5. CAMPIONAMENTI E PROCEDURE ANALITICHE
5.1 Campionamento
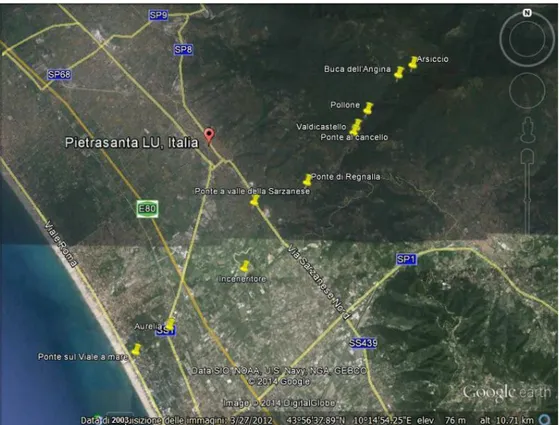
Le stazioni di campionamento delle acque, in questo lavoro di Tesi, comprendono l’intera asta fluviale del torrente Baccatoio, dalla discarica della Miniera del Monte Arsiccio fino alla foce. Sono state eseguite diverse campagne, con particolare continuità nel periodo che va da dicembre 2013 fino a gennaio 2014.
Figura 5.1: Localizzazione delle stazioni di campionamento delle acque superficiali del Torrente Baccatoio.
La localizzazione dei punti di campionamento delle acque superficiali è riportata nella Figura 5.1.
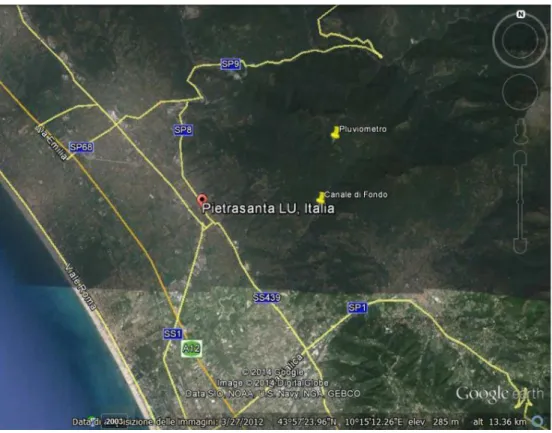
Oltre alle acque del Torrente Baccatoio sono state campionate anche le acque dei drenaggi delle principali gallerie minerarie e alcuni degli stillicidi ed aree di accumulo in galleria nelle miniere del Pollone e di Monte Arsiccio; è stato inoltre campionato il Canale di Fondo (Figura 5.2), un affluente di destra che come si è detto incontra il Baccatoio nel paese di Valdicastello. Inoltre, a Sant’Anna di Stazzema è stato posizionato un pluviometro per la raccolta delle acque meteoriche da gennaio 2014 fino a luglio 2014 (Figura 5.2).
In alcune stazioni lungo il corso del Baccatoio sono stati effettuati campionamenti ripetuti in diverse condizioni di portata. In dettaglio, le stazioni sono: stazione poco a valle della SS 439 Sarzanese: campioni CDMW7,CDMW7-2,CDMW7-3 raccolti
l’11/12/2013; campione CDMW23, raccolto il 21/1/2014; campione CDMW38, raccolto il 12/6/2014 (Tabella 5.1).
Figura 5.2: Localizzazione del Canale di Fondo e del pluviometro.
Stazione abitato di Valdicastello, nella parte a monte dell’abitato: campioni CDMW8, CDMW8-2, CDMW21, CDMW28, CDMW34, CDMW37, CDMW39 (Tabella 5.1).
In regime di piena, il 21/1/2014, sono stati raccolti campioni da stazioni distribuite lungo l’intero corso del Baccatoio, dalla zona immediatamente a valle della discarica del M.te Arsiccio fino alla foce. Comprendono anche il campionamento alle stazioni di Valdicastello e vicino alla Statale SS 439 Sarzanese prima menzionate. I campioni, progressivamente dalla zona a monte alla foce, sono: CDMW18-19 (Figure 5.3 e 5.4), il CDMW20 e CDMW21 (Figura 5.5), il CDMW22, il CDMW23(Figura 5.6), i CDMW24-25 ed infine il campione CDMW26(Figura 5.7).
