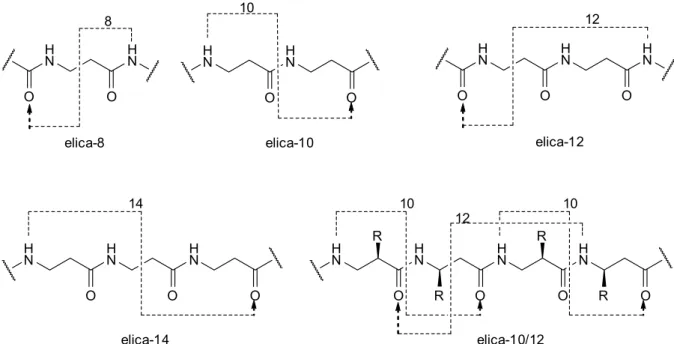
PARTE
TEORICA
Introduzione
1 Gli α-amminoacidi
Gli α-amminoacidi, che più correttamente dovrebbero essere chiamati α-amminocarbossi-acidi, sono caratterizzati dalla presenza di un gruppo amminico e di un gruppo carbossilico legati al medesimo atomo di carbonio, detto carbonio α (Figura 1). La loro formula di struttura generale è H2NCHRCOOH, nella quale R rappresenta un generico sostituente
organico più o meno strutturalmente complesso: si può andare da un semplice atomo di idrogeno, nel caso della glicina, a gruppi alchileteroarilici biciclici, come nel caso del triptofano, a strutture ancora più complesse.
NH2
R H
COOH
Figura 1 Proiezione di Fischer generica di un α-amminoacido proteinogenico.
Gli α-amminoacidi sono i building-block di peptidi e proteine, nei quali sono concatenati per mezzo di un legame ammidico, comunemente detto peptidico.
Esistono 22* amminoacidi proteinogenici, ossia identificati in proteine, ai quali risultano essere associati uno o più codoni (una tripletta di basi azotate) nel codice genetico standard (Figura 2) H2N COOH H H H2N COOH H H2N COOH H H2N COOH H H2N COOH H N H COOH Glicina
(Gly/G) L-Alanina(Ala/A) L-Valina(Val/V) L-Leucina(Leu/L) L-Isoleucina(Ile/I) L-Prolina(Pro/P) H
H
H2N COOH H H2N COOH H H2N COOH H H2N COOH H H2N COOH H H2N COOH H L-Fenilalanina
(Phe/F) L-Triptofano(Trp/W) L-Metionina(Met/M) L-Asparagina(Asn/N) L-Glutammina(Gln/Q) L-Serina(Ser/S) H N S O H2N H2N O OH H2N COOH H H2N COOH H H2N COOH H H2N COOH H H2N COOH H H2N COOH H HO H L-Treonina
(Thr/T) L-Tirosina(Tyr/Y) L-Cisteina(Cys/C) L-selenocisteina(Sec/U) L-lisina(Lys/K) L-Pirrolisina(Pyl/O) OH SH SeH H2N HN N O H H2N COOH H H2N COOH H H2N COOH H H2N COOH H NH NH2 HN L-Arginina
(Arg/R) L-Istidina(His/H) L-acido aspartico(Asp/D) L-acido glutammico(Glu/E) N
HN HO
O
OH O
Figura 2 Formule di struttura dei 22 amminoacidi proteinogenici.
2 β- e γ-amminoacidi
Un quesito molto rilevante, da un punto di vista di cultura chimica, affrontato da vari gruppi di ricerca in maniera sistematica, a partire dal 1996, è il seguente: Che cosa accade se uno o due gruppi CH2 vengono introdotti in ogni building-block amminoacidico nella catena
di un peptide o di una proteina, a dare gli omologhi degli α-amminoacidi proteinogenici? Ancora: qual è l’effetto dell’isomerizzazione di ciascun building-block in un peptide cosicché il gruppo amminico sia spostato dalla posizione 2 alla 3 ? Le alterazioni strutturali alle quali ci si è riferiti sono presentate in Figura 3.
CO H N H R NH R O α-peptide Da L-amminoacidi H-Xaa-OH COOH H2N H R CH2 β-peptidi CH2 H N H R CO N H R O CH2 CH2 H R H N CO β3-omoamminoacidi H-β3hXaa-OH H N O R β2-omoamminoacidi H-β2hXaa-OH γ-peptidi CH2 H2N H R CH2 COOH COOH CH2 H R CH2 H2N CH2 H N H R CH2 CO N H R O γ4-bis(omoamminoacidi) H-γ4hhXaa-OH H N R CO CH2 H R H N O γ3-bis(omoamminoacidi) H-γ3hhXaa-OH CO CH2 H R H N γ2-bis(omoamminoacidi) H-γ2hhXaa-OH N H R O COOH H2N H R β-isoamminocidi CH2 H2N H R COOH COOH H2N H CH2 R α-amminoacidi
H-Xaa-OH β-isoamminoacidiH-βiXaa-OH
(Lo spostamento del gruppo NH2nonè tuttavia possibile in Gly, Ser, Cys, Thr)
Figura 3 Formule di struttura e nomenclatura per le abbreviazioni utilizzate per i β- e γ-peptidi derivanti dai 22 amminoacidi proteinogenici naturali. Una descrizione puntuale della nomenclatura dei building-block omologati è fornita nel testo.
Nel caso dei β-omo- e dei γ-omo-omoamminoacidi*, le catene laterali rimangono inalterate e la posizione del residuo è individuata dall’apice (β2, β3, γ2, γ3, γ4), in presenza di più di un residuo sono inseriti più numeri all’apice (es. β2,2, β2,3, γ2,4
, γ2,3,4). L’amminoacido
monomologato viene individuato mediante la lettera minuscola h [5], per un building-block non protetto alle estremità si utilizza la scrittura H-βnhXaa-OH (con n = 2 o 3), analogamente per un γ-ammino acido H-γnhhXaa-OH (con n = 2, 3 o 4).
I β-iso-amminoacidi, derivanti dallo scambio formale del gruppo α-NH2 con uno dei due
β-H, sono abbreviati H-βiXaa-OH†.
3 β-amminoacidi in natura
In natura sono presenti più β-amminoacidi che α-amminoacidi proteinogenici, per non parlare dei γ-amminoacidi, identificati tal quali, o come building-block in prodotti naturali, più o meno strutturalmente complessi‡.
Un insieme non completo di β-amminoacidi naturali, con relativa origine e riferimenti, è mostrato in Figura 4; nella successiva Figura 5 sono mostrate le formule di struttura di alcuni composti naturali contenenti β- e γ-amminoacidi quali building-block [14, 24, 31, 39-41] (per uno anche la relativa struttura cristallina ai raggi-X). Come è facile notare, esiste un’ampia variabilità strutturale e di gruppi funzionali.
Probabilmente il più comune, e forse più importante, β-amminoacido è la H-βhGly-OH, o ‘β-alanina’ (un amminoacido “essenziale”, componente dell’acido pantotenico, del coenzima A, e della carnosina nel tessuto muscolare). Come è possibile verificare
*
I termini “homophenylalanine” e “homopeptide” sono stati usati per la prima volta da Ondetti et al. [3]. Al contrario le espressioni “β-“ e “γ-peptide” sono state introdotte da Dado e Gellman [4].
† Esiste pure la possibilità di avere un altro tipo di β-iso-amminoacidi, derivante dallo “scambio” del
carbossile con uno dei due atomi β-H; ad esempio, R-CH2-CH(NH2)-COOH → R-CH(COOH)-CH2NH2 (un β2
-amminoacido, la cui catena laterale è omologa inferiore rispetto a quella dell’amminoacido proteinogenico) o prolina → 3-carbossipirrolidina.
‡
mediante una ricerca sul Cambridge Structural Database, l’angolo diedro (N–CH2–CH2–
CO) mostra un’ampia variabilità; la flessibilità conformazionale derivante rende non solo la
H2N
COOH R
COOH
Asp(R = H) [8]
nella microcisitina E (R = Me) [9] H2N CONH2 COOH Asn[8] H2N COOH βhGly[10] (= βAla) ('beta-alanina') nell'acido pantotenico [10] nella carnosina [11] nella anserina [11] nel coenzima A [10] nella esocolina [12] nella nefilatossina [13] dall'uracile [11] dalla teonellapeptolide [14] H2N COOH β2hAla (=βiAib) nella criptoficina [15] dalla timina [11] H2N COOH β3hPgl Ar = Ph: nell'astinene [18] nella nicastrina [19] Ar = 4-OH-C6H4: nei geodiamolidi [20] nelle ocratossine [21] Ar H2N COOH β3hVal (= βiLeu) dalla leucina [16] dall'acido 3-ossocapronico [17] H2N COOH (S)-β2hAla dalla valina [11], [22] Me3N NHR CO2 emeriamina (R = H) [23] emericedina A (R = Ac) H2N COOH (CH2)3NH2 β3hOrn[16] (= βiLys) nelle streptotricine [24] H2N COOH β3hAib
nella fenammide [25] H2N COOH NH2 acido-2,3-diamminocapronico (R = H) [26] ed acido butanoico (R = Me) [27] nella ciclocinammide A [28] R H2N O OH R3 R4 R1 R2 RHN COOMe βhGly(α-metilene) R = Me(CH2)nCO (n = 14 - 18) [29] R = Me(CH2)nOCO, R = Me(CH2)nCH(OH)CO (n = 11 - 13) [29b] H2N COOH R OH R = Ph nel tassolo [19] R = Bn(2S,3R)-β3hPhe(α-OH) nella bestatina [30] R = H (2S)-β3hGly(α-OH) nella ciclocinammide A [28] (2R)-β3hGly(α-OH) nelle cheramammidi [31] H2N COOH R O R = Meβ3hAla(α-cheto) nelle euristatine [32] R =iBuβ3hIle(α-cheto) nelle cheramammidi [31] NH2 COOH H cispentacene [33] N NH2 R H H O nicotinammide [10] N H COOH β3hPro acido-2-(2-pirrolidinil)acetico [34] HN O OH N H COOH OH O NH2 [35] N H COOH R nella siastatina [36] nella cocaina [10] N H R O X nelle ergotamine [37] nell'LSD [37] O H2N COOH H ossetina [38] N O S NH2 COOH OH H H tienamicina [39]
β-alanina un ideale building-block per eliche e ripiegamenti, ma anche per
l’incoroporazione in segmenti peptidici lineari (Figura 5).
