4
5 CLASSIFICAZIONE TRASPORTATORI ATP-DIPENDENTI
Le ATP-binding cassette transporters (trasportatori ABC), fanno parte di una superfamiglia proteica molto diffusa in diversi tipi di organismi. Dall’espressione di 49 geni nell’uomo si originano ben 7 sottoclassi di proteine ABC : ABC A, ABC B, ABC C, ABC D, ABC E, ABC F, ABC G. Sono proteine transmembrana capaci di regolare il passaggio di diverse tipologie di sostanze attraverso numerose barriere fisiologiche :
-Barriera Emato-Encefalica (BEE) -Placenta
-Intestino -Stomaco -Fegato
Per poter svolgere tale funzione, necessitano di ATP (adenosina trifosfato) quale fonte di energia. Per questo motivo, questi trasportatori sono, a tutti gli effetti, delle “pompe di membrana”. Le proteine ABC esercitano una funzione protettiva contrastando l’accumulo di molte sostanze potenzialmente pericolose o per un determinato distretto fisiologico o per la sopravvivenza della singola cellula. Tali trasportatori possiedono la capacità di interagire con una vasta gamma di substrati, strutturalmente e funzionalmente, molto diversi fra loro (per esempio, metaboliti, lipidi, steroli, farmaci). Tra le numerose proteine trasportatrici ABC, la ABC B1, nota anche come Glicoproteina-P (P-gp), ha suscitato un grande interesse a causa del suo ruolo in alcune patologie neuro-degenerative (Parkinson,Alzheimer) e tumorali. In questo senso, l’aspetto sul quale la ricerca si sta concentrando negli ultimi anni è, oltre alla caratterizzazione delle sue funzioni e della sua struttura, la valutazione della sua espressione in condizioni fisiologiche e patologiche. Potenzialmente, potrebbe rappresentare uno dei bersagli principali sia per quanto riguarda la diagnosi che il trattamento di patologie neurodegenerative e tumorali. I vantaggi che si potrebbero riscontrare sono svariati e riguardano : una migliore diagnosi delle patologie e l’aumento della biodisponibilità dei farmaci con conseguente miglioramento del profilo terapeutico e riduzione di fenomeni di resistenza.
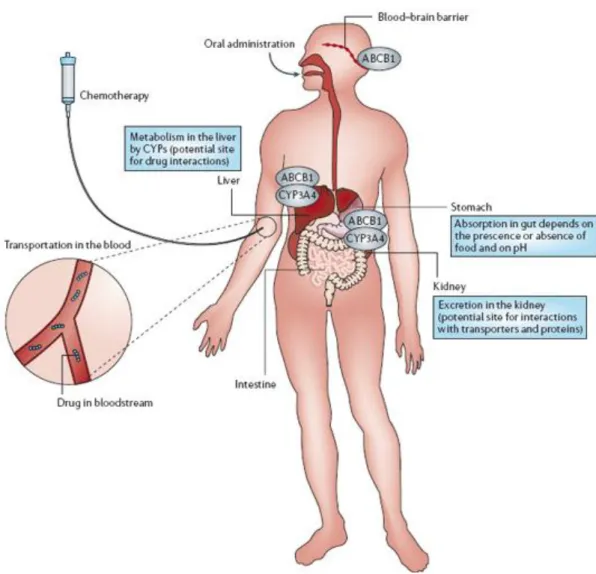
6 Figura 1.Localizzazione della P-gp nell'organismo
7
P-GLICOPROTEINA (P-gp)
1.ESPRESSIONE DELLA P-gp E CONTROLLO DELLA SUA ATTIVITÀ
Sia l’espressione che l’attività della P-gp sono influenzate da numerosissimi segnali, oltre che fisiologici anche patologici, che possono o aumentare o ridurre la sua attività di trasporto. Per la P-gp, è noto come la sua espressione sia influenzata dall’azione di ben 49 geni, fra i quali, il più importante è sicuramente l’MDR1, il cui analogo umano è l’hMDR1 : essi agiscono da promotori1. La regolazione avviene grazie all’azione di molte altre proteine che regolano il suo processo trascrizionale. Finora, sono stati caratterizzati diversi elementi della zona del promotore come la