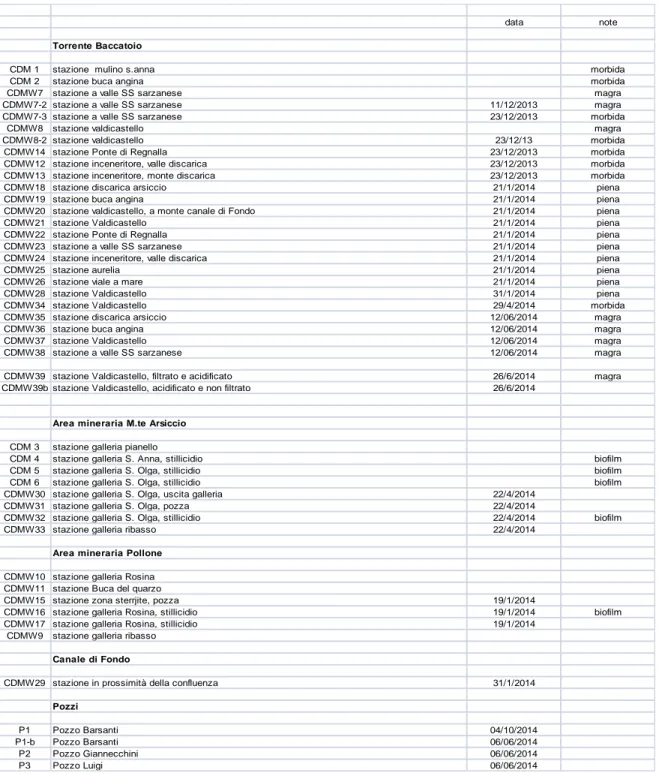
L’insieme dei campioni, distinto tra aree minerarie e acque superficiali, è riportato schematicamente nella Tabella 5.1 più avanti.
Figura 5.3: Immagine della Discarica del Monte Arsiccio del 21 gennaio 2014 (CDMW18).
Figura 5.5: Immagine Baccatoio a Valdicastello, 21 gennaio 2014 (CDMW21).
Figura 5.7: Immagine del Fosso di Motrone al ponte sul viale a mare, vicino alla foce, 21 gennaio 2014 (CDMW26).
In regime di morbida sono stati raccolti i campioni al Mulino Sant’Anna (Figura 5.8), alla Buca dell’Angina (Figura 5.9), nella stazione di Valdicastello, a monte dell’inceneritore e a valle di questo in data 23/12/2013.
Figura 5.9: Immagine del campionamento in morbida allla Buca dell’Angina (CDM2).
Sono stati inoltre campionate le acque nell’alto corso del Baccatoio, immediatamente a valle della discarica del M.te Arsiccio e località Buca dell’Angina il 12/6/2014, rispettivamente CDMW35 e CDMW36.
Per quanto riguarda le altre acque superficiali, è stato campionato il Canale di Fondo in prossimità dell’intersezione con il Torrente Baccatoio il 31/1/2014, campione CDMW29 (Figura 5.10).
Figura 5.10: Immagine del Canale di Fondo (CDMW29).
Per quanto riguarda i drenaggi dalle gallerie minerarie, questi sono stati raccolti nella loro varietà di forme (drenaggi, stillicidi dalla volta, stillicidi da biofilm, pozze) sia nella zona mineraria del M.te Arsiccio (campioni CDM3-4-5-6; CDMW30-31-32-33) (Figura 5.11) che del Pollone (campioni CDMW9-10-11-15-16-17).
Figura 5.11: Immagine campionamento nella Galleria S. Anna, stillicidio da biofilm (CDM4).
Infine sono stati campionati dei pozzi che si trovano nel conoide alluvionale di Pietrasanta, campioni P1, P2, P3 e P4 in data 6 Giugno 2014 (Figura 5.12).
Figura 5.12: Stazioni di campionamento dei pozzi.
Questi pozzi si trovano nell’area vicino al Torrente Baccatoio e all’inceneritore, punto di campionamento. Le loro profondità variano da 10 m del pozzo P2 a 35 m quelli rimanenti. Lo scopo dello studio delle acque dei pozzi è stato quello di vedere se le
caratteristiche del Torrente Baccatoio si riflettevano anche sulle acque dei pozzi che drenano le falde del conoide.