E’ da notare che β-amminoacidi si trovano nella struttura peptidica di composti altamente attivi fisiologicamente (Figure 4 e 5), frequentemente isolati da piante od organismi marini [42]. La biosintesi ha luogo in complessi enzimatici modulari, le peptido-sintetasi non ribosomiali (NRPS) [43], (comparabili alle polichetido-sintetasi (PKSs) [44]), come è stato dimostrato dal sequenziamento di numerosi geni e frammenti di geni NRPS in batteri e funghi. La macchina NPRS è in grado di far reagire non solo gli amminoacidi proteinogenici, ma anche numerosi α-amminoacidi non-proteinogenici, incluso quelli di configurazione D, N-metil amminoacidi, β-, γ-, δ- ed ε-derivati amminoacidici, nonché vari idrossiacidi (incluso l’acido β-idrossibutanoico); più di 300 substrati sono stati identificati attualmente [43]. I prodotti delle NPRS, che spesso sono di tipo macrociclico, mostrano numerose attività biologiche, tra di essi sono infatti stati riconosciuti composti tensidioattivi, sideroforici, antibiotici, fungicidi, antivirali, citotossici, neurotossici, nefrotossici, cancerostatici ed immunosoppressivi.
La formazione di legami β-peptidici in una proteina, mediante riarrangiamento di un residuo di acido aspartico od asparagina (Schema 1A), è una reazione non-enzimatica che procede spontaneamente in alcune regioni caratteristiche, come segmenti Asn-Gly, Asn-Ser, Asp-Gly ed Asn-His (tempo di emivita da poche ore ad un mese a 37°C). Questo processo di invecchiamento proteico è un’alterazione strutturale, che può essere accompagnata da perdita di attività e funzione; è stato pure individuato nelle placche formate da frammenti di peptide β-amiloide nel cervello di pazienti affetti dal morbo di
Alzheimer. Per la riduzione dei danni fisiologici, cagionati dalla formazione di isoaspartato,
esiste un enzima riparatore, la L-isoaspartato-(D-aspartato)-O-metil-transferasi (PIMT), identificata in numerosi organismi e della quale è disponibile pure la struttura a raggi X [45].
cromoforo della chedarcidina (antitumorale ed antibiotico) cheramamidi (citotossici) polimissina B2 (antibiotico) nourseotricine (n = 1 - 7) (antibiotici, nefrotossici) microcistine (epatossiche) teonellapeptolide (citotossico)
Figura 5 Alcuni prodotti naturali contenenti building-block β- e γ-amminoacidici. La cheradicina appartiene alla classe degli endini e contiene residui di acido-β-ammino-β-arilpropanoico. La microcistina è stata isolata da un cianobatterio; questa famiglia di composti contiene, oltre ad un residuo di acido-β-amminodecadienoico, un’unità di acido aspartico legata in modo β- ed una di acido glutammico in modo γ-. La teonella-peptolide rappresenta un esempio di cheramamide; i composti precedenti sono peculiari per la presenza di tre ulteriori tipi di β-amminoacidi funzionalizzati (un α-idrossi-, un α-ammino- ed un un α-cheto-β-amminoacido), l’ultima specie citata presenta tre residui di βhGly, che allo stato cristallino sono presenti in
conformazione tipo gauche attorno al legame C-C centrale. La polimissina-B2 è stata estratta dal Bacillus
polymixya e contiene un totale di sei building-block di acido-α,γ-diamminobutanoico (ossia ornitina), cinque dei quali sono concatenati con un legame α-ammidico ed uno δ-ammidico; quattro sono parte di un macrociclo. Le nourseotricine e le streptotricine posseggono un ampio spettro di attività (contro virus, batteri, funghi, insetti e pesci), ma sono pure tossiche per mammiferi; la catena di βhLys con il legame ε-peptidico è tipica.
Nel metabolismo dei mammiferi, esistono diverse vie attraverso le quali β-amminoacidi non-proteinogenici possono essere prodotti (Schema 1):
Schema 1 A) Asparagina β-peptidica in proteine [45] e B) β-amminoacidi come intermedi metabolici da basi nucleiche [46] o C) e D) da α-amminoacidi [16,22,46.] H N N H H N O O O Rn Rn+2 Z O spontaneo enzima PIMT H N N H COOH O Rn O N H O Rn+2 -Xaa-Asp-Yaa- (Z = OH) -Xaa-Asn-Yaa- (Z = NH2) -Xaa-(L-isoaspartato)-Yaa-A) B) HN N H O O uracile (R = H) timina (R = Me) NAPDH diidrouracil deidrogenasi R HN N H O O R +H2O, -CO2,-NH3 diidropirimidinasi β−ureidopropionasi HO H2N O R C) H2N COOH L-valina transaminasi deidrogenasi deidrogenasi idratasi idrolasi deidrogenasi transaminasi H2N COOH (S)-H-β2hAla-OH D) R NH2 COOH R = Ph R =iPr R = NH2(CH2)3 fenilalanina- leucina-lisina- 2,3-amminomutasi R COOH NH2 H-βiPhe-OH H-βiLeu-OH H-βiLys-OH (= β3hPgl) (= β3hVal) (= β3hOrn) .
- degradazione delle basi azotate pirimidimiche (uracile, citosina e timina) in β-alanina (H-βhGly-OH) od in acido (R)-3-ammino-2-metilpropanoico (H-β2hAla-OH) [46] (Schema 1B);
- formazione dell’acido (S)-3-ammino-2-metilpropanoico dalla valina per mezzo di una successione di reazioni enzima-catalizzate [22] [46] (Schema 1C);
- amminazione riduttiva dell’acido 4-metil-3-ossopentanoico (‘acido β-ossoisocaproni-co’) nella H-βhVal-OH [17];
- riarrangiamento, da parte di una 2,3-amminomutasi, di un α-amminoacido nel corrispettivo β-isoamminoacido (Schema 1D) [16, 47-8].
4 Lavori di letteratura datati sui β-peptidi: troppi o troppo pochi residui L’idea di incorporare un β-amminoacido in una catena α-peptidica “normale” non è recente: nel 1928, E. Abherhalden, uno studente di Emil Fischer [49], considerava questa una potenziale strategia per accrescere la stabilità enzimatica di un peptide [46], quantunque tale opinione non rimase incontrastata [50]. Da allora sono stati i chimici farmaceutici che, in ricerche decennali di sostanze peptidiche attive – come ad esempio ormoni peptidici – aventi proprietà superiori rispetto alle controparti puramente α-peptidiche, hanno preparato e testato peptidi contenenti singoli amminoacidi omologati in varie posizioni [46]; la letteratura dei brevetti e le riviste di chimica medicinale sono piene di esempi*.
Baralam e Karle, assieme ad i rispettivi collaboratori, hanno recentemente condotto alcune
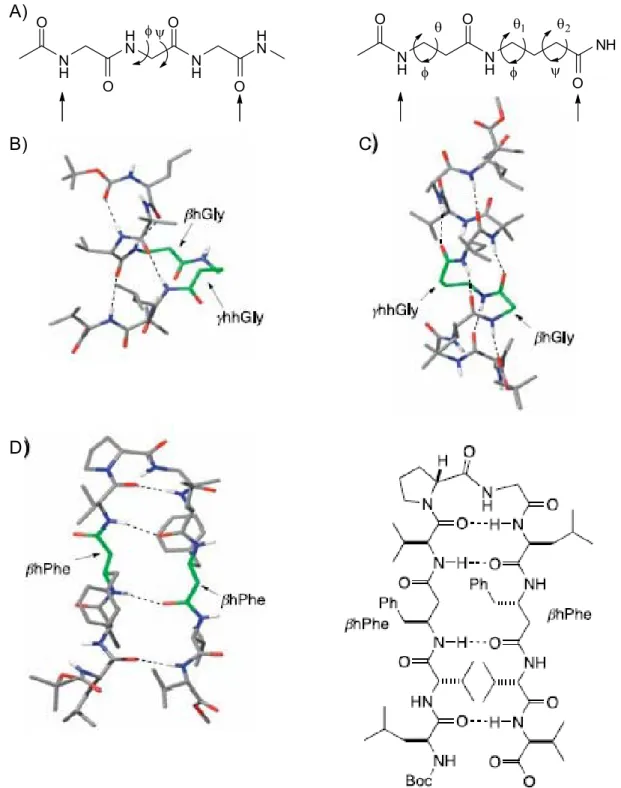
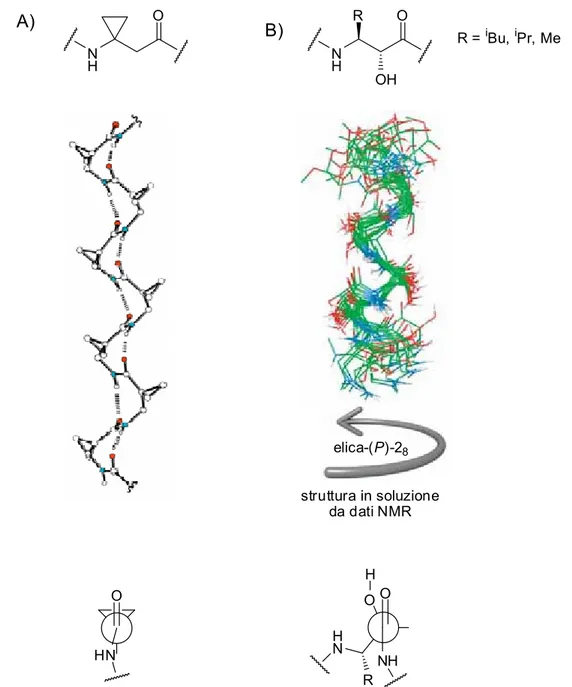
virtuose “mutazioni” sistematiche di questo tipo, pubblicando la struttura di eliche e ripiegamenti peptidici, recanti amminoacidi omologati di questo tipo incorporati nella catena cosicché venga preservata, od incrementata, la stabilità della corrispettiva struttura secondaria allo stato solido (ed in soluzione), come nel caso delle struttura peptidiche contenenti βhGly-, γhhGly-, β3hPhe- mostrate nella seguente Figura 6.