GC-box, la Y-box, l’elemento p53, l’elemento X del recettore del Pregnano (PXR)1. Questi sono siti di legame per diversi fattori di trascrizione i quali rispondono a diversi stimoli ambientali come : l’ipossia, gli xenobiotici, l’infiammazione, lo stress ossidativo. Inoltre, anche meccanismi epigenetici, quali l’acetilazione degli istoni o la metilazione del DNA, influiscono pesantemente sulla espressione dell’ hMDR11
. È, poi, possibile un controllo post-traduzionale, grazie all’azione di numerosi segnali intracellulari, che, senza influenzare direttamente l’espressione del gene, possono dar luogo a numerosi meccanismi di modulazione1. Ad esempio, la fosfo-/defosforilazioe della proteina,la degradazione,la sua associazione con altre proteine di membrana. A monte dell’estremità 5’ dell’hMDR1, si trova un complesso regolatore, formato da diversi siti di legame per il recettore nucleare ligando-dipendente : il PXR o recettore “X” del pregnano1. Questo è in grado di mediare l’induzione della trascrizione su
MDR11. Il PXR, fa parte della superfamiglia dei fattori di trascrizione ligando-dipendenti. È attivato, oltre che dagli steroidi naturali (es., pregnenolone, progesterone) e sintetici (antiglicocorticoidi e glucicocorticoidi), anche da molti altri xenobiotici (farmaci, sostanze tossiche). I suoi substrati variano a seconda della sua localizzazione nei vari distretti dell’organismo. Per quanto riguarda la sua relazione con la P-gp, vari studi hanno evidenziato un’azione diretta del PXR sia sull’espressione che sull’attività di trasporto della proteina1
. È stato evidenziato che l’espressione della P-gp, nell’epilessia e nell’ictus, aumenti, ma non è ancora chiaro se tale aumento sia la diretta conseguenza dell’attivazione del PXR o di altri recettori nucleari da parte di ligandi endogeni o farmaci1.
8 2.STRUTTURA DELLA P-GP
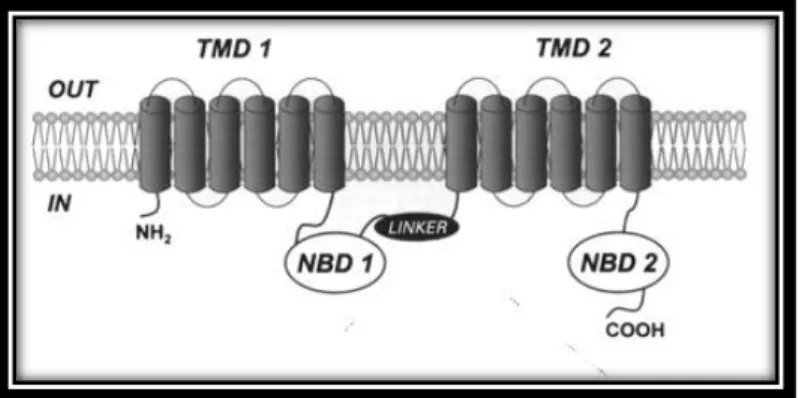
Tutti i transporters ABC esistono sottoforma di monomeri, la cui unità funzionale di base formata da 4 domini fondamentali suddivisibili in :
2 domini Trans-membrana (TMD), che costituiscono l’estremità N-terminale 2 domini di Legame Nucleotidici (NBD), che costituiscono l’estremità
C-terminale
Sono, inoltre, presenti un linker ed una cavità transmembranaria2
I TMD sono costituiti da 12 α-eliche transmembrana ed attraversano il doppio strato lipidico 6 volte3.
Figura 2.Struttura 3D P-gp. Le 2 subunità colorate in giallo e turchese.