Tabella 5.1. Campioni, descrizione delle stazioni e note
data note
Torrente Baccatoio
CDM 1 stazione mulino s.anna morbida
CDM 2 stazione buca angina morbida
CDMW7 stazione a valle SS sarzanese magra
CDMW7-2 stazione a valle SS sarzanese 11/12/2013 magra CDMW7-3 stazione a valle SS sarzanese 23/12/2013 morbida
CDMW8 stazione valdicastello magra
CDMW8-2 stazione valdicastello 23/12/13 morbida
CDMW14 stazione Ponte di Regnalla 23/12/2013 morbida
CDMW12 stazione inceneritore, valle discarica 23/12/2013 morbida CDMW13 stazione inceneritore, monte discarica 23/12/2013 morbida
CDMW18 stazione discarica arsiccio 21/1/2014 piena
CDMW19 stazione buca angina 21/1/2014 piena
CDMW20 stazione valdicastello, a monte canale di Fondo 21/1/2014 piena
CDMW21 stazione Valdicastello 21/1/2014 piena
CDMW22 stazione Ponte di Regnalla 21/1/2014 piena
CDMW23 stazione a valle SS sarzanese 21/1/2014 piena CDMW24 stazione inceneritore, valle discarica 21/1/2014 piena
CDMW25 stazione aurelia 21/1/2014 piena
CDMW26 stazione viale a mare 21/1/2014 piena
CDMW28 stazione Valdicastello 31/1/2014 piena
CDMW34 stazione Valdicastello 29/4/2014 morbida
CDMW35 stazione discarica arsiccio 12/06/2014 magra
CDMW36 stazione buca angina 12/06/2014 magra
CDMW37 stazione Valdicastello 12/06/2014 magra
CDMW38 stazione a valle SS sarzanese 12/06/2014 magra CDMW39 stazione Valdicastello, filtrato e acidificato 26/6/2014 magra CDMW39b stazione Valdicastello, acidificato e non filtrato 26/6/2014
Area mineraria M.te Arsiccio CDM 3 stazione galleria pianello
CDM 4 stazione galleria S. Anna, stillicidio biofilm CDM 5 stazione galleria S. Olga, stillicidio biofilm CDM 6 stazione galleria S. Olga, stillicidio biofilm CDMW30 stazione galleria S. Olga, uscita galleria 22/4/2014
CDMW31 stazione galleria S. Olga, pozza 22/4/2014
CDMW32 stazione galleria S. Olga, stillicidio 22/4/2014 biofilm
CDMW33 stazione galleria ribasso 22/4/2014
Area mineraria Pollone CDMW10 stazione galleria Rosina CDMW11 stazione Buca del quarzo
CDMW15 stazione zona sterrjite, pozza 19/1/2014
CDMW16 stazione galleria Rosina, stillicidio 19/1/2014 biofilm CDMW17 stazione galleria Rosina, stillicidio 19/1/2014
CDMW9 stazione galleria ribasso Canale di Fondo
CDMW29 stazione in prossimità della confluenza 31/1/2014 Pozzi
P1 Pozzo Barsanti 04/10/2014
P1-b Pozzo Barsanti 06/06/2014
P2 Pozzo Giannecchini 06/06/2014
P3 Pozzo Luigi 06/06/2014
I campioni per le analisi di laboratorio sono stati raccolti in contenitori di polietilene; filtrati in campagna con filtri di nylon a 0.45 µm per l’analisi degli anioni; filtrati e stabilizzati con acido nitrico ultrapuro per l’analisi dei cationi maggiori e degli elementi
in traccia; filtrati per le analisi isotopiche dello stronzio e tal quali per le determinazioni isotopiche di ossigeno e idrogeno.
In aggiunta, nella stazione posta nell’abitato di Valdicastello e alla Buca dell’Angina, parte alta del corso del Torrente Baccatoio, erano state poste due sonde CTD per la misura in continua (intervalli di un’ora) della conducibilità elettrica, la temperatura e livello dell’acqua, quest’ultimo parametro, comunque, non compensato per le variazioni di pressione atmosferica, nell’alveo attivo del Torrente E’ da segnalare che una terza sonda CTD è stata posta nell’abitato di Valdicastello dopo che le due sonde precedenti erano state distrutte da un evento improvviso di piena nel gennaio 2014.