N H H N N H H N N H NH NH O O O O O O O φ ψ φ θ φ ψ θ1 θ2 A) B) C) D)
Figura 6 Strutture di peptidi ibridi composti da α-, β- o γ-amminoacidi. A) Un segmento di (Gly)3 in una
catena peptidica può essere sostituito da (βhGly-γhhGly) senza che il pattern di legame 1,9 tra accettore (NH) e donatore (CO) ne risulti pregiudicato. B) L’ottapeptide Boc-Leu-Aib-Val-βhGly-γhhGly-Leu-Aib-Val-OMe esiste allo stato cristallino come un elica, con sole piccole differenze strutturali rispetto all’analogo α-peptidico “puro”. C) L’undecapeptide Boc-Leu-Aib-Val-βhGly-γhhGly-Leu-Aib-Val-Ala-Leu-Aib-OMe presenta una struttura elicoidale sia in soluzione che allo stato cristallino. D) Un ripiegamento ottenuto in un decapeptide, nel quale sono presenti due amminoacidi omologati (βhPhe 3 ed 8).
β-peptidi ‘puri’ si trovano in articoli particolarmente datati di letteratura [53-8]. Drey et al.
β3,3hAib, (R)- ed (S)-β3hAla tra il 1970 ed il 1986, per i quali, tuttavia, non sono state specificate né le strutture secondarie né le proprietà fisiologiche; lo stesso per oligomeri β-peptidici di acido L-aspartico [57].
Inoltre, i chimici dei polimeri non hanno esaminato solo il Nylon-6, il polimero derivante dall’ε-caprolattame, ma pure il Nylon-5, -4 e -3, ottenuti rispettivamente mediante polimerizzazione dei corrispettivi δ-, γ-, β-lattami [59], nonostante la preparazione e le proprietà dei polimeri β-peptidici fossero assolutamente non competitive con quella del
Nylon-6 o del Nylon-6,6. Polimeri ottenuti dal β-lattame dell’acido-3-amminobutanoico
[60-61] e dal monoestere dell’acido aspartico [62-4], racemi od enantiopuri, o prodotti di policondensazione del 4-nitrofenil-3-pirrolidina-3-carbossilato [65], con masse molecolari dell’ordine di 105 Da, sono stati studiati nei lavori di Bestian, Graf e collaboratori [59],
Kovacs et al., Schmidt [60], Yuki et al. [62,65] e Goodman e collaboratori [61,66] negli anni
1960 e 1970. Gli spettri CD ed NMR forniscono indicazioni dell’esistenza di una struttura ordinata, la cui natura (elica destrorsa o sinistrorsa, catena lineare, foglietto planare) appariva controversa.
Studi di diffrazione a raggi-X di fibre allungate dell’estere dell’acido poliaspartico, condotti dal gruppo di Subirana e Muñoz Guerra hanno fornito dati strutturali –supportati da modelli molecolari – di catene polimeriche β-peptidiche: numerose eliche sono state identificate, in dipendenza dalla natura del raggruppamento estereo (COOR), tra le quali ad esempio un’elica 13/4 [64] (Figura 7).
Il fatto che la catena polimerica β-peptidica adotti una struttura secondaria non è di per sé sorprendente: anche polimeri ‘achirali’ semplici, come la poliformaldeide od il polietilene ossido, presentano una conformazione elicoidale (come ad esempio il polipropilene isotattico (elica-31) od il poliestere dell’acido β-idrossibutirrico (PHB)). In assenza della
NMR in soluzione [67]. Peraltro, un polimero ad alta massa molecolare (distribuzione gaussiana di masse molecolari) non presenta la medesima struttura di un oligomero, analogamente costituito, o di un composto macromolecolare uniforme (com’è ad esempio una proteina od un polipeptide), l’influenza da parte del grado di polimerizzazione e della tatticità risulta essere drammatica.
H N H OH O m n polimero di Ny lon-m (m = 1 - 6) H N H OH O n polimero dell'acido (S)-3-amminobutanoico (H-β3hAla-OH) H N H OH O n O O R H polimero dell'estere dell'acido L-aspartico H N OH H O n polimero dell'acido (R)-pirrolidin-3-carbossilico ('β-prolina') poli-acido-3- pirrolidin-carbossilico poli-β3hAla (film) elica-174(α-Me) elica-134(α-Pr)
Figura 7 Polimeri composti da building-block β-amminoacidici. Spettri CD e due strutture ottenute da studi diffrattometrici a raggi-X su fibre allungate di derivati di acido poliaspartico. Le poliammidi composte da α-, β- γ-, δ-, od ε-amminoacidi sono note come n-nylon; esse sono preparate dai corrispettivi lattami, proprio come i polimeri β-peptidici sintetizzati dall’acido-β-amminobutanoico e dall’emiestere dell’acido aspartico.Il polimero della β-prolina è ottenuto per condensazione del 4-nitrofenilpirrolidin-3-carbossilato. Lo spettro CD suggerisce l’esistenza di strutture secondarie chirali. Esperimenti di diffrazione a raggi-X condotti su fibre β-peptidiche allungate dell’estere dell’acido poliaspartico forniscono dati strutturali dai quali è possibile dedurre diverse conformazioni elicoidali ((P)-134 e (P)-174, in nomenclatura cristallografica, ossia 13 o 17 residui ogni
Bibliografia
[1] A. Böck, K. Forchhammer, J. Heider, W. Leinfelder, G. Sawers, B. Veprek, F. Zinoni, Mol. Microbiology 1991, 5, 515.
[2] B. Hao, W. Gong, T. K. Ferguson, C. M. James, J. A. Krzycki, M. K. Chan, Science 2002, 296, 1462. [3] M. A. Ondetti, J. Pluščec, E. R. Weaver, N. Williams, E. F. Sabo, O. Kocy, in “Chemistry and Biology of Peptides, Proceedings of the Third American Peptide Symposium”, Ed. J.Meierhofer, Ann Arbor Science, Ann Arbor, Michigan, 1972, pp. 525.
[4] G. P. Dado, S. H. Gellman, J. Am. Chem. Soc. 1994, 116, 1054.
[5]S. A. W. Gruner, V. Truffault, G. Voll, E. Locardi, M. Stöckle, H. Kessler, Chem. – Eur. J. 2002, 8, 4365.
[6] M. Brenner, Diss. ETH No. 14409, ETH-Zürich 2001.
[7] J. Podlech, Angew. Chem. Int. Ed. 1999, 38, 477-8; J. Podlech, Cell. Mol. Life Sci. 2001, 58, 44.
[8] G. C. Barrett, D. T. Elmore, ‘Amino Acids and Peptides’, Cambridge University Press, Cambridge, 1998. [9] R. Luukkainen, K. Sivonen, M. Namikoshi, M. Färdig, K. L. Rinehart, S. I. Niemelä, Appl. Environ. Microbiol. 1993, 59, 2204.
[10] Lehrbücher der Medizin, der Biologie, der Biochemie, der Medizinalchemie und der Organischen Chemie.
[11] S. Abele, D. Seebach, Eur. J. Org. Chem. 2000, 1. [12] L. Dong, M. J. Miller, J. Org. Chem. 2002, 67, 4759.
[13] M. Kobayashi, S. Aoki, N. Ohyabu, M. Kurosu, W. Wang, I. Kitagawa, Tetrahedron Lett. 1994, 35, 7969. [14] M. Doi, T. Ishida, M. Kobayashi, Y. Katsuya, Y. Mezaki, M. Sasaki, A. Terashima, T. Taniguchi, C. Tanaka, Biopolymers 2000, 54, 27.
[15] M. Kobayashi, M. Kurosu, N. Ohyabu, W. Wang, S. Fujii, I. Kitagawa, Chem. Pharm. Bull. 1994, 42, 2196.
[16] J. M. Poston, J. Biol. Chem. 1976, 251, 1859. [17] J. M. Poston, J. Biol. Chem. 1984, 259, 2059.
[18] H. Morita, S. Nagashima, Y. Uchiumi, O. Kuroki, K. Takeya, H. Itokawa, Chem. Pharm. Bull. 1996, 44, 1026.
[19] L. Ettouati, A. Ahond, O. Convert, C. Poupat, P. Potier, Bull. Soc. Chim. Fr. 1989, 687.
[20] W. F. Tinto, A. J. Lough, S. McLean, W. F. Reynolds, M. Yu, W. R. Chan, Tetrahedron 1998, 54, 4451. [21] R. D. Wei, F. S. Chu, Experientia 1974, 30, 174.
[22] K. Tanaka, L. E. Rosenberg, ‘Disorders of Branched Chain Amino Acids and Organic Metabolism’, in ‘The Metabolic and Molecular Based of Inherited Disease’, Eds. J. B. Stanbury, J. B. Wyngaardem, D. S.
Fredrickson, J. L. Goldstein, M. S. Brown, 5th edn., McGraw-Hill, New York, 1983, 440.
[23] S. Shinigawa, T. Kanamaru, S. Harada, M. Asai, H. Okazaki, J. Med. Chem. 1987, 30, 1458. [24] S. Kusumoto, Y. Kambayashi, S. Imaoka, K. Shima, T. Shiba, J. Antibiotics, 1982, 35, 925.
[25] N. S. Makkar, T. E. Nickson, M. Tran, N. Biest, M. Miller-Widemann, J.Lawson, C. I. McGary, R. Stonard, J. Antibiot. 1995, 48, 369.
[26] A. S. Seneviratne, L. Fowden, Phytochemistry 1968, 1039.
[27] G. J. Shaw, P. J. Ellingham, A. Bingham, G. J. Wright, Phytochemistry, 1982, 1635. [28] W. D. Clark, T. Corbett, F. Valeriote, P. Crews, J. Am. Chem. Soc. 1997, 119, 9285. [29] a) Y. Kashman, L. Fishelson, I. Ne’eman, Tetrahedron 1973, 29, 3655.