Nella loro porzione intramembranaria, questi domini sono costituiti da sequenze amminoacidiche molto poco conservate che presentano una estrema variabilità strutturale. Tale caratteristica fa sì che essi siano in grado di funzionare come siti di riconoscimento e di traslocazione nei confronti di una vastissima gamma di substrati. È, inoltre, presente una larga cavità a livello dell’interfaccia fra questi due domini transmembranari che consente alle sostanze idrofile di essere estruse facilmente senza che vi sia bisogno di un particolare grado d’affinità3
9 Figura 3.Cavità centrale TMD della P-gp vista dal versante extracellulare
Figura 4.Posizione cavità centrale TMD nella P-gp
10 I domini NBD sono localizzati sul versante citoplasmatico e sono caratterizzati da sequenze altamente conservate. Formano i siti che catalizzano sia il legame che l’idrolisi dell’ATP. Ogni NBD presenta due siti di legame per l’ATP che però non possono operare contemporaneamente. Nella maggior parte dei trasportatori ABC, inclusa la P-gp, si riscontra la medesima struttura-tipo : TMD-NBD-TMD-NBD, un singolo polipeptide multi-dominio che rappresenta l’unità trasportatrice4,5
3.SITI DI LEGAME DELLA P-gp
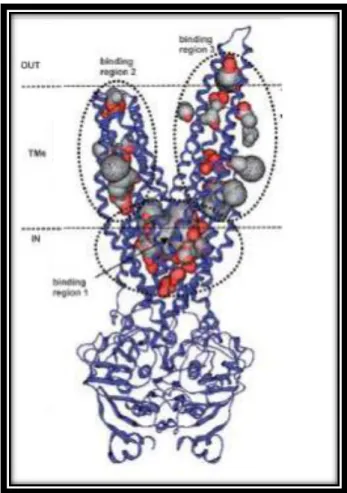
Data l’enorme varietà di substrati che interagiscono con la P-gp, è logico supporre dell’esistenza di diversi siti di legame, ognuno dei quali consente il riconoscimento ed il legame con diverse tipologie strutturali e/o funzionali di molecole. Sono stati identificati, finora, 4 siti diversi che presentano alcune proprietà caratterizzanti :
I siti da I a III sono adibiti al trasporto dei substrati, come vinblastina o paclitaxel, e all’interazione con modulatori, quali XR9576 e XR90516.Il sito IV, invece, ha solo attività di regolazione. Su di esso, la nicardipina, agisce come un modulatore.
I quattro siti di legame, sono in grado di comunicare allostericamente fra loro. Quando un sito forma un legame con il proprio ligando, si innesca un riarrangiamento strutturale a livello degli altri tre che determina una riduzione della loro affinità per i rispettivi ligandi6,7.
11 Figura 6.Siti di legame della P-gp con i substrati
4.MECCANISMO DEL TRASPORTO
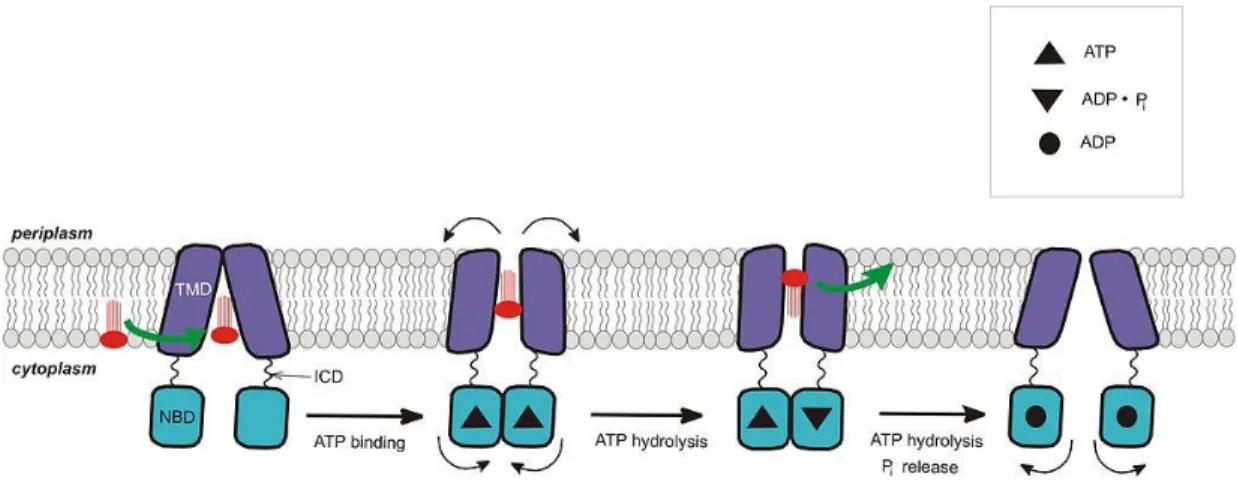
Il meccanismo grazie al quale la P-gp è in grado di estrudere gli xenobiotici dalla cellula è caratterizzata dall’alternanza di 2 stadi associati a due conformazioni diverse fra loro :
1) Conformazione con un orientamento prevalente verso il lume della cellula, nella quale il sito di legame per i substrati è libero ed accessibile (inward-facing conformation)