5.2 Analisi in situ
In ciascuna stazione di campionamento è stata misurata la temperatura dell’acqua, eventualmente riferita anche alla temperatura dell’aria. Sono stati misurati inoltre il pH, il potenziale di ossido-riduzione (Eh) e la conducibilità elettrica (riportata a 20 °C) tramite uno strumento Delta Ohm HD2156.1 corredato degli opportuni elettrodi. Le incertezze analitiche sono di ±0.1 °C per la temperatura, ±0.05 unità di pH per il pH; ±0.01 V per Eh; 1% per la conducibilità elettrica. Il valore di Eh misurato è stato riportato al corrispondente valore relativo all’elettrodo standard di idrogeno (SHE) misurando contemporaneamente al campione la soluzione di Zobell. Al momento dei campionamenti è stata inoltre misurata la quantità di ossigeno disciolto (mg/L), dopo opportuna correzione per la salinità quando necessario e la saturazione in ossigeno (%) tramite uno strumento Delta Ohm HD2109.1 con sonda polarografica. L’incertezza è ±0.02 mg/L per la concentrazione di ossigeno disciolto. L’alcalinità è stata determinata in situ o immediatamente in laboratorio tramite titolazione acidimetrica utilizzando HCl 0.1N e arancio di metile come indicatore. I valori di pH misurati escludono la presenza significativa di ioni carbonato, e tutta l’alcaninità è stata attribuita a ioni bicarbonato. Infatti, le abbondanze relative degli ioni bicarbonato e carbonato, a partire dalla dissociazione dell’acido carbonico prodotto dall’interazione del CO2(g) con H2O ed in funzione del pH, sono descrivibili da una serie di reazioni. L'acido carbonico è un acido debole. La prima fase per la sua formazione è la dissoluzione di CO2 (g) in acqua secondo la reazione:
All'equilibrio si ha:
Una volta in soluzione, CO2 (aq) reagisce con acqua per formare acido carbonico:
La costante di equilibrio a 25 ° C è:
La costante di equilibrio KCO2 è una misura della solubilità della CO2(g) in acqua.
La maggior parte della CO2 disciolta è in realtà presente come CO2(aq); solo una piccola quantità è effettivamente presente come H2CO3, acido carbonico.
L'acido carbonico è un acido debole che si dissocia in base alla reazione:
la costante di dissociazione a 25 ° C e 1 bar è:
il bicarbonato si può dissociare:
2 2 2 CO CO CO p a K 46 . 1 * 10 2 3 2 2 CO CO H CO p a K 35 . 6 * 1 10 3 2 3 CO H H HCO a a a K
Riarrangiando l'espressione per K1 come il rapporto tra K1 e aH+:
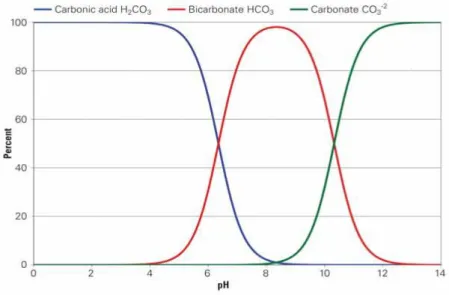
Questa relazione mostra che, quando pH = PK1, le attività di acido carbonico e bicarbonato sono uguali. Quando il pH < circa 4 circa è presente la sola forma H2CO3; a pH neutri o prossimi alla neutralità domina HCO3 (Figura 5.13).
Figura 5.13: Digramma delle differenti attività dei carbonati in funzione del pH.
Segue, a pH> 9.5 circa, la formazione in quantità significativa di ione carbonato (Figura 5.13).
In alcuni campioni, aventi pH acido, è presente quindi solo acido carbonico non dissociato.
5.3 Analisi degli ioni maggiori: cromatografica ionica
Con il termine cromatografia si indica genericamente un insieme di tecniche che hanno lo scopo di separare una miscela nei suoi componenti, per permetterne il
33 . 10 2 10 3 2 3 HCO H CO a a a K * 1 3 2 3 CO H HCO H a a a K
riconoscimento qualitativo e quantitativo. Queste tecniche sono basate sulla distribuzione differenziale dei vari componenti fra due fasi: una chiamata fase
stazionaria e l’altra detta fase mobile o eluente, che fluisce in continuo attraverso la fase
stazionaria. Il principio su cui si basa questo tipo di cromatografia, nel caso di separazioni ioniche, è l'attrazione che si verifica tra molecole cariche di segno opposto. Le separazioni a scambio ionico sono condotte in colonne impaccate con una resina scambiatrice di ioni. Esistono due tipi di resine: gli scambiatori anionici e gli scambiatori cationici. La differenza tra i due tipi di resina sta nella composizione chimica della fase stazionaria che costituisce la colonna a scambio ionico. Quelle di tipo cationico possiedono gruppi carichi negativamente come ad esempio SO3- , mentre quelle a scambio anionico possiedono gruppi carichi positivamente come ad esempio NH4+. Le prime attraggono, selettivamente, molecole cariche positivamente; viceversa le seconde. La tecnica della cromatografia ionica applicata alla fase acquosa permette di determinare gli ioni maggiori (generalmente Ca2+, Mg2+, K+, Na+ per i cationi e SO42-, Cl-, NO3- per gli anioni) in modo sequenziale, rispetto al tempo e dato un flusso a pressione costante di eluente. I singoli analiti vengono eluiti in tempi successivi e determinati da un rivelatore conduttometrico previa soppressione chimica o