[30] H. Suda, T. Takita, T. Aoyagi, H. Umezawa, J. Antibiot. 1976, 29, 100.
[31] H. Uemoto, Y. Yahiro, H. Shingemori, M. Tsuda, T. Takao, Y. Shimonishi, J. Kobayashi, Tetrahedron 1998, 54, 6719.
[32] H. H. Wasserman, A. K. Petersen, J. Org. Chem. 1997, 62, 8972.
[33] T. Iawamoto, E. Tsujii, M. Ezaki, A. Fujie, S. Hasimoto, M. Okuhara, M. Kohsaka, H. Imanaka, K. Kawabata, Y. Inamoto, K. Sakane, J. Antibiot. 1990, 43, 1.
[34] C. M. Paßreiter, Phytochemistry 1992, 31, 4135.
[35] P. A. Tayler, H. K. Schnoes, R. D. Durbin, Biochim. Biophys. Acta 1972, 286, 107.
[36] Y. Kawase, M. Takahashi, T. Takatsu, M. Arai, M. Nakajima, K. Tanzawa, J. Antibiot. 1996, 49, 61. [37] G. A. Cordell, ‘Introduction to Alkaloids: A Biogenetic Approach’, John Wiley, New York, 1981.
[38] S. Omura, M. Murata, N. Imamura, Y. Iwai, H. Tanaka, A. Furusaki, T. Matsumoto, J. Antibiot. 1984, 37, 1324.
[39] W. Dürckheimer, J. Blumbach, R. Lattrell, K. H. Scheumann, Angew. Chem. Int. Ed. 1985, 97, 180. [40] S. Kawata, S. Ashlizawa, M. Hirama, J. Am. Chem. Soc. 1997, 119, 12012.
[41] J. A. Orwa, C. Govaerts, R. Busson, E. Roets, A. Van Schepdael, J. Hoogmartens, J. Chromatogr. A 2001, 912, 369.
[42] G. Cardillo, C. Tomasini, Chem. Soc. Rew. 1996, 25, 117.
[43] H. Kleinkauf, H. von Döhren, Eur. J. Biochem. 1996, 236, 335; S. Donadio, P. Monciardini, M. Sosia, Natural Product Rep. 2007, 24, 1073.
[45] M. M. Skinner, J. M. Puvathingal, R. L. Walter, A. M. Friedman, Structure 2000, 8, 1189; N. Thapar, S. C. Griffith, T. O. Yeates, S. Clarke, J. Biol. Chem. 2001, 277, 1058; J. L. Radkiewicz, H. Zipse, S. Clarke, K. N. Houk, J. Am. Chem. Soc. 2001, 123, 3499; Y.-M. Kuo, T. A. Kokjohn, T. G. Beach, L. I. Sue, D. Brune, J. C. Lopez, W. M. Kalback, D. Abramowski, C. Sturchler-Pierrat, J. Biol. Chem. 2001, 276, 12991.
[46] O. W. Griffith, Ann. Rew. Biochem. 1986, 55, 855.
[47] P. A. Frey, G. H. Reed, Arch. Biochem. Biophys. 2000, 382, 6. R. LoBrutto, V. Bandarian, O. T. Magnusson, X. Chen, V. L. Schramm, G. H. Reed, Biochemistry 2001, 40, 9.
[48] L. Zhao, S. Borisova, S.-M. Yeung, H.-W. Liu, J. Am. Chem. Soc. 2001, 123, 7909. [49] H. T. Hanson, E. L. Smith, J. Biol. Chem. 1948, 175, 833.
[50] M. Rodriguez, P. Fulcrand, J. Laur, A. Aumelas, J. P. Bali, J. Martinez, J. Med. Chem. 1989, 32, 522. D. L. Steer, R. A. Lew, P. Perlmutter, A. I. Smith, M.-I. Aguilar, J. Peptide Sci. 2000, 6, 470.
[51] H. N. Gopi, G. Ravindra, P. P. Pal, P. Pattanaik, H. Balaram, P. Balaram, FEBS Lett. 2003, 535, 175. [52] M.-I. Aguilar, A. W. Purcell, R. Devi, R. Lew, J. Rossjohn, A. I. Smith, P. Perlmutter, Org. Biomol. Chem. 2007, 5, 2884.
[53] C. N. C. Drey, J. Lowbridge, R. J. Ridge, J. Chem. Soc. Perkin Trans. 1 1973, 2001. [54] C. N. C. Drey, E. Mtetwa, J. Chem. Soc. Perkin Trans. 1 1982, 1587.
[55] D. N. J. White, C. Morrow, P. J. Cox, C. N. C. Drey, J. Lowbridge J. Chem. Soc. Perkin Trans. 2 1982, 239.
[56] C. N. C. Drey, ‘The Chemistry and Biochemistry of β-Amino Acids’, in ‘Chemistry and Biochemistry of Amino Acids, Peptides and Proteins, Vol. 4’, Ed. B. Weinstein, Marcel Dekker, New York, 1977, 241.
[57] M. Kajtár, M. Hollósi, Z. Reidi, Acta Chim. Acad. Sci. Hung. 1976, 88, 301.
[58] T. Weller, L. Alig, M. H. Müller, W. C. Kouns, B. Steiner, Drug Future 1994, 19, 461.
[59] R. Graf, G. Lohaus, K. Börner, E. Schmidt, H. Bestian, Angew. Chem. Int. Ed. 1962, 1, 481; H. Bestian, Angew. Chem. Int. Ed. 1968, 7, 278.
[60] E. Schmidt, Angew. Makromol. Chem. 1970, 14, 185.
[61] F. Chen, G. Lepore, M. Goodman, Macromolecules 1974, 7, 779. [62] H. Yuki, Y. Taketani, J. Polym. Sci. Part B: Polym. Lett. 1972, 10, 373.
[63] J. M. Fernández-Santín, J. Aymamí, A. Rodríguez-Galán, S. Muñoz-Guerra, J. A. Subirana, Nature 1984, 311, 53.
[66] D. F. Bradely, M. Goodman, A. Felix, R. Records, Biopolymers 1966, 4, 607. [67] G. Monaco, A. Zambelli, Macromol. Chem. and Phys. 2005, 206, 203.
Capitolo 1
Sintesi e purificazione di peptidi
In questo capitolo sarà trattata, da una prospettiva storica e con particolare riferimento allo
“stato dell’arte”, la sintesi e la purificazione di α-peptidi. Nella parte finale, la prospettiva
sarà spostata sui β-peptidi.
1.1 Confronto dei metodi di ottenimento di peptidi e proteine
I peptidi e le proteine svolgono un ruolo fondamentale in numerosi processi biologici e fisiologici, per questa ragione sono stati oggetto di numerosi studi. Il primo step, affinché ciò sia possibile, consiste nell’isolamento della specie in quantità e grado di purezza accettabile; a tale scopo esistono sostanzialmente tre metodi:
- isolamento della proteina nativa da sorgenti naturali;
- tecniche ricombinanti per l’espressione di proteine in microrganismi; - sintesi chimica.
Ciascuna presenta ovviamente dei limiti e dei vantaggi, ad esempio nel caso dell’isolamento da fonti naturali, la quantità di peptide desiderato potrebbe essere consistentemente scarsa (anche dell’ordine dei nanogrammi) ed inoltre potrebbero essere necessarie complesse e tediose procedure di purificazione. Al contrario, la sintesi chimica offre la possibilità di ottenere in quantitativi significativi il peptide desiderato a grado di purezza elevato. D’altronde i tempi richiesti per una sintesi chimica di un peptide/proteina, utilizzando anche le tecniche più avanzate, non sono assolutamente comparabili con la biosintesi in vivo, per la quale sono richiesti, anche nel caso di proteine particolarmente complesse strutturalmente, tempi dell’ordine del secondo, al più minuti. La sintesi peptidica offre però l’incomparabile ventaggio di poter incorporare nella sequenza amminoacidi non naturali. Dalla prima sintesi di un dipeptide nel 1901 ad opera di Emil
Fischer, la sintesi peptidica ha compiuto enormi progressi, tanto che oggigiorno è possibile sintetizzare ordinariamente proteine composte da ben 200 amminoacidi.
1.2 Sintesi peptidica di α-peptidi
La formazione di un legame peptidico tra due amminoacidi, a dare un dipeptide, potrebbe apparire come un processo chimico molto semplice: formazione di un legame ammidico (peptidico) accompagnata dall’eliminazione di acqua (Schema 1.1).
Schema 1.1 Formazione di un legame peptidico (ammidico) tra due generici amminoacidi.
H2N OH O R1 H2N OH O R2 - H2O H2N O R1 H N OH O R2 legame peptidico dipeptide
La formazione del legame peptidico in condizioni blande può essere tuttavia ottenuta solo dopo attivazione del gruppo carbossilico dell’amminoacido (A); il secondo (B) attacca nucleofilicamente il composto carbossi-attivato, con la conseguente formazione del dipeptide A-B (Schema 1.2).
Schema 1.2 Coupling peptidico mediante attivazione generica del partner di reazione carbossilico.
H2N X O R1 H2N OH O R2 - HX H2N O R1 H N OH O R2 (A) componente carbossi-attivato (B) componente amminico (A-B) dipeptide X = gruppo attivante
La pubblicazione nel 1901, da parte di E. Fischer (con E. Forneau) [1], della preparazione del dipeptide glicilglicina, per idrolisi della dichetopiperazina della glicina, è considerata l’inizio della chimica peptidica. Ad onor del vero, T. Curtius aveva sintetizzato e caratterizzato, circa 20 anni prima, durante i suoi studi di dottorato con H. Kolbe, il primo
dipeptide N-protetto, la benzoilglicilglicina, per mezzo del trattamento del sale d’argento della glicina con cloruro di benzoile [2] (Schema 1.3).
Schema 1.3 La prima sintesi di un dipeptide di T. Curtius ed in seguito E. Fischer.
