- 25 -
Gli Oppioidi
2.1 Oppioidi: classificazione
Gli oppioidi sono una classe farmaceutica morfino-simili che si lega a recettori specifici dell’organismo. L’oppio è ottenuto dall’essicazione del lattice delle capsule immature (materiale brunastro e gommoso) del Papaver somniferum e la morfina è una dei suoi principali alcaloidi che ha fatto da prototipo per altri oppioidi di sintesi come il metadone e il fentanyl. L’oppio è stato usato in Medicina Veterinaria per il sollievo del dolore già nel Papiro di Ebers, scritto intorno al 1500 a.C. nell’antico Egitto (Adams, 1999).
I recettori per gli oppioidi sono localizzati sopratutto a livello delle lamine I e II delle corna dorsali del midollo spinale e a livello cerebrale (amigdala, locus ceruleus, talamo, ipotalamo), ma anche in corrispondenza della capsula articolare e a livello tissutale. Nel tessuto infiammato si ha un maggior numero di recettori per gli oppioidi (Bufalari et al., 2012).
Ad oggi esistono cinque differenti tipi di recettori: mu (µ), kappa (ҡ), delta (δ), sigma (σ), epsilon (ε). Alcuni di questi sono a loro volta suddivisi in sottotipi. L’International Union of Pharmacologhy (IUPHAR) raggruppa i recettori e i loro sottotipi con un’altra nomenclatura: OP1 (recettori δ, δı, δ2), OP2 (recettori ҡ, ҡı, ҡ2, ҡз ), OP3 (µ, µı, µ2, µз), ORL1(recettori σ e ε).
Gli oppioidi sono impiegati in medicina veterinaria e umana per il trattamento del dolore, da lieve a moderato fino anche a quello più severo.
- 26 -
2.2 Azione farmacologica degli oppioidi e uso clinico
La risposta di tutti i sottotipi recettoriali degli oppioidi non è stata pienamente chiarita, ma per esempio, i µı sono ritenuti responsabili dell’analgesia sovraspinale perché sono numerosi a livello della sostanza grigia periacqueduttale. Se stimolati, attivano delle vie discendenti inibitorie che modulano la trasmissione del dolore all’altezza del corno dorsale nel midollo spinale. I recettori µ2 determinano una depressione respiratoria e µз, invece, sembrano partecipare a un’azione immunosoppressiva dei globuli bianchi e sono attivati sia da morfina sia da codeina (Bufalari, 2012).
I sottotipi δı e δ2 interagiscono anche con oppoidi endogeni (metencefalina, leucoencefalina, dinorfina ed endorfine).
Il recettore ORL-1 (Opioid Receptor Like 1), scoperto nel 1994, ha affinità per la nocicettina che modula il meccanismo dell’analgesia e dell’iperalgesia. L’attivazione dei recettori ҡ riduce il trasporto di ioni Ca2+
a livello della membrana presinaptica interferendo con il rilascio di alcuni neurotrasmettitori quali l’acetilcolina, la dopamina, la norepinefrina e la sostanza P, rilasciata nei neuroni afferenti a livello delle corna dorsali midollari in seguito a uno stimolo dolorifico periferico. L’inibizione del rilascio cellulare di acetilcolina potrebbe essere un importante processo implicato nell’analgesia indotta dagli oppioidi.
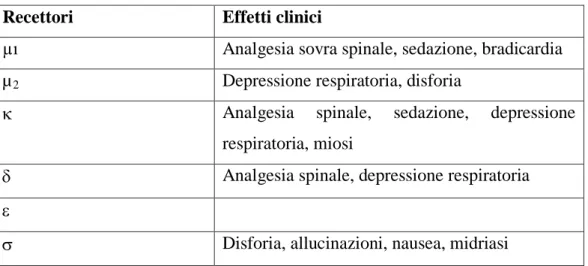
Recettori Effetti clinici
µı Analgesia sovra spinale, sedazione, bradicardia
µ2 Depressione respiratoria, disforia
Analgesia spinale, sedazione, depressione
respiratoria, miosi
Analgesia spinale, depressione respiratoria
Disforia, allucinazioni, nausea, midriasi
- 27 -
Il legame oppiode-recettore determina delle risposte dose-dipendenti: bradicardia, depressione respiratoria, ipotermia, diminuzione del tono gastrointestinale con costipazione e ileo, a dosi ripetute, raramente spasmo dei dotti biliari e pancreatici, variazioni nella produzione ormonale: rilascio di ormone antidiuretico (ADH), somatotropo (SH) e prolattina (PRL) e inibizione di ormone follicolostimolante (FSH) e luteinizzante (LH).