elettrochimica della conducibilità elettrica dell’eluente. Dall’integrazione delle aree dei singoli picchi cromatografici si ricavano le concentrazioni dei cationi sopra elencati mediante confronto con curve di calibrazione ottenute iniettando, nelle medesime condizioni sperimentali adottate per i campioni, soluzioni a concentrazioni note comprese nel campo di indagine dell’analita. Le tecniche cromatografiche sono sempre distruttive (anche se in senso strettamente analitico possono in alcuni casi essere non distruttive), in quanto operano esclusivamente su campioni in soluzione (o in fase vapore): i materiali oggetto di analisi vanno quindi disciolti in un opportuno solvente, nel caso siano solidi. Nel caso di analisi di acque naturali, come per le applicazioni effettuate in questo lavoro di Tesi, il campione è stato opportunamente diluito, ove necessario, in modo da non saturare i siti di scambio della fase stazionaria. Per l’analisi sono sufficienti da 1 ml a 1 μl di soluzione. Le basi del procedimento sono (Figura 5.14):
• il campione è introdotto nella fase mobile e può essere un gas o un liquido (come nel caso di analisi di acque);
• la fase mobile viene fatta eluire in continuo attraverso la fase stazionaria, che è immiscibile nell’eluente, alla opportuna pressione; mediamente circa 2000 psi anche se differenziata per cationi ed anioni;
• la fase stazionaria (liquida o solida) si trova all’interno di una colonna;
• la fase mobile e la fase stazionaria sono scelte in modo che i componenti della miscela da separare si distribuiscano tra le due fasi; i componenti più affini alla fase stazionaria eluiranno più lentamente attraverso la colonna, mentre i componenti più affini alla fase mobile si sposteranno più velocemente nel fronte cromatografico;
• la separazione dei componenti avviene in quanto ogni sostanza ha una distribuzione caratteristica tra le due fasi (costante di ripartizione Kd= Cs/Cm, dove Cs è la concentrazione di fase solida (mg/kg) mentre Cm è la concentrazione del liquido (mg/L).
Ponendo all’uscita della colonna un rivelatore che misuri la concentrazione del soluto nell’eluato e riportando il segnale in funzione del tempo si può ottenere un
cromatogramma. La posizione dei picchi sull’asse dei tempi, o tempo di ritenzione, serve
per identificare i componenti del campione. L’area sottesa dai picchi è proporzionale alla quantità di ogni singolo componente e può essere utilizzata a scopo quantitativo.
Più in dettaglio, i meccanismi di separazione impiegati in cromatografia sono: • adsorbimento
• ripartizione
• scambio ionico
• esclusione
• affinità
Quella utilizzata in questo studio è la cromatografia con scambio ionico dove la fase stazionaria è costituita da un polimero inerte contenente siti attivi ionizzati o ionizzabili, i cui contro-ioni possono essere scambiati con altri ioni aventi carica dello stesso segno. Il meccanismo di separazione è basato sulla competizione per i siti di scambio tra gli ioni presenti nella fase mobile e quelli presenti nel campione.
In base allo stato fisico della mobile possiamo classificare le tecniche cromatografiche come segue:
• Cromatografia Liquida (LC): la fase mobile è un liquido nel quale siano solubili i componenti della miscela da separare; la fase stazionaria deve essere insolubile nella fase mobile;
• Gascromatografia (GC): la fase mobile è un gas che funge da carrier per i componenti della miscela;
• Cromatografia fluida supercritica (SFC): la fase mobile è un fluido supercritico, con proprietà intermedie tra un liquido e un gas.
Le analisi di laboratorio sono state eseguite presso il laboratorio di chimica dell’Istituto di Geoscienze E Georisorse (IGG) del CNR di Pisa, sotto la supervisione del dott. Massimo Guidi e presso Università di Trieste, sotto la supervisone della dottoressa Francesca Slejko. I metodi analitici utilizzati comprendono cromatografia ionica per la determinazione degli ioni maggiori in fase acquosa.
L’analisi degli anioni delle acque è stata effettuata per vari campioni diluiti in modo diverso a seconda della necessità. L’analisi prevede l’uso di cinque standard, i quali servono per la calibrazione dello strumento (Figura 5.15):
• S1 con concentrazioni [F-]=0.20, [Cl-]=0.30, [NO3-]=1.00, [PO43-]= 1.5, [SO42-]=1.5; • S2 con concentrazioni [F-]=0.40, [Cl-]=0.60, [NO3-]=2.00, [PO43-]= 3.00, [SO42-]=3.00; • S3 con concentrazioni [F-]=0.80, [Cl-]=1.20, [NO3-]=4.00, [PO 3-]= 6.00, [SO42-]=6.00; • S4 con concentrazioni [F-]=2.00, [Cl-]=3.00, [NO3-]=10.00, [PO43-]= 15.00, [SO42-]=15.00; • S5 con concentrazioni [F-]=20.00, [Cl-]=30.00, [NO3-]=100.00, [PO43-]= 150.00, [SO 42-]=150.00.