2) Conformazione con un orientamento prevalente verso l’ambiente extracellulare,con esposizione di una “tasca di estrusione” (outward-facing conformation)
12 Figura 7.Meccanismo d'azione generale della P-gp
La discriminante che determina la presenza dell’una o dell’altra conformazione è il legame con l’ATP. Quando la proteina lega l’ATP, si formano delle strette interazioni fra gli NBDs che a loro volta determinano un cambiamento conformazionale a livello dei TMDs che consente il passaggio da“inward” ad
“outward”. Particolarmente coinvolto è il sesto paio di eliche di entrambi i TMDs. In
questo stato,i substrati legati possono essere estrusi,in base alla loro idrofobicità,o nell’ambiente acquoso extracellulare o nel foglietto esterno del doppio strato lipidico. Ovviamente,quando si verifica l’idrolisi dell’ATP ad ADP, si ha un ritorno alla conformazione “inward”, che consente alla proteina di legare un altro substrato. Teoricamente, il numero massimo di substrati che possono essere trasportati contemporaneamente è 2. Ma ciò che limita effettivamente il loro numero, è il Peso Molecolare (PM). Due molecole con ridotto (PM) possono essere riconosciute come una e, quindi, essere trasportate contemporaneamente. Viceversa, una molecola con PM alto, può essere riconosciuta come due molecole e, di conseguenza, trasportata singolarmente3. Finora sono stati ipotizzati 3 meccanismi plausibili di traslocazione della P-gp :
A. Modello a poro : Le sostanze da espellere legano la P-gp sul versante citosolico della membrana e sono portate al di fuori della cellula attraverso un canale proteico
B. Modello a Flippasi : La P-gp,“flippando”,sposta le sostanze all’esterno della cellula attraverso la membrana contro gradiente di concentrazione.
13 C. Modello Vacuum cleaner : La P-gp riconosce le molecole nel doppio strato lipidico, le quali passano attraverso la membrana entrando da un sito di membrana ed uscendo da una cavità centrale6.
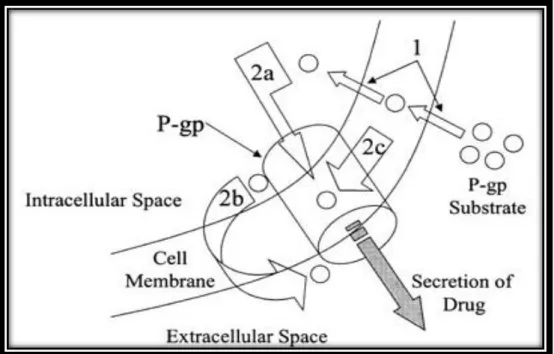
Figura 8. 1)diffusione passiva dello xenobiotico attraverso la membrana. 2a)formazione di un canale idrofobico (Poro) tra lo spazio intracellulare ed extracellulare. 2b)Flippasi. Lo xenobiotico è capovolto dal foglietto interno a quello esterno della membrana cellulare. 2c)"Vacuum cleaner model". Lo xenobiotico interagisce con la P-gp nel doppio strato lipidico ed è successivamente estrusa.
5.RUOLO DELLA P-GP NELLE DIVERSE MEMBRANE FISIOLOGICHE
La P-gp è estremamente diffusa in moltissimi distretti fisiologici.