Theodor Curtius (1857-1928) H2N OAg O Cl O 2 2 O N H H N OH O O COOH 2AgCl Emil Fischer (1852-1919) HN NH O O HCl conc Δ H3N H N OH O Cl O
Il lavoro intensivo di T. Curtius con i diazocomposti ha condotto allo sviluppo del primo metodo pratico di sintesi peptidica: il metodo di coupling azidico, utilizzato con successo nella sintesi di peptidi benzoilglicinici di una definita lunghezza (Schema 1.4) [3]. E.
Fischer, al contrario, sviluppò un metodo di coupling peptidico basato sull’uso di acilcloruri,
preparati dal corrispettivo amminoacido libero utilizzando PCl5 e cloruro di acetile come
solvente [4].
I problemi maggiori di ambedue i metodi derivavano dalle difficoltà di reperire all’epoca L-amminoacidi enantiopuri e dall’assenza di gruppi protettivi facilmente rimovibili, il cui utilizzo è imprescindibile nella sintesi peptidica moderna.
Schema 1.4 Sintesi di benzoilpentaglicine per mezzo della metodologia di coupling azidico. O N H N3 O H2N H N O O O - NH3 O N H H N O N H O O O 1)NH2NH2 O N H H N O N H N3 O O NH2CH2CONHCH2COOC2H5 O N H H N O N H O O H N N H O O O 2) HNO2
Ad esempio, riallacciandoci al caso del dipeptide generico A-B, se la funzione amminica del componente carbossi-attivato (A) non è protetta, la formazione del legame peptidico ha luogo in modo incontrollato (Schema 1.5): vari peptidi lineari e ciclici sono formati come sottoprodotti indesiderati, assieme al composto target A-B.
Schema 1.5 Rappresentazione schematica delle possibili reazioni secondarie in una sintesi peptidica coinvolgente un componente carbossilico Nα-non protetto (A) ed un componente amminico (B).
A + A A A A A + B A A B A A A A A A + B + A A A A A A A A A + B A A B A A A A A B + A + A etc.
1.2.1 Gruppi protettivi: evoluzione storica
Da quanto appurato nel precedente paragrafo, è necessario nel corso della sintesi peptidica che tutti i gruppi funzionali non coinvolti nella formazione del legame peptidico siano bloccati, temporaneamente e reversibilmente. Il primo, storico, gruppo protettivo è stato introdotto nel 1931, da M. Bergmann (precedentemente studente nel gruppo di E.
Fischer) e L. Zervas; si trattava del gruppo carbobenzossi (Cbz, o semplicemente Z,
Figura 1.1), raggruppamento protettivo amminico temporaneo.
O N
H O
COOH R1
Figura 1.1 α-amminoacido protetto all’estremità N-terminale, in evidenza il gruppo protettivo Z.
L’introduzione della protezione al gruppo amminico consentiva di risolvere parte dei problemi della sintesi peptidica, furono di fatti sintetizzati negli anni successivi alcuni piccoli peptidi come il glutatione [5], la carnosina [6] e l’ossitocina [7]. M. Bergmann e L.
Zervas dimostrarono, peraltro, che l’uso del gruppo protettivo Cbz previene la
racemizzazione durante la formazione del cloruro acilico, mentre con gli amminoacidi N-acil ed N-benzoil protetti, la reazione conduceva ad una quasi completa racemizzazione. E’ stato successivamente provato che la stabilità configurazionale è una proprietà generale dei gruppi protettivi uretanici.
Nel 1957 venne introdotto, da parte di L. A. Carpino [8], e di F. C. McKay e N. F. Albertson [9], un nuovo gruppo protettivo, labile in ambiente acido: il gruppo tert-butilossicarbonile (Boc) (Figura 1.2). Esso risultava essere stabile all’idrogenazione, alla riduzione di Birch, alle basi forti, dunque totalmente ortogonale al gruppo Cbz (o ad eventuali modifiche di esso) ed anche ad esteri ed eteri benzilici. L’introduzione di questo nuovo raggruppamento ampliò consistentemente l’arsenale dei gruppi protettivi disponibili all’epoca. La
tra i quali l’esempio più spettacolare è senz’altro quello dell’ormone β-corticotropino Adenocorticotrofico (ACTH), un ormone suino costituito da 39 residui, nel 1963 da parte di
R. Schwyzer e P. Sieber [10]. O N H O COOH R1
Figura 1.2 α-amminoacido protetto all’estremità N-terminale, in evidenza il gruppo protettivo Boc.
1.2.2 Gruppi protettivi: “stato dell’arte”
L’introduzione di gruppi protettivi rende la formazione di ciascun legame peptidico un processo a tre steps (Schema 1.6).
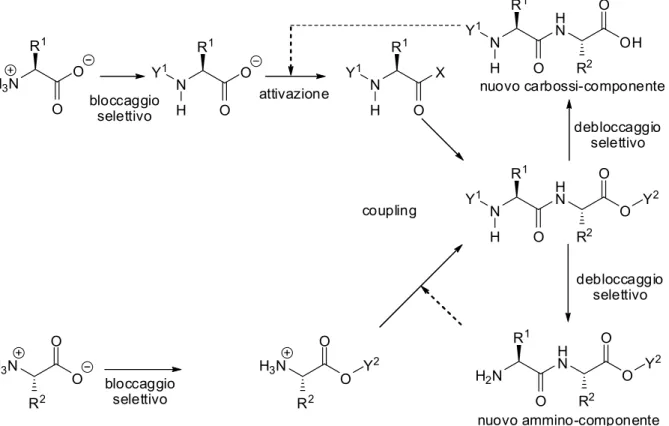
Schema 1.6 Il processo multistep di sintesi peptidica. Y1 = gruppo protettivo amminico; Y2 = gruppo protettivo carbossilico; R1, R2 = catene laterali amminoacidiche.
H3N R1 O O N R1 O O Y1 H N R1 O X Y1 H bloccaggio selettivo attivazione N R1 O H N Y1 H O Y 2 O R2 N R1 O H N Y1 H OH O R2 debloccaggio selettivo nuovo carbossi-componente coupling debloccaggio selettivo H2N R1 O H N O Y 2 O R2 bloccaggio selettivo H3N O O R2 H3N O O R2 Y2 nuovo ammino-componente
1. Nel primo step è richiesta la preparazione di un amminoacido parzialmente protetto, dopo la quale non è più presente la struttura zwitterionica.
2. Il secondo step, la formazione del legame peptidico, prevede due sotto-step. L’amminoacido N-protetto deve essere attivato al gruppo carbossilico allo scopo di essere convertito in un intermedio reattivo, successivamente la formazione del
legame peptidico ha luogo. La reazione di coupling può essere condotta one-pot oppure in due step separati e consecutivi.
3. Nel terzo step di reazione, è compiuto il cleavage selettivo o totale dei gruppi protettivi. Nonostante la deprotezione totale sia richiesta solo quando la catena peptidica è stata assemblata del tutto, il debloccaggio selettivo è necessario per continuare la sintesi peptidica.
La sintesi peptidica è ulteriormente complicata dal fatto che ben undici amminoacidi proteinogenici (Ser, Thr, Tyr, His, Cys, Sec, Lys, Pyl, Arg, Asp e Glu) hanno gruppi funzionali nella catena laterale che richiedono una protezione selettiva. Dato i diversi requisiti di selettività, una distinzione deve essere compiuta tra gruppi protettivi intermedi (temporanei o transienti) e semipermanenti. I primi sono impiegati per la protezione delle funzioni amminiche e carbossiliche coinvolte nella successiva formazione del legame peptidico; questi gruppi debbono essere rimossi in condizioni tali da non coinvolgere i legami peptidici già presenti od i gruppi semipermanenti alle catene laterali amminoacidiche. Tra gli altri requisiti di un gruppo protettivo temporaneo, vi sono la necessità di non avere la racemizzazione durante le operazioni richieste (introduzione, coupling, cleveage), la possibilità di avere intermedi stabili e caratterizzabili, nonché un’idonea solubilità nel mezzo di reazione. Al contrario, i gruppi protettivi semipermanenti vengono sottoposti a cleveage generalmente solo al termine della sintesi peptidica, occasionalmente in step intermedi. Una descrizione dei principali gruppi protettivi è presentata nei paragrafi seguenti.
L’attivazione del componente carbossilico e la seguente formazione del legame peptidico dovrebbero, in condizioni ideali, procedere a velocità elevata e senza racemizzazione o formazione di sottoprodotti.
Affinché sia possibile il cleavage selettivo dei gruppi protettivi, la strategia sintetica richiede infatti un’accurata pianificazione. In dipendenza dalla strategia scelta, possono essere rimossi selettivamente od il gruppo protettivo del raggruppamento Nα-amminico o quello della funzione carbossilica. Il termine “strategia” si riferisce alla sequenza di reazioni di condensazione dei singoli amminoacidi a produrre il peptide. Generalmente, è compiuta una distinzione tra tra sintesi step a step e coupling di frammenti (condensazione di frammenti). Quando la sintesi peptidica è compiuta in soluzione (spesso indicata some “sintesi convenzionale”), ad esempio con sequenze complesse, la crescita del peptide è compiuta tramite l’assemblaggio di piccoli frammenti, approccio imprescindibile per molecole target ad elevato peso molecolare. La pianificazione di un’opportuna strategia richiede anche la scelta di un adatta combinazione di gruppi protettivi e l’applicazione del miglior metodo di coupling per la condensazione dei diversi segmenti.
Gli schemi protettivi adottati si basano sia sulla differente velocità delle reazioni di deprotezione, che sul diverso meccanismo nel caso di gruppi protettivi ortogonali.
Ortogonalità significa che un sottoinsieme di gruppi protettivi in una data molecola può
essere rimosso selettivamente ed indipendentemente in certe condizioni, mentre gli altri gruppi presenti rimangono di fatto intatti. Spesso l’ortogonalità si ha grazie a diversi meccanismi di reazione, soprattutto tra gruppi semipermanenti e temporanei, e generalmente essa consente la possibilità di compiere le trasformazioni in condizioni più blande. Ad esempio l’utilizzo del gruppo Boc, quale raggruppamento protettivo temporaneo, esclude l’utilizzo di gruppi protettivi alle catene laterali che abbiano analoga labilità in condizioni acide, conseguentemente l’ortogonalità può essere raggiunta solo mediante l’impiego di gruppi resistenti nelle condizioni di cleavage del Boc, come ad esempio il gruppo benzilico. Questo schema protettivo è noto come Boc/Bn. Od ancora, il gruppo protettivo temporaneo Fmoc (che esaminerò più in dettaglio successivamente) è rimosso mediante trattamento con basi; in questo caso le catene laterali ed il gruppo
carbossi terminale possono essere protetti mediante gruppi tert-butile, quindi questo schema portettivo ortogonale è indicato come Fmoc/tBu.
