat S, Fizames C, Bent-ley DR, et al: A physical map of 30,000 human genes. Science 282:744–746 (1998).
Halachmi N, Lev Z: The Sec1 family: a novel family of proteins involved in synaptic transmission and general secretion. Review. J Neurochem 66:889– 897 (1996).
Lennon G, Auffray C, Polymeropoulos M, Soares MB: The I.M.A.G.E. Consortium: an integrated molec-ular analysis of genomes and their expression. Ge-nomics 33:151–152 (1998).
Pevsner J, Hsu SC, Hyde PS, Scheller RH: Mammalian homologues of yeast vacuolar protein sorting (vps) genes implicated in Golgi-to-lysosome trafficking. Gene 183:7–14 (1996).
Rieder SE, Emr SD: A novel RING finger protein com-plex essential for a late step in protein transport to the yeast vacuole. Mol Biol Cell 8:2307–2327 (1997).
Stewart EA, McKusick KB, Aggarwal A, Bajorek E, Brady S, Chu A, Fang N, Hadley D, Harris M, Hus-sain S, Lee R, Maratukulam A, O’Connor K, Per-kins S, Piercy M, Qin F, Reif T, Sanders C, She X, Sun WL, Tabar P, Voyticky S, Cowles S, Fan JB, Cox DR, et al: An STS-based radiation hybrid map of the human genome. Genome Res 7:422–433 (1997).
Tellam JT, James DE, Stevens TH, Piper RC: Identifi-cation of a mammalian Golgi Sec1p-like protein, mVps45. J biol Chem 272:6187–6193 (1997). Wendland B, Emr SD, Riezman H: Protein traffic in
the yeast endocytic and vacuolar protein sorting pathways. Curr Opin Cell Biol 10:513–522. (1998).
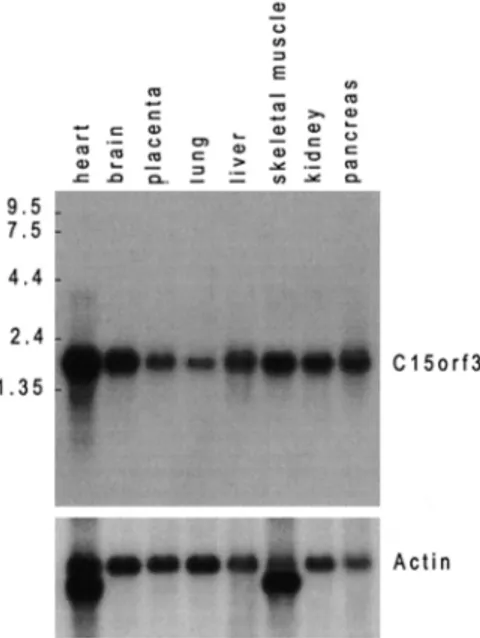
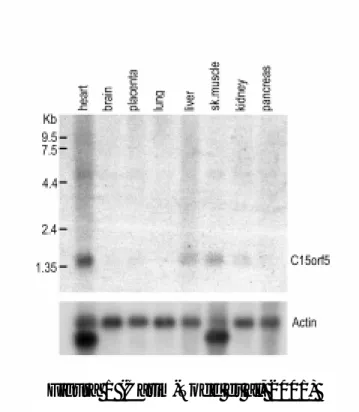
III. Identificació i anàlisi de l’expressió del gen C15orf3
La publicació següent constitueix un exemple d’identificació de gens completament nous. Es tracta de seqüències amb pauta de lectura oberta sense homologia a cap seqüència coneguda en aquell moment que permetès inferir la seva funció o implicació en processos cel.lulars concrets. L’article descriu el mapatge de C15orf3 a 15q21.1-21.2, excloent-lo de la regió q24-q26 objecte d’estudi en la Part II d’aquesta tesi. Tot i no presentar similaritat amb cap família de gens ja coneguts, el patró d’expressió ubicu de C15orf3 suggeria un paper universal i general necessari a totes les cèl.lules humanes. Es van identificar els gens ortòlegs a rata i ratolí confirmant que C15orf3 és membre d’una nova família de proteïnes encara no caracteritzades.