Per l’analisi degli anioni F-, Cl-, NO3-, PO43- e SO42- è stata utilizzata una colonna Ion Pac AS23 (Dionex).
Figura 5.14: Schema del cromatografo ionico.
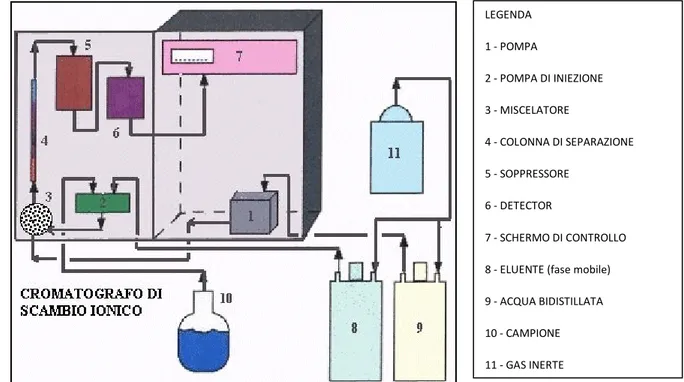
Prima di effettuare le analisi cromatografiche, bisogna inserire nello strumento, tramite una siringa, circa 5 ml di acqua milli-Q per ricondizionare il circuito. Successivamente si possono iniettare nel cromatografo ionico le soluzioni di standard (sopra descritte), tramite l’uso una siringa, aspirando 5 ml di soluzione ed iniettate nell’apposito foro. Una volta tarato lo strumento, facendo passare nel cromatografo l’acqua ed i campioni standard, si possono inserire 5 ml dei campioni di acque presi in considerazione (Figura 5.16). LEGENDA 1 - POMPA 2 - POMPA DI INIEZIONE 3 - MISCELATORE 4 - COLONNA DI SEPARAZIONE 5 - SOPPRESSORE 6 - DETECTOR 7 - SCHERMO DI CONTROLLO 8 - ELUENTE (fase mobile) 9 - ACQUA BIDISTILLATA 10 - CAMPIONE 11 - GAS INERTE
Figura 5.15: Cromatogramma degli anioni, per la costruzione della curva di calibrazione, per lo Standard 5.
L’analisi delle acque per i cationi è stata effettuata, anche questa, per vari campioni diluiti in modo diverso a seconda della necessità. L’analisi prevede l’uso di quattro standard, i quali servono per la calibrazione dello strumento (Figura 5.17):
• S1 con concentrazioni [Na +]=1.00; [K +]=0.20, [Mg 2+]=2.00, [Ca 2+]=5.00 ; • S2 con concentrazioni [Na +]=2.00, [K +]=0.40, [Mg 2+]=4.00, [Ca 2+]=10.00; • S3 con concentrazioni [Na +]=5.00, [K +]=1.00, [Mg 2+]=10.00, [Ca 2+]=25.00; • S4 con concentrazioni [Na +]=10.00, [K +]=2.00, [Mg 2+]=20.00, [Ca 2+]=50.00.
Per l’analisi dei cationi, Na+ , K+ ,Ca2+, Mg2+ è stata usata una colonna CS12A (Dionex). Il procedimento è uguale a quello degli anioni (Figura 5.18).
Figura 5.18: Cromatogramma dei cationi del campione CDMW 21.
5.4 Analisi degli elementi in traccia: ICP-MS
Il Plasma ad accoppiamento induttivo - spettrometria di massa o ICP-MS è una tecnica analitica utilizzata per le determinazioni elementari. La tecnica è stata introdotta nel 1983 e ha guadagnato l'accettazione generale in molti tipi di laboratori. L’ICP-MS ha molti vantaggi rispetto ad altre tecniche di analisi elementare come l'assorbimento atomico e spettrometria ad emissione ottica ( ICP-OES ) tra cui:
Limiti di rilevabilità per la maggior parte degli elementi uguali o migliori di quelli
ottenuti con la spettroscopia di assorbimento atomico in fornetto di grafite(GFAAS)
La capacità di gestire matrici semplici e complesse con un minimo di interferenze
della matrice a causa della temperatura elevata della sorgente ICP
La capacità di ottenere determinazioni isotopiche, pur limitatamente ad alcuni
casi.