14
5.A BARRIERA EMATO-ENCEFALICA
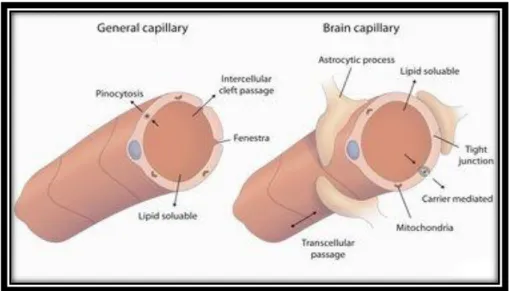
Da tempo è noto come, sia l’estrusione che l’ingresso delle sostanze a livello del cervello, sia finemente regolata dalla Barriera Emato-Encefalica. La BEE, è costituita fondamentalmente da cellule endoteliali che si trovano a livello dei micro-vasi che presentano un diametro variabile da 3 ad 8-μm. Esse sono caratterizzate dalla presenza di giunzioni strette fra loro. L’integrità fisica della BEE è assicurata da astrociti, da periciti e da matrice extracellulare1,8.
Figura 10.Differenze strutturali fra un capillare cerebrale ed uno comune
La BEE è in grado di impedire l’ingresso degli xenobiotici filtrandoli dal distretto cerebrale. Tale impedimento è dovuto alle proprietà chimico-fisiche degli xenobiotici. Alcuni studi hanno, infatti, dimostrato che tale impedimento è dovuto alle proprietà chimico-fisiche degli xenobiotici : solo le sostanze dotate di sufficienti caratteristiche lipofile sono in grado di attraversare la BEE per diffusione passiva9. La lipofilia, però, non l’unico fattore che influenza tale capacità. Infatti,è stato verificato come alcuni farmaci,particolarmente lipofili, non fossero in grado di attraversare efficacemente la BEE. Si è considerato, come altro fattore discriminante, la capacità di formare legami ad idrogeno. È stata, poi, verificata una correlazione negativa fra la capacità di penetrazione della BEE da parte delle sostanze lipofile ed il numero totale di legami ad idrogeno. Tutte le sostanze che interagiscono con molti legami ad idrogeno, sono caratterizzate da una bassa permeabilità di membrana10.
15 Ultimo fattore, ma non meno importante, è rappresentato dalle dimensioni della molecola che, aumentando, ne limitano il passaggio attraverso la BEE11.
Ricapitolando, tre sembrano essere i fattori che potrebbero influenzare la capacità di penetrazione della BEE da parte di una sostanza:
Lipofilia
Legami ad idrogeno Dimensioni.
Comunque, la scarsa concentrazione cerebrale di diverse sostanze, anche molto lipofile, non è esclusivamente dovuta a questi tre fattori limitanti. In questo senso, vi è un importante contributo della P-gp che riconoscendo lo xenobiotico, ne catalizza l’immediata espulsione da tale distretto. Ne consegue che l’accumulo cerebrale degli xenobiotici, quindi anche dei farmaci, sia estremamente difficoltosa e ridotta. In questo contesto, è stato rilevato il coinvolgimento della P-gp in numerosi processi biologici e patologici. Essa sembra avere azione immuno-modulatoria, poiché regola la secrezione di citochine da parte dei linfociti T e la migrazione delle cellule dendritiche verso i nodi linfatici, dai quali dipende l’immunità cellulo-mediata13
. Poi, analisi di laboratorio, sembrano indicare il suo coinvolgimento nell’inibizione dell’apoptosi14
. Ciò è stato suggerito a causa della sua azione nella regolazione della secrezione delle citochine, dei lipidi di segnalazione e del pH intracellulare che sembrerebbero essere tutti meccanismi attraverso i quali, la P-gp, influenzi la resistenza apoptotica cellulare15.