Alcune specie (cavallo, gatto, maiale e ruminanti) possono mostrare tachicardia, ansia, ipercinesi e nel cavallo può manifestarsi anche sudorazione. A dosi farmacologiche, l’effetto disforico è possibile nel cavallo e nel gatto che hanno una concentrazione di recettori a livello dell’amigdala e della corteccia frontale due volte minore rispetto al cane e ai primati. L’eccitazione sembra dovuta a un maggior rilascio dalle cellule di norepinefrina e dopamina: infatti, l’associazione in premedicazione di oppiodi con farmaci che inibiscono il rilascio presinaptico di dopamina (acepromazina) o di noradrenalina (xilazina o α2 agonisti adrenergici) ne riduce l’effetto (Green et al., 2002).
Una volta legati ai recettori, gli oppioidi producono degli effetti che possono essere utilizzati come un successivo metodo di classificazione di questi farmaci. Si può quindi parlare di: agonista, agonista parziale, agonista-antagonista, antagonista.
Gli agonisti, o agonisti puri, raggruppano gli analgesici oppioidi con un’elevata affinità recettoriale in grado di produrre un effetto analgesico maggiore quanto maggiore è la dose del farmaco. Di questo gruppo fanno parte: morfina, metadone, fentanyl, alfentanyl, sufentanyl, remifentanyl, petidina, idromorfone.
Gli agonisti parziali hanno un’elevata affinità recettoriale, come gli agonisti puri, ma all’aumentare della dose non determinano un incremento dell’analgesia come invece accade per i primi. Oltre la dose massima si raggiunge un valore soglia detto “plateau” sopra il quale non si può incrementare l’effetto analgesico. Sono agonisti parziali la buprenorfina e la codeina. In particolare, il ruolo della buprenorfina a livello recettoriale è controverso: è considerata un parziale agonista µ e un’antagonista dei ҡ, anche se vi sono numerose opinioni in merito. Essa è utilizzata nel cane soprattutto per l’analgesia post operatoria poiché è 25-50 volte più potente della morfina e garantisce un’analgesia della durata di 6-8 ore. Il suo forte legame con i
- 28 -
recettori rende difficile un successivo antagonismo da parte di altri oppioidi. Tuttavia, la sua attività nei confronti di oppioidi agonisti puri non è stata completamente valutata in ogni specie (Goyenechea et al., 2006).
Il butorfanolo è l’unico agonista-antagonista, a indicare l’agonismo per i recettori ҡ e l’antagonismo per i recettori µ (secondo alcuni autori è agonista parziale) (Bufalari, 2012).
Infine, il naloxone è un farmaco antagonista puro dei recettori µ e meno degli altri recettori (i recettori σ sono particolari perché non rispondono al naloxone). Il suo meccanismo risolve la depressione respiratoria ed elimina l’analgesia, provoca tachicardia, ipertensione, disaritmie e possibile edema polmonare. Il naloxone ha un onset molto breve, di 2-5 minuti e può essere somministrato per via parenterale a dosi comprese tra 0,01-0,04 mg/Kg. La durata è breve, spesso inferiore a quella del farmaco da spiazzare ed è quindi necessario ripeterne la somministrazione dopo venti minuti dalla prima iniezione (Bufalari, 2012). Per antagonizzare parzialmente le difficoltà respiratorie provocate da un agonista puro, senza eliminare l’analgesia, possono essere somministrate basse dosi di butorfanolo (0,05 mg/Kg ev) prima dell’uso del naloxone (Bufalari, 2012).