1.2.2.1Gruppi Nα-ammino protettivi
Gruppi protettivi di questo genere possono essere utilizzati per tutti i gruppi Nα -amminoacidici di amminoacidi proteinogenici (incluso quello della prolina), per quelli Nω
(come nel caso delle catene laterali della lisina e dell’ornitina) e pure, se necessario, per il bloccaggio temporaneo di idrazidi di N-acil-amminoacidi ed altri gruppi funzionali. In linea di principio, un gruppo amminico può essere bloccato reversibilmente per aciliazione, alchiliazione ed acil-alchiliazione. Sono stati sviluppati pure gruppi protettivi basati sul fosforo e sullo zolfo, mentre la formazione di sali di ammonio non offre una protezione genuina durante le operazioni di sintesi peptidica. Alcune centinaia di gruppi protettivi sono stati sviluppate nel corso degli ultimi decenni, a conferma della non esistenza attuale di un gruppo protettivo amminico universale. La classificazione dei gruppi può essere compiuta su base strutturale/sulle condizioni di cleveage: acidolisi, cleveage basico, riduzione/ossidazione, sostituzione nucleofila, fotolisi. Dato che la maggior parte dei peptidi risultano essere sufficientemente stabili in condizioni moderatamente acide, gruppi protettivi amminici, con diverse labilità verso agenti acidi debloccanti, sono preferiti. In alternativa, il gruppo, labile in amibiente basico, 9-fluorenilmetossicarbonile (Fmoc) ha trovato ampie applicazioni. E’ stato stimato che in pratica l’80% di tutte le sintesi utilizza essenzialmente questi due metodi di debloccaggio. E’ possibile pure il cleavage per idrogenolisi con certi gruppi protettivi. I limiti dell’impiego di raggruppamenti strutturalmen-te semplici come acili (ad esempio acetili, benzoili, etc.) risiede nel fatto che sono molto simili a legami peptidici, ciò rende il loro cleavage selettivo molto difficle, se non praticamente impossibile. Per questa ragione sono stati sviluppati gruppi protettitivi di tipo uretanico, oggigiorno i più comuni e diffusi; tra di essi meritano una trattazione più
- gruppo benzilossicarbonile: Ad esso è già stato fatto brevemente cenno nella sezione 1.2.1. L’introduzione del raggruppamento viene compiuta per reazione con il
benzilcloroformiato sotto le condizioni di Schotten-Bauman in presenza di NaOH, NaHCO3
o MgO, prevalentemente in solventi organico-acquosi sotto agitazione vigorosa (Schema 1.7). Esteri amminoacidici possono essere convertiti in derivati Cbz protetti in cloroformio.
Schema 1.7 Condizioni di reazione e reagenti più utilizzati per l’introduzione del gruppo protettivo Cbz.
O Cl O + H2N COOH R O HN O COOH R H2N COOH R O O O NO2 + Cbz-Xaa-OH
La protezione selettiva nel caso di amminoacidi recanti gruppi funzionali in catena laterale richiede di frequente condizioni di reazione speciali: l’arginina può essere, ad esempio, selettivamente Nα-monoprotetta utilizzando il benzilcloroformiato in una soluzione di
NaHCO3-NaOH tamponata a pH 9-11 [12]. Di sovente i cbz-amminoacidi sono ottenuti in
forma cristallina con alte rese, anche se talvolta si ottengono prodotti oleosi, ricristallizzabili come sali trattando con dicicloesilammina. Il benzil-4-nitrofenilcarbonato (Schema 1.7) ed altri derivati analoghi attivati possono essere utilizzati per introdurre il raggruppamento Cbz, la cui rimozione può essere compiuta per acidolisi, trattando con HBr in acido acetico, HBr liquido, acido trifluoroacetico bollente (TFA), HBr in TFA, HF liquido o per idrogenazione catalitica (utilizzando diversi catalizzatori in un ampio ventaglio di condizioni di reazione) (Schema 1.8). L’idrogenazione catalitica procede tranquillamen-te in solventi organici (ad esempio, acido acetico, alcoli, dimetilformammide (DMF)) od in soluzioni acquose organiche con nero di palladio, Pd/C o Pd/BaSO4. Gli unici sottoprodotti
in tali condizioni sono toluene e CO2. Il consumo di H2 e/o lo sviluppo di CO2 possono
essere idoneamente monitorati allo scopo di analizzare l’evoluzione della reazione.
Schema 1.8 Condizioni di cleveage del gruppo benzilossicarbonile.
O HN O COOH R Cbz-Xaa-OH Na/NH3liq. H2N COOH R H2/Pd Na2CO3 CO2 CO2 HBr/AcOH H2N COOH R Br H3N COOH R Br
Nonostante il cleveage idrogenolitico del gruppo Z sia fallimentare in presenza anche di un solo residuo di cisteina (o di una cistina), la reazione può aver luogo con successo in presenza di BF3 · OEt2. L’acidolisi è compiuta preferibilmente utilizzando HBr 2N in acido
acetico, sebbene altre varianti (ad es. HCl, HI) e solventi (ad es. diossano, nitrometano, TFA, CCl4) siano state suggerite. Utilizzando questo metodo standard, sono possibili
reazioni secondarie, ad esempio: O-acetilazioni sono state riportate alla treonina od alla serina, mentre S-transesterificazioni sono possibili in presenza di metionina. I building-block triptofano o nitroarginina possono essere chimicamente distrutti in queste condizioni, come d’altronde esteri benzilici o gruppi carbossammidici possono subire cleveage in queste condizioni. Sono stati descritti circa 20 varianti del gruppo benzilossicarbonile (ad es. 4-metossiCbz [13, 14], 2-nitroCbz e 4,5-metossi-2-nitroCbz [15], 3,5-dimetossiCbz
modificarne la stabilità agli agenti acidi, per poter avere a disposizione strategie più versatili e selettive.
- gruppo tert-butossicarbonile: Il gruppo Boc riveste un ruolo molto importante, al pari di Z
e Fmoc. La tert-butossicarbonile azide (Schema 1.9) è un reagente di aciliazione eccellente, reagisce con sali di amminoacidi in miscele acqua-diossano in presenza di ammine terziarie od ossidi di magnesio, o sotto pH controllato con NaOH, da 2 a 4N, a dare amminoacidi Boc-protetti (Boc-Xaa-OH), ma anche con esteri amminoacidici in piridina.
Schema 1.9 Condizioni di reazione e reagenti più utilizzati per l’introduzione del gruppo protettivo Boc.
O N3 O + H2N COOH R O N H O COOH R HN3 O O O + H2N COOH R CO2 tBuOH Boc-Xaa-OH
Nonostante l’uso dell’azide sia molto efficiente, sono stati sviluppati altri metodi, principalmente in ragione della natura esplosiva delle azidi. Il fluoruro di tert-butossicarbonile, ottenuto dal fluoruro di clorocarbonile e dal tert-butanolo a -25°C ha attratto particolare attenzione, perché consente di ottenere l’amminoacido Boc-protetto in alte rese in condizioni pH controllato. Il tert-butil-(S)-4,6-dimetilpirimidil-tiocarbonato, il
2-tert-butossicarbonilossimmino-2-fenilacetonitrile e, preferibilmente, il 2-tert-butildicarbonato
(Boc2O) sono utilizzati anch’essi come reagenti. Sali amminoacidici possono essere
utilizzati in soluzione acquosa con additivi solubilizzanti, quali diossano o tetraidrofurano. Il gruppo protettivo Boc è compatibile con la maggior parte dei metodi di coupling per la sintesi peptidica, può essere rimosso in condizioni acidolitiche blande ed è resistente
all’idrogenazione catalitica, all’idrolisi alcalina ed alla riduzione con Na/ammoniaca liquida. HCl in acido acetico, diossano, etere ed acetato di etile sono importanti reagenti di cleveage. Un metodo di cleveage frequentemente utilizzato è il trattamento con TFA non-acquoso a 0°C, che consente la facile rimozione del gruppo Boc (Schema 1.10).
Schema 1.10 Meccanismo di cleveage acidolitico del gruppo tert-butossicarbonile.
O N H O COOH R Boc-Xaa-OH + H O N H OH COOH R + CO2 H H2N COOH R +
tert-butilazioni indesiderate possono aver luogo, durante l’acidolisi, ai siti nucleofilici della
catena peptidica, come ad esempio l’anello indolico del triptofano od il gruppo tioetereo della metionina. I carbocationi intermedi, responsabili di tali reazioni scecondarie, possono essere intrappolati utilizzando scravengers quali l’anisolo od il tioanisolo. Ulteriori reagenti debloccanti sono BF3 · OEt2, acido-2-mercaptosolfonico, TFA acquoso od acido formico al
98%. Reazioni pulite non sono state comunque osservate in alcun caso con questo tipo di reagenti (utilizzabili in presenza di gruppi protettivi quali Cbz od altri analoghi).
Analoghi modificati del gruppo tert-butossicarbonile sono stati descritti in numerosi occasioni, tra i più importanti vi sono il gruppo α,α-dimetil-3,5-dimetossibenzoilcarbonile [19] (rimuovibile pure fotoliticamente), il (4-bifenilil)-propossicarbonile [20,21] ed il 2-cianoBoc [22], allontanabile per β-eliminazione in condizioni debolmente basiche.