Un ICP-MS combina un ICP ad alta temperatura (plasma accoppiato induttivamente) con uno spettrometro di massa. La sorgente ICP converte gli atomi degli elementi del
campione a ioni. Questi ioni sono quindi separati da uno spettrometro di massa quadrupolare e rilevati.
Il campione viene tipicamente introdotto nel plasma ICP come aerosol, aspirando una soluzione, tramite un nebulizzatore, o utilizzando un laser per convertire direttamente campioni solidi in un aerosol. Una volta che l'aerosol campione viene introdotto nella torcia ICP, gli elementi vengono convertiti prima in atomi gassosi e poi ionizzati.
Una volta che gli elementi del campione vengono convertiti in ioni, vengono accelerati nello spettrometro di massa tramite i coni di interfaccia. La regione di interfaccia nella ICP-MS trasmette gli ioni che viaggiano nel flusso campione argon a pressione atmosferica (1-2 torr) nella regione di bassa pressione dello spettrometro di massa (<1 x 10 -5 torr). Questo viene fatto attraverso la regione intermedia dove il vuoto è stato creato dai due coni di interfaccia, il campionatore e skimmer (Figura 5.19). I coni campionatori e skimmer sono dischi di metallo con un piccolo foro (~ 1 mm) al centro. Lo scopo di questi coni è di campionare la porzione centrale del fascio ionico proveniente dalla torcia. A causa dei piccoli diametri dei fori nel campionatore e skimmer coni, l’ ICP-MS ha alcune limitazioni per quanto riguarda la quantità di solidi totali disciolti nei campioni da analizzare. In generale, si raccomanda che i campioni non abbiano più dello 0,2% solidi totali disciolti (TDS) per ottimizzare le prestazioni dello strumento e la stabilità. Se vengono eseguiti campioni con livelli molto elevati di TDS, gli orifizi dei coni si ostruiscono, causando una diminuzione della sensibilità e capacità di rilevamento e per cui il sistema deve essere spento per manutenzione. Questo è il motivo per cui molti tipi di campioni, compreso il terreno e campioni di roccia devono essere diluiti prima di essere analizzati.
Gli ioni dalla sorgente ICP sono poi focalizzati da lenti elettrostatiche. Diversi tipi di sistemi ICP-MS hanno tipi di sistemi di lenti variabili. Il più semplice utilizza una sola lente, mentre i sistemi più complessi possono contenere fino a 12 lenti.
Una volta che gli ioni entrano nello spettrometro di massa, sono separati dal loro rapporto massa-carica. Il tipo più comunemente usato è il filtro di massa a quadrupolo. In questo tipo, a 4 aste (circa 1 cm di diametro e 15-20 cm di lunghezza) disposte come in Figura 5.20 sono applicate tensioni CA e CC a coppie opposte di aste. Il risultato è che un filtro elettrostatico permette il passaggio di ioni con un dato rapporto massa-carica (m/e) al rilevatore in un dato istante di tempo.
Figura 5.20: Schema dello spettrometro di massa a quadrupolo.
Una volta che gli ioni sono stati separati sulla base del loro rapporto massa/carica, devono essere misurati da un rivelatore. Lo scopo fondamentale del rivelatore è di tradurre il numero di ioni che colpiscono il rivelatore in un segnale elettrico che può essere misurato e correlato al numero di atomi di tale elemento nel campione mediante l'uso di standard di calibrazione. La maggior parte dei rivelatori utilizzano moltiplicatori di elettroni o fotomoltiplicatori.
Le analisi sono state effettuate presso l’ Università di Pisa sotto la supervisione del Professor Massimo D’Orazio. Lo strumento utilizzato è il NexION 300X della Perkin Elmer. I detection limits sono riportati in Tabella 5.2.
Tabella 5.2: Detection limits dei principali ioni per quanto riguarda il NexION 300X.
Mo Ag Cd Sb Hg Tl Pb V Cr Mn Co Ni Cu Zn As
5.5 Analisi isotopiche: spettrometria di massa
Su alcuni dei campioni di acque superficiali, drenaggi e sulle acque meteoriche sono stati determinati i rapporti isotopici D/H e 18O/16O. Su una parte dei campioni del torrente Baccatoio è stata inoltre determinato il rapporto 87Sr/86Sr (comunemente detto: composizione isotopica dello stronzio).
Le determinazioni sono state effettuate tramire IRMS (isotope-ratio mass spectrometry) per ossigeno e idrogeno e tramite TIMS (thermal ionization mass spectrometry) per stronzio.
Sia nella metodologia IRMS che TIMS, gli spettrometri permettono di separare ioni in fase gassosa sulla base del loro rapporto massa/carica per azione di campi magnetici, a differenza dello spettrometro a quadrupolo descritto in precedenza per la tecnica ICP-MS che utilizza campi elettrici.