2.3 Oppioidi impiegati nello studio clinico
Morfina
La morfina è l’alcaloide dell’oppio e la molecola di riferimento nel gruppo degli analgesici morfinici. Prende il suo nome da Morfeo, il dio del sonno (Trescot et al., 2008).
Ha una preferenza per i recettori µ e ha un’azione analgesica centrale, spinale e sovra-spinale, in particolare è efficace per un’elevata nocicezione (viscerale, muscolare, osteoarticolare, cancerogena), ma non per i dolori neuropatici. La sua durata è di circa 2-4 ore nel cane, e leggermente più lunga nel gatto (3-6 ore).
La formula utilizzata in veterinaria per le preparazioni iniettabili (im, sc, locale/ locoregionale) è la morfina cloridrato. La via endovenosa è altresì
- 29 -
indicata nei libri, ma meno praticata perché una somministrazione eccessivamente veloce potrebbe portare a una liberazione d’istamina da parte dei mastociti. Il rilascio dell’istamina sembra dovuto ad una reazione idiosincrasica di alcuni mastociti che non coinvolge gli anticorpi IgE, come invece accade nella liberazione d’istamina in seguito a una forma d’ipersensibilità immunomediata (ipersensibilità di tipo I). Questa reazione non-allergica può causare dilatazione venosa, aumento della permeabilità vascolare, contrazione della muscolatura liscia con possibile broncospasmo, secrezione di ghiandole mucose. La somministrazione di naloxone non è in grado di arrestare le reazioni avverse date dall’istamina e questo dimostrerebbe che il rapporto istamina-morfina non dipende da un legame recettoriale (Fanny Li, 2006).
La morfina è una molecola molto idrofilica: il suo passaggio attraverso la membrana encefalica, lipofila, è più lento rispetto ad altri oppioidi. Questa proprietà permette di sfruttare la morfina per una lunga analgesia spinale che può arrivare anche a 16-24 ore con dosaggi da 0,1 a 0,2 mg/kg. La comparsa dell’effetto analgesico si ha dopo circa trenta minuti (Bufalari, 2012).
La morfina è un farmaco antitussigeno e ha azione emetica perché stimola direttamente il centro del vomito (CTZ Chemoreceptor Trigger Zone). Il gatto necessita di dosaggi di morfina 740 volte più elevati rispetto a quelli che stimolano il vomito nel cane.
Il cane è meno sensibile all’azione miotica della morfina rispetto all’uomo. Per avere una miosi massimale nel cane sono necessarie dosi parenterali di morfina superiori a 2 mg/kg (Adams, 1999).
Il metabolismo della morfina è epatico e renale ed è principalmente eliminata coniugata con le urine.
Può interferire con alcuni farmaci che hanno la stessa azione farmacocinetica, per esempio riduce o annulla l’effetto dei diuretici, potenzia l’azione miorilassante dei bloccanti neuromuscolari, e aggrava l’emorragia da dicumarolo e di altri anticoagulanti. La co-somministrazione con rifampicina causa una diminuzione della concentrazione e dell’attività della morfina e del suo metabolita.
- 30 - Fentanyl
Le vie attraverso cui è possibile somministrare il fentanyl sono numerose. La biodisponibilità orale-enterale è lieve, quindi il fentanyl è preferibilmente ideato come farmaco iniettabile (via endovenosa, sottocutanea, intramuscolare, epidurale - spinale). Nell’uomo, per il trattamento del dolore cancerogeno (dolore cronico) sono state approvate delle formulazioni di supporto ad altri oppiacei in forma di spray nasale o orale transmucosale, come stick (Actiq®) o dispositivi patch (Effentora®, Abstral®, Onsolis®), al momento non adattabili o utilizzati per l’analgesia dei piccoli animali (Kukanich et al., 2012).
Il fentanyl presenta delle caratteristiche differenti rispetto alla morfina: ha una potenza 100 volte superiore e una maggiore liposolubilità.