- gruppo 9-fluorenilmetossicarbonile: il gruppo è stato introdotto nel 1970 da L. A. Carpino e G. Y. Han [23]. Esso è rimuovibile in condizioni basiche blande, offrendo la possibilità di uno schema protettivo ortogonale in presenza di raggruppamenti suscettibili ad acidolisi. La particolare sensibilità dei Fmoc-Xaa-OH verso ammine secondarie consente il debloccaggio anche con soluzioni diluite di piperidina o dietilammina in DMF (Schema
rimuovere il raggruppamento Fmoc in pochi secondi a temperatura ambiente; altri reagenti, come l’1,8-diazabiciclo[5.4.0]undecene (DBU) o lo ione fluoruro, rappresentano valide alternative. La reazione procede con un meccanismo E1cB (eliminazione), con iniziale estrazione del protone a dare l’anione dibenzociclopentadienile stabilizzato, che evolve nel dibenzofulvene, il quale reagisce in fine con la piperidina, a dare un addotto stabile come sottoprodotto della reazione di debloccaggio (Schema 1.11). Nel caso il cleveage venga operato in assenza di ammine primarie o secondarie, è fortemente raccomandata l’aggiunta di un 2% di piperidina, all’uopo di intrappolare il dibenzofulvene.
Schema 1.11 Meccanismo di rimozione del gruppo Fmoc.
O X O + H2N COOH R O HN O COOH R O HN O COOH R H N H O HN O COOH R N H H O HN O COOH R H CO2 N + H2N COOH R X = Cl,O N O O
Sotto queste condizioni né il gruppo Boc né il Cbz sono attaccati; allo scopo di prevenire il cleveage prematuro nelle condizioni del coupling peptidico, l’addizione di un additivo debolmente acido, come l’1-idrossibenzotriazolo, è fortemente raccomandata. Anche l’addizione di CaCl2 incrementa la longevità del gruppo Fmoc, che in queste condizioni può
esteri carbossi-terminali [24]. Interessante è il fatto che il gruppo Fmoc non sia del tutto inerte nelle condizioni standard di cleveage idrogenolitico.
I reagenti Fmoc-Cl e Fmoc-OSu (9-fluorenilmetil-N-succinimmidilcarbonato) sono I più utilizzati per la formazione dell’amminoacido Fmoc-protetto. Il gruppo è molto utilizzato anche per sintesi su fase solida, nonostante le prime applicazioni siano comparse solo molti anni dopo la sua introduzione. La stabilità in condizioni acide e le condizioni di cleveage debolmente basiche rendono il gruppo Fmoc compatibile con gruppi protettivi semipermanenti quali il tert-butile, anche per sintesi peptidica su fase solida (SPPS). Inoltre, il legame con una porzione aromatica di grosse dimensioni, rende la purificazione del derivato amminoacidico più semplice [25].
Anche per il gruppo Fmoc sono stati sviluppati analoghi, recanti in particolare la funzionalità solfonilica, aventi una stabilità superiore alle basi: il gruppo 2-(4-nitrofenilsolfonil)etossicarbonile [26-28], il 2-(tert-butilsolfonil)-2-propenilossicarbonile [29], l’1,1-diossobenzo[b]-tien-2-ilmetossicarbonile [30,31] ed il 2-(metilsolfonil)-3-fenil-2-propenilossicarbonile [32].
- gruppi protettivi ortogonali al Boc ed al Fmoc: sicuramente il più importante è l’Nα -allilossicarbonile (Aloc) [33-5], il quale può essere utilizzato nella sintesi di derivati labili o peptidi ciclici. La sua deprotezione (Schema 1.12) ha luogo mediante il trasferimento del gruppo allilico, Pd(0) catalizzata, a vari nucleofili (ad esempio ammine, complessi ammino-borani, carbossilati, organosilani, etc.).
Schema 1.12 Meccanismo di cleveage del gruppo Aloc.
O HN COOH O R Pd(PPh3)4 Pd Ph3P PPh3 O HN O Pd(PPh3)4 Nuc H R COOH
- gruppi protettivi carbossiammidici: sono riportati i due esempi più comuni in Figura 1.3. R1 N H OH O O R2 R1=H, CF 3
Figura 1.3 Amminoacido protetto con gruppo (carbossi)ammidico.
- gruppi protettivi solfonammidici e solfenammidici (Figura 1.4, Ar = raggruppamento aromatico od eteroaromatico più o meno strutturalmente complesso, [36-40])
Ar S N H OH O R O O Ar S N H OH O R
Figura 1.4 Amminoacido protetto da un raggruppamento solfonammidico/solfenammidico.
- gruppi protettivi “alchilici” (Figura 1.5) [41-4].
H N COOH R2 R1 Alk O O R3 H N COOH Ph R3 Ph Ph H N COOH R3 N COOH R3 R1, R2= Ar, Alk
Figura 1.5 Amminoacidi protetti con gruppi alchilici più o meno strutturalmente complessi.
1.2.2.2 Protezione al gruppo Cα-carbossilico
Esistono solo poche eccezioni in letteratura [45-7] allo schema generale di assemblaggio di un peptide, dall’estremità C-terminale a quella N-terminale (direzione C→N), conseguentemente la protezione all’estremità C-terminale non è stata oggetto di sviluppi considerevoli come l’altra. Il gruppo protettivo al raggruppamento Cα-carbossi deve essere
ortogonale al gruppo protettivo temporaneo all’Nα sia in soluzione, che nel caso del linker impiegato in fase solida (come sarà esaminato nel Paragrafo 1.2.6).
In linea di principio è possibile compiere sintesi peptidica senza una completa protezione al gruppo carbossilico: è sufficiente la formazione di sali, in genere di metalli alcalini od alcalino terrosi, a pH basico, che presentano la possibilità di reagire in acqua, o diossano
acquoso, con il corrispondente partner carbossi-attivato. Il corrispondente carbossilato di una base terziaria (N-metilmorfolina, N-etilpiperidina, trietilammina, 1,1,3,3-tetrametilguani-dina, etc.) svolge un ruolo fondamentale per la sintesi peptidica in solventi organici, specialmente nel caso della DMF. Il metodo dell’anidride mista e quello azidico sono, oltre a quello dell’estere attivato, i migliori per il coupling salino. Un grande vantaggio è che, dopo la reazione di coupling, il gruppo carbossilico può essere liberato per acidificazione. Questo protocollo non è tuttavia universalmente applicabile, in ragione di rischi di reazioni secondarie, problemi di solubilità e/o di protezione dei raggruppamenti.
E’ possibile operare una suddivisione in due classi per quel che riguarda i gruppi carbossi-protettivi, mostrata in Schema 1.13:
Schema 1.13 Classificazione dei gruppi protettivi al carbossile.
gruppi carbossi-protettivi
gruppi protettivi "reali"
gruppi protettivi tipo esteri od ammidici
gruppi protettivi "tattici" gruppi protettivi "attivati" gruppi protettivi "attivabili"
- gruppi protettivi “veri”: possono essere debloccati normalmente al termine della sintesi peptidica, a rigenerare il raggruppamento carbossile libero;
- gruppi protettivi “tattici”: impiegabili in strategie sintetiche speciali, si tratta di derivati attivati per amminolisi al termine della sintesi, o dopo un’idonea trasformazione chimica. - esteri: sono attualmente utilizzati esteri di alcoli primari, secondari o terziari. Esteri metilici ed etilici, utilizzati pure da Curtius e Fischer, sono agevolmente ottenibili per reazione di un amminoacido con il corrispettivo alcol in presenza di cloruro di tionile a basse temperature (è presumibile il passaggio attraverso un intermedio alchilclorosolfinato AlkO-SO-Cl). La rimozione di questi semplici gruppi protettivi è operata per blanda idrolisi
alcalina in acqua o solventi organici, quali diossano, metanolo, etanolo, acetone o DMF, necessari a solubilizzare i materiali di partenza. Altri gruppi comunemente utilizzati sono esteri benzilici (ottenibili per esterificazione diretta dell’alcol benzilico in presenza di un catalizzatore acido –acido para-toluensolfonico, HCl, acido benzensolofonico, acido polifosforico- e rimuovibili per idrolisi alcalina, idrogenazione catalitica, trattamento con HBr sat. in acido acetico, o HF liquido, od ancora Na/NH3 liq.) e tert-butilici (preparati per
addizione di isobutene in conzioni acide o transesterificazione con l’acetato di tert-butile e rimuovibili per acidolisi con TFA, HCl/acido acetico, BF3 · OEt2/acido acetico).
Un’importante innovazione dovuta a Carpino [48] è stata l’introduzione del gruppo diciclo-propilmetile, debloccabile per semplice trattamento con TFA in CH2Cl2 all’1%, condizione a
cui resistono anche gruppi semipermanenti, come ad esempio i tert-butile.
Questi gruppi prottettivi possono essere adottati pure nel caso di gruppi ω-carbossilici. - ammidi ed idrazidi: L’utilizzo di idrazidi è fondamentale qualora sia stato scelto di attivare il gruppo carbossilico come azide [49]; il seguente Schema 1.14 chiarisce meglio il ruolo di questo gruppo “tattico”.
L’utilizzo di carbossiammidi quali gruppi protettivi presenta dei limiti soprattutto in fase di idrolisi: pur essendo superiore la labilità rispetto ad un legame peptidico, è molto difficile ottenere un cleveage selettivo.
1.2.2.3 Protezione della catena laterale
L’influenza da parte della catena laterale sulla reattività del raggruppamento α-amminico ed α-carbossilico risulta essere notevole, tanto che è frequentemente necessaria la protezione delle funzionalità più reattive e dunque maggiormente suscettibili ad indesiderate reazioni secondarie. Soprattutto in presenza di gruppi amminici od ω-carbossilici possono sorgere problemi di competizione: una funzionalizzazione selettiva con un gruppo protettivo non è affatto un problema banale in questa circostanza. Gruppi semipermanenti sono necessari pure per la funzione tiolica della cisteina o quella
guadinica dell’arginina, anche se talvolta, utilizzando condizioni di reazione specifiche, è possibile minimizzare le reazioni secondarie dovute alle catene laterali. Quando si lavora su fase solida, è preferibile ad ogni modo utilizzare tutti i gruppi protettivi necessari.
Schema 1.14 Gruppi idrazidici differentemente protetti sono potenzialmente gruppi protettivi al carbossile.