L’equazione base della spettrometria di massa a settore magnetico è:
Dove r rappresenta il raggio di deflessione che subisce lo ione attraversando il campo magnetico in funzione della propria massa (m) e carica (e; considerando che vengono analizzate cariche +1) data l’intensità del campo magnetico (B) e la differenza di potenziale con cui lo ione è accelerato nel campo magnetico stesso (V), che determina la sua energia cinetica.
In generale, uno spettrometro di massa è costituito da:
- sorgente: è la parte in cui avviene il processo di ionizzazione. Nel caso delle analisi isotopiche di ossigeno e idrogeno della molecola d’acqua, e considerando gli spettrometri a doppio sistema di ingresso, il campione e il gas utilizzato come standard di riferimento, vengono alternativamente introdotti, nella sorgente. Le molecole del gas (CO2 per la determinazione dei rapporti di 18O/16O, SO2 per, H2 per D/H), sottoposte all'interno della sorgente ad un intenso bombardamento di elettroni prodotti da un filamento incandescente, subiscono un processo di ionizzazione ed una conseguente accelerazione e focalizzazione per mezzo di opportuni campi elettrici. Nel caso della analisi di metalli, come lo stronzio, tramite TIMS, gli ioni sono prodotti per riscaldamento dopo essere stati depositati sulla superficie metallica di un filamento. Per
2 / 1 2 2 eB mV r
effetto termoionico, il campione si ionizza ed il gas ionizzato è accelerato e focalizzato come descritto.
-magnete: ha il compito di deviare gli ioni su traiettorie circolari sulla base del loro rapporto massa/carica.
- analizzatore (detto anche tubo di volo): è la parte che collega tra loro la sorgente ed i collettori, dove avviene la deflessione.
- sistema di collettori (uno o più): rappresentano il punto di arrivo degli ioni ed è costituito da un Faraday Cup che tramite un’alta resistenza trasforma il segnale di ioni in corrente elettrica.
- sistema di pompaggio: assicura il necessario alto vuoto all’interno della sorgente ed analizzatore Il sistema di pompaggio è generalmente realizzato accoppiando una pompa turbomolecolare ed una pompa rotativa a doppio stadio.
Nel caso delle analisi isotopiche di O-H, durante la misura, lo spettrometro esegue più volte il confronto tra i due gas immessi e alla fine del ciclo fornisce un valore medio delle differenze relative ai confronti eseguiti.
Per l’analisi della composizione isotopica dell’ossigeno e dell’idrogeno nelle acque è stata utilizzata una metodologia che consiste nel portare il campione liquido in equilibrio isotopico con un gas puro, CO2 nel caso dell’ossigeno, e H2 nel caso dell’idrogeno.
Nel caso delle analisi isotopiche dello Sr il rapporto 87Sr/86Sr viene corretto per il frazionamento isotopico che avviene in fase di emissione sulla base del rapporto 86Sr/88Sr posto 0.1194. L’interferenza con l’isobaro 87Rb è stata minimizzata tramite procedure di separazione cromatografica in laboratorio e successivamente manipolando la temperatura del filamento durante l’emissione in sorgente. Non è stato necessario applicare alcuna correzione
per l’interferente.
Data la difficoltà nell’ effettuare una determinazione accurate delle abbondanza assolute, si fa
in genere riferimento ad abbondanze relative, utilizzando come termine di paragone il rapporto isotopico determinato in una sostanza standard di riferimento. Da qui si introduce la composizione isotopica δ‰ ,definita come la deviazione in parti per mille del rapporto isotopico di un campione rispetto ad uno standard di riferimento, la sua notazione è:
δ‰ =[(Rc-R(stand)/R(stand)] x 1000
In particolare vengono studiati i rapporti tra 18O e 16O, e tra 2H e 1H; dal momento che per
indicare l’isotopo dell’idrogeno con massa 2 di usa il termine deuterio (D), in genere si trovano indicati i due principali isotopi dell’idrogeno con H e D. È quindi possibile definire le composizioni isotopiche di ossigeno e idrogeno come:
Le analisi sono state effettuate, per quanto riguarda gli isotopi dell’ossigeno e dell’idrogeno presso l’Università degli Studi di Trieste, sotto la supervisione della Dr.ssa Barbara Stenni e della Dr.ssa Marzia Michelini; per quanto riguarda gli isotopi dello stronzio le analisi sono state fatte presso l’Università la Sapienza di Roma, sotto la supervisione della Prof.ssa Francesca Castorina.