La potenza è una caratteristica del farmaco che indica la sua affinità per i recettori. Di conseguenza, maggiore è la potenza e minore sarà la dose necessaria per produrre l’effetto.
La liposolubilità, invece, contraddistingue la cinetica e la distribuzione del farmaco che mostra una rapida efficacia (onset di 2-4 minuti) e un’azione brevissima (15-20 minuti).
Il fentanyl si lega in elevata percentuale alle proteine plasmatiche e subisce un’elevata ridistribuzione tissutale, il che comporta una certa variabilità nella velocità di eliminazione del farmaco (circa 10-20 ml/min/kg). I suoi metaboliti sono escreti soprattutto con le urine (Bufalari, 2012).
Per prolungare la breve analgesia e la rapida clearance che contraddistingue il fentanyl dopo una singola somministrazione, è stata ideata una formulazione in cerotto transdermico (Duragesic®), per umana, in deroga per uso veterinario. La sua particolare formulazione permette al principio attivo, lipofilo, di diffondersi attraverso la matrice lipidica dello strato corneo. Il farmaco contiene come principio attivo il fentanyl a diverse concentrazioni (12,5, 25, 50, 75,100 µg/h) e raggiunge una concentrazione plasmatica analgesica detta “efficace” di 1-2 ng/ml dopo 12-24 ore dall’applicazione nel cane, e dopo 7 ore nel gatto. Da ciò si evince che è necessario somministrare altri analgesici per avere un’immediata copertura dolorifica (Kukanich, 2012).
- 31 -
L’effetto analgesico del cerotto dura più di 72 ore sia nel cane che nel gatto e, in quest’ultimo, l’analgesia prodotta è equivalente a quella data da più somministrazioni di butorfanolo (Hofmeister, 2004).
La minima concentrazione plasmatica efficace (MEC), intesa come la minima concentrazione plasmatica di analgesico che è sufficiente a prevenire un’altra richiesta di farmaco, è stata stimata nell’uomo in media intorno a 0,63 ng/ml, dopo una somministrazione endovenosa di fentanyl a 20 µg/h. Nel cane la valutazione del dolore è diversamente interpretabile ed è più difficile definire un valore analgesico preciso. Alcuni studi hanno stimato un range efficace attendibile tra 0,4 e 1,28 ng/ml (Kukanich, 2012).
A secondo del cane la variabilità nella concentrazione plasmatica è del 30% e, a dosi diverse, non si nota una differente concentrazione plasmatica. Infatti, con 75 µg/h e 100 µg/h i valori sono stati di 1,4 e 1,2 ng/ml rispettivamente (Kukanich, 2012).
Detto ciò va rilevato che una singola dose o concentrazione di fentanyl non fornisce lo stesso grado di analgesia in tutti gli animali e questo è il motivo per cui esiste una grande variabilità di risposta a una stessa dose del farmaco. In infusione continua endovenosa a 10 µg/kg/h, dopo un primo bolo da 10 µg/kg, il fentanyl mantiene una concentrazione plasmatica efficace di 1 ng/ml per tutta la durata dell’infusione; dopo 3 ore la sua concentrazione tende a decrescere.
- 32 -
Figura 2.1: concentrazione plasmatica di fentanyl dopo somministrazione endovenosa (Recuvyra nella pratica quotidiana, Briganti e Staffieri).
Rispetto alla somministrazione in boli, la via transdermica permette un continuo rilascio di fentanyl che rende stabile la concentrazione plasmatica, evita il metabolismo epatico che avviene con la somministrazione orale e transrettale, e riduce la necessità di dosi ripetute (Sano et al., 2006).
Tuttavia, lo stesso Hofmeister che ha paragonato l’analgesia del fentanyl cerotto con quella della butorfanolo in boli, riporta che l’utilizzo transdermico nei piccoli animali ha causato irritazioni cutanee e un uptake del principio attivo variabile e non prevedibile.