R N H NH2 O R N3 O coupling azidico R N H O H N O O R N H O H N O O R N H O H N Ph Ph Ph
- protezione della funzionalità guanidinica: nonostante normalmente tal gruppo sia protetto grazie alla protonazione, a causa del suo spiccato carattere basico, la bassa solubilità nei solventi organici che ne consegue è un grosso ostacolo per la sintesi peptidica. Una parziale aciliazione del gruppo guanidinico può aver luogo in tali circostanze, a dare, nel caso di attacco intramolecolare, un δ-lattame (Schema 1.15).
Schema 1.15 Formazione di un δ-lattame da derivati argininici attivati al carbossile.
R2HN N H OH NH O NHR1 R2HN N H X NH O NHR1 N R2HN NH O NHR1
Derivati di bromuro di argininio possono essere utilizzati in linea teorica come partner amminici nella sintesi peptidica: ad esempio il sistema Nα-benzilossicarbonile protetto può
essere facilmente ottenuto per trattamento della Z-Arg-OH con HBr in metanolo 1.4 N e successiva precipitazione con etere assoluto.
Idealmento, tutti i tre atomi di azoto del gruppo guanidinio dovrebbero essere protetti, ma la maggior parte delle strategie si basano o sulla Nω-protezione o sulla Nω-, Nδ -diprotezio-ne. Non è stato individuato tuttora un gruppo protettivo ideale, quelli attualmente impiegati si suddividono in quattro categorie: gruppi nitro (stabili al TFA, all’HBr/AcOH ed agli alcali, rimuovibile per trattamento con HF o riduzione od idrogenolisi catalitica), uretanici [50-2], arensolfonilici [53-7] e di tipo tritilico, seppure i derivati di quest’ultimo tipo presentino una limitata solubilità in solventi organici, che conseguentemente ne riduce l’impiego.
Un approccio scaltro ed elegante prevede l’introduzione del gruppo guanidinico solo al termine del completamenteo della sintesi peptidica, su residui ornitinici, per trattamento con un idoneo reattivo, generalmente 1-ammino-3,5-dimetilpirazolo (Schema 1.16).
Schema 1.16 Introduzione di un residuo di argininina per mezzo della guanidilazione nucleofilica di ornitina.
NH2 O H2N N NH NH + N N H O NH NH2 NH
- protezione del gruppo ω-amminico: Acidi diamminocarbossilici, quali la lisina o l’ornitina, richiedono spesso protezioni ortogonali per i gruppi α ed ω-amminici. Ad esempio, nella lisina il gruppo ε-amminico è più basico e nucleofilico rispetto a quello α-amminico e può dunque reagire preferibilmente in condizioni opportunamente selettive (Schema 1.17): può essere infatti convertito in un’ε-immina per trattamento con benzaldeide in presenza di 1 eq. di LiOH. Successivamente in condizioni basiche il gruppo α-amminico può essere selettivamente aciliato e di seguito il gruppo imminico può essere rimosso a dare, ad esempio, Z-Lys(H)-OH, per la quale è possibile una protezione ortogonale alla funzionalità
ε, trattando di seguito con Boc2O, da cui si ottiene Z-Lys(Boc)-OH. In un approccio
disattivati per mezzo della formazione di un chelato (Schema 1.17) con ioni Cu(II), in tali condizioni il gruppo ε-amminico può dunque reagire selettivamente.
Schema 1.17 Strategie per la protezione selettiva del gruppo α-amminico od ε-amminico della lisina.
H2N COOH NH2 N COOH NH2 H2N COO NH2 Cu2+ a) Z-Cl/NaOH b) HCl conc. c) Δ H2N COOH N Z H N H COOH NH2 Boc
Nello schema Boc, la funzionalità ε-amminica della lisina è generalmente protetta con il gruppo 2-clorobenzilossicarbonile [Z(2-Cl)], che presenta una labilità in ambiente acido 60 volte inferiore rispetto al gruppo Z; ciò previene l’accidentale rimozione nel corso della deprotezione acidolitica del gruppo temporaneo Boc nel corso dell’assemblamento della catena.
Nell’approccio Fmoc, la protezione della catena laterale col gruppo Boc è ottimale, come il raggruppamento allilossicarbonile (Aloc), pienamente ortogonale ad entrambi.
- protezione del gruppo ω-carbossilico: una protezione ortogonale è richiesta nella maggior parte dei casi per gli acidi amminodicarbossilici, quali l’acido aspartico ed il glutammico. La formazione del peptide isoaspartilico, attraverso il corrispettivo intermedio succinimmidico, è la reazione secondaria più probabile della catena laterale dell’estere dell’acido aspartico. Essa ha luogo mediante un attacco nucleofilico alla catena laterale, con l’apertura finale dell’anello da parte di un nucleofilo (ad es. acqua od ammine, come la piperidina). Il risultato è una miscela di peptidi α- e β-aspartilici (Schema 1.18), un
fenomeno che si verifica pure nel corso dell’invecchiamento di proteine contenenti aspartato nei sistemi biologici, a cui si è già fatto riferimento nel Paragrafo 3 dell’Introduzione.
Schema 1.18 Ciclizzazione indesiderata di un residuo di acido aspartico e conseguente formazione di una miscela di α- e β-peptidi. O R N N H Y O O RH ROH N R N H Y O O Nuc Nuc NHR N H Y O O + NHR Nuc N H Y O O
L’acido glutammico viene generalmente protetto alla catena laterale con gli stessi raggruppamenti utilizzati per l’acido aspartico, sebbene i suoi derivati siano meno inclini alla ciclizzazione precedentemente descritta (anche se durante il cleveage con HF nella schema Boc, si osserva talvolta la disidratazione dello ione acile ed altre trasformazioni secondarie).
È possibile ottenere un’esterificazione regioselettiva, catalizzata da acidi, del gruppo ω-carbossilico (ad es. trattando con l’alcol benzilico) per trattamento con H2SO4 (Schema
1.19); l’estere è poi selettivamente proteggibile alla funzione Nα, con la formazione di un
derivato uretanico. Il protocollo è pure estendibile a derivati dell’acido aspartico.
In alternativa, l’acido aspartico Z-protetto può essere convertito in un’anidride ciclica per trattamento con acido acetico (Schema 1.19); dopo alcolisi e separazione dei due regioisomeri ottenuti, l’α-estere Asp(OH)-OR è trasformato in un derivato tert-butilico Z-Asp(OtBu)-OR, che è poi saponificato a dare Z-Asp(OtBu)-OR; infine il gruppo Z può essere scambiato con altri gruppi protettivi, ad esempio Fmoc.
Schema 1.19 Protocollo per l’α-esterificazione selettiva di acido glutammico. HOOC COOH NH2 COOH NH2 O BnO COOH N O BnO Y H HOOC COOH N Z H O O O N Z H N Z H O O R HO O + N Z H OH O O O R N Z H O O R O O tBu N Z H OH O O O tBu
- protezione del gruppo funzionale tiolico: la presenza di cisteina in una catena peptidica complica notevolmente la sintesi: può essere necessario che il gruppo tiolico rimanga libero o che sia coinvolto nella formazione di un ponte disolfuro, regioselettivamente, intra- od intermolecolarmente.
L’elevata nucleofilicità e facilità di ossidazione, nonché il carattere acido, del gruppo tiolico della cisteina, richiedono il bloccaggio selettivo e semipermanente della funzionalità nel corso delle operazioni di sintesi; di fatti sono stati descritti in letteratura più di settanta tipologie di gruppi protettivi, in varie monografie [50,58].
La prima, storica, protezione, come S-benzile, introdotta da Vignaud nel 1930, nella sintesi dell’ossitocina, cui si è già fatto riferimento, ha rappresentato un notevole passo avanti. Le drastiche condizioni di cleveage (riduzione con Na/NH3) e l’alta inclinazione
all’epimerizzazione hanno condotto tuttavia allo sviluppo di alcune varianti strutturalmente modificate (4-metilbenziltio [59], 4-metossibenziltio [60], 2,4,6-trimetossibenziltio [61]). Altri
trimetilacetammidometiltio [66], che consentono, all’atto della deprotezione, un coupling ossidativo simultaneo, a dare la corrispettiva cistina. In alternativa, è possibile la protezione mediante il gruppo solfonico (-SO3-) [67], che offre elevata stabilità ed una
semplice procedura di rimozione, riducendo con acido tioglicolico a pH 5.
Nonostante la presenza di un gruppo protettivo, i residui di cisteina possono essere coinvolti in indesiderate reazioni secondarie: β-eliminazioni a dare deidroalanine (soprattutto in condizioni basiche, con la conseguente addizione coniugata di un nucleofilo, come ad esempio la piperidina), ossidazioni od alchiliazioni. Protezioni di tipo
S-acilico hanno poca rilevanza pratica, in quanto è entropicamente favorita la migrazione
dell’acile dall’atomo di zolfo a quello di azoto (come vedremo di seguito nel paragrafo dedicato alla ligazione naturale).
- protezione del gruppo imidazolico: L’istidina è uno dei building-block più problematici nella sintesi peptidica [68]: reazioni secondarie, come N-aciliazioni, racemizzazioni, formazioni di imidazolidi ciclici, N-guanilazioni, possono coinvolgere facilmente l’anello imidazolico se non protetto. Alcuni derivati istidinici presentano una solubilità scarsa nei solventi normalmente utilizzati per la sintesi peptidica. Tutti questi problemi sono eliminabili o riducibili per mezzo di un mascheramento reversibile. Tra i gruppi più utilizzati vi sono il benzile [69], seppur di difficile cleveage, ma modificabile strutturalmente (2,4-dinitrobenzile [70], rimuovibile per tiolisi od idrazinolisi). Gruppi protettivi di tipo uretanico, Z o Boc [71], sono compatibili con lo schema protettivo temporaneo Fmoc, anche se presentano una labilità elevata e possono dare indesiderate reazioni di trasferimento di acile. In alternativa è possibile utilizzare pure il tritile con questo schema [72,73]. Il gruppo 4-toluensolfonile [74], rimuovibile per trattamento con HF liquido od acido trifluorometansolfonico (CF3SO3H), è utilizzabile nello schema Boc, col quale è compatibile