Il PK del cerotto, inteso come il pH al quale il 50% del farmaco si trova in forma ionizzata (idrosolubile) e il 50% in quella non ionizzata (liposolubile), dipende dal flusso ematico e dalla temperatura cutanea. L’anestesia con isofluorano può ridurre del 50% la concentrazione plasmatica del cerotto, probabilmente in seguito alla vasodilatazione data dall’alogenato che determinerebbe un minor afflusso sanguigno a livello cutaneo o un’ipotermia con conseguente minor assorbimento del farmaco (Kukanich, 2012).
- 33 -
Una ricerca condotta su come gli oppiodi possano alterare la temperatura nei gatti, ha descritto un rialzo della temperatura basale negli animali trattati con buprenorfina, butorfanolo, chetamina e idromorfone, da soli o in associazione con ketamina o isofluorano (Posner et al., 2010). L’effetto del cerotto di fentanyl sulla temperatura corporea, invece, determinerebbe una diminuzione nella produzione di calore e un aumento nei meccanismi di dispersione dello stesso attraverso una maggiore ventilazione. In letteratura non ci sono però dati di controllo della temperatura per 96 ore successive alla chirurgia dopo l’utilizzo del cerotto transdermico di fentanyl, quindi la minore temperatura può essere attribuita agli oppioidi, agli anestetici o ad altre cause (Kukanich, 2012). I differenti metodi per riscaldare un animale e la temperatura ambientale possono influire su una corretta misurazione della temperatura nei pazienti per il periodo intra e post-operatorio, ma sull’argomento vi sono ancora pochi studi in medicina veterinaria.
Recuvyra
Recuvyra è il nome commerciale di una soluzione transdermica di fentanyl (50 mg/ml) registrata per il cane. Il farmaco, come suggerito dalla casa produttrice, è stato ideato per il controllo del dolore associato a interventi chirurgici ortopedici e dei tessuti molli.
Il liquido si presenta limpido, incolore o giallo chiaro. È somministrato con un apposito applicatore nella regione scapolare dorsale, a dosi di 0,5 ml direttamente sulla pelle spostando il pelo, senza tosare il cane. L’applicatore a “U” distribuisce la quantità di farmaco in due punti distinti e, se la dose è maggiore di 0,5 ml, lo stesso volume deve essere distanziato di 2,5 cm dal sito precedente.
- 34 -
Figura 2.2: Recuvyra e applicatore.
Studi condotti sul farmaco hanno dimostrato che la concentrazione plasmatica del fentanyl tende a crescere più rapidamente nell’applicazione dorsale a livello scapolare, mentre persiste più a lungo nella regione addominale ventrale. Tale differenza è probabilmente dovuta a diverse caratteristiche dell’epidermide tra la regione dorsale e quella ventrale, in particolare in merito allo spessore della cute, al numero di follicoli piliferi, alla secrezione sebacea e all’afflusso sanguigno (Kukanich, 2012).
Oltre al principio attivo, Recuvyra contiene anche due componenti che coadiuvano l’azione del fentanyl stesso: l’alcol isopropilico e l’octil salicilato. L’alcol è un solvente che determina il passaggio del fentanyl nello strato corneo ed evapora velocemente. L’octil salicilato aumenta la velocità di assorbimento del principio attivo e crea un deposito all’interno della matrice lipidica.
Il fentanyl, in seguito, diffonde attraverso la cute in maniera passiva secondo un gradiente di concentrazione.
Recuvyra raggiunge la concentrazione considerata analgesica (1 ng/ml) entro 4 ore dalla somministrazione e la sua azione antidolorifica si protrae per i quattro giorni successivi all’applicazione. La concentrazione plasmatica massima compresa tra 0,7 e 4,7 ng/ml viene raggiunta entro 10-18 ore.
Esso è metabolizzato dal fegato ed escreto con le urine. Tuttavia, il profilo farmacocinetico del farmaco si distingue dalle altre formulazioni contenenti il fentanyl: infatti, il suo periodo di assorbimento è relativamente più lungo rispetto alla sua eliminazione, secondo una cinetica definita flip-flop. Di
- 35 -
conseguenza, la parte terminale della curva concentrazione-tempo è determinata dall’assorbimento del farmaco e non dalla sua eliminazione.
Il cerotto di fentanyl alla concentrazione di 75 µg/h in cani di peso di 26 ± 3,5 kg, così come una somministrazione di fentanyl in bolo a 5 µg/kg EV seguita da un’infusione a 0,5µg/kg/h, diminuiscono la MAC dell’isofluorano del 37 % se paragonati ad animali normotermici che hanno ricevuto solo un somministrazione placebo (Kukanich, 2012). Allo stesso modo Recuvyra ha un effetto sparing in grado di ridurre la MAC di anestetici inalatori, in modo dose-dipendente (Kukanich, 2012).
Gli effetti del fentanyl sulle funzioni vitali sono tutti dose-dipendente: minima ipotensione, diminuzione della frequenza respiratoria transitoria e marginale, ipotermia (Savides, 2012). A livello cardiovascolare determina poche variazioni di frequenza e di ritmo cardiaco, si osserva bradicardia, ma contemporaneamente si ha un aumento della gittata sistolica che mantiene costante la gittata cardiaca e la pressione media. La frequenza respiratoria diminuisce del 30% al massimo nelle prime 48 ore dalla somministrazione della soluzione di fentanyl in cani che ricevono una dose di farmaco da tre fino a cinque volte superiore a quella raccomandata (Savides, 2012).
La termoregolazione viene influenzata a livello centrale, producendo meno calore e aumentando la dispersione dello stesso (Kukanich, 2012). Importante è la differenza tra la soluzione transdermica e il cerotto di fentanyl in merito alla temperatura corporea: nel cerotto un’ipotermia può causare un abbassamento della concentrazione plasmatica del farmaco che, invece, non avviene con Recuvyra.
Un eccessiva dose del farmaco può influenzare negativamente l’appetito e la sete dell’animale con una transitoria riduzione di peso corporeo, determinare sedazione, oliguria e stitichezza. (Kukanich, 2012).
Recuvyra è stato studiato per l’immissione in commercio per i cani di età superiore ai 6 mesi e di peso superiore ai 3 kg, in animali sani, non in gravidanza. La somministrazione del farmaco richiede l’uso dei guanti da parte dell’operatore perché la soluzione impiega 5 minuti per asciugarsi e, prima di questo tempo, il liquido potrebbe passare attraverso la cute dell’uomo determinando delle reazioni avverse.
- 36 -
Il naloxone si è dimostrato efficace per il trattamento di un sovradosaggio di Recuvyra cinque volte superiore (13 mg/kg) a quello ritenuto appropriato (2,6 mg/kg). I cani reclutati nello studio erano 24 e tutti di razza Beagle; tutti hanno ricevuto naloxone IM dopo un’ora fino a 8 ore dopo il trattamento con Recuvyra e di questi, 16 sono stati trattati con 160 µg/kg, mentre 8 con un dosaggio di 40 µg/kg.
Entrambi i dosaggi sono stati sicuri ed efficaci per contrastare gli effetti collaterali della soluzione di fentanyl transdermica. Tuttavia, il dosaggio di naloxone a 160 µg/kg si è dimostrato più rilevante per un maggiore aumento della temperatura e della frequenza cardiaca, ma anche per una probabilità di sedazione tre volte inferiore rispetto al dosaggio di 40 µg/kg. La sedazione dopo l’utilizzo dell’antagonista accade per l’effetto sedativo del fentanyl che si ripresenta dopo 1-3 ore dal trattamento col naloxone (Freise, 2012).
I bambini di peso uguale o inferiore a 15 kg non devono toccare i cani trattati per le 72 ore successive all’applicazione del Recuvyra, quindi per cani di peso uguale o superiore ai 20 kg, è indicata l’ospedalizzazione.
Il farmaco non rientra nel registro di carico e scarico di stupefacenti e per l’approvvigionamento necessita semplicemente di una ricetta in triplice copia non ripetibile, secondo il D.P.R. 309/90.