Altro aspetto da considerare è la sua up-regulation durante fenomeni patologici neurodegenerativi. Questo potrebbe rappresentare un meccanismo protettivo, riscontrabile sulle cellule sane, contro la citotossicità determinata dal rilascio di sostanze endogene. Un esempio, riguarda il glutammato, un neurotrasmettitore eccitatore ma anche eccito-tossico, il quale è un substrato della P-gp. Analisi su campioni di cervello affetti da fenomeni epilettici hanno dimostrato che alla iperespressione della P-gp, si accompagni una mancata espressione di diversi marcatori e proteine pro-apoptotiche, come la p53 e la p21. Ciò, è la prova del ruolo che la P-gp gioca nella protezione nei confronti della morte cellulare per apoptosi12. Un altro importante ruolo della P-gp è stato riscontrato nell’insorgenza della patologia di AD, in quanto sembra essere coinvolta nella sua patogenesi16.
16
5.B-PLACENTA
La placenta è un organo che consente la comunicazione fra il sistema circolatorio della madre e quello del feto,mantenendoli,nel contempo,separati. È importante sia per il corretto mantenimento della gravidanza che per lo sviluppo del feto. Le sue funzioni più importanti riguardano la regolazione dello scambio di sostanze,non solo nutrienti,fra la madre ed il feto e la rimozione,da quest’ultimo,dei prodotti di scarto. È,inoltre,considerata il primo organo fetale ad essere esposto a sostanze esogene come gli xenobiotici17. È risaputo,poi,che qualsiasi sostanza chimica somministrata alla madre,è capace di permeare,in una certa maniera,attraverso essa e raggiungere il feto stesso,attraverso i classici meccanismi riscontrati a livello delle membrane biologiche. Si sfruttano i meccanismi di diffusione passiva e mediato da trasportatori, nel caso in cui la sostanza esogena sia strutturalmente simile ai tipici substrati dei trasportatori per i nutrienti.
Negli ultimi tempi,è stato sempre più chiaro il coinvolgimento delle proteine ABC ed anche della P-gp,nella regolazione dell’assorbimento,della distribuzione e del metabolismo degli xenobiotici18.
5.C-FEGATO ED INTESTINO
Dal punto di vista farmacologico sia fegato che intestino rappresentano due organi importanti, poiché influenzano pesantemente il metabolismo degli xenobiotici e la loro farmacocinetica. In questi due organi sia l’espressione che l’attività del citocromo-P3A4 e della P-gp sembrano essere correlate2. Entrambi questi elementi facilitano l’eliminazione degli xenobiotici in diversa maniera. Il citocromo, essendo responsabile del metabolismo degli xenobiotici, ne aumenta il carattere idrofilo e la solubilità nei fluidi extracorporei come le urine. La P-gp, estrudendo attivamente gli xenobiotici, ne riduce le concentrazioni plasmatiche6.
17 6.MULTIDRUG RESISTANCE (MDR)
L insorgenza di fenomeni di resistenza in un organismo nei confronti di sostanze chimiche e/o farmaci di varia natura strutturale e funzionale è definito multidrug resistence (MDR). L’esposizione ad un singolo farmaco, può determinare fenomeni di resistenza multipla nei confronti di farmaci aventi diverse struttura chimica e diversi meccanismi d’azione. Gli organismi che manifestano MDR possono essere cellule alterate in seguito ad una determinata patologia, ma anche cellule neoplastiche.
6.1-MECCANISMI DI RESISTENZA
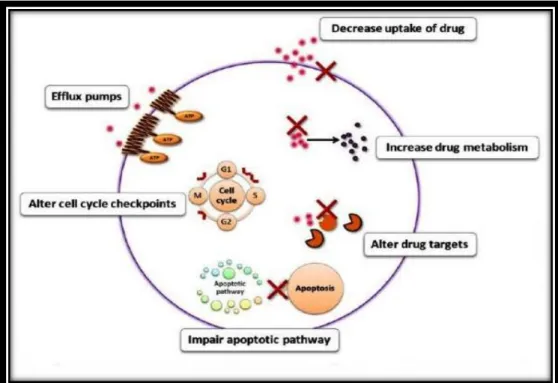
Questa capacità che le rende contemporaneamente resistenti all’azione di più farmaci, rappresenta l’ostacolo principale per avere un’efficace chemioterapia. Alla base di questi fenomeni vi è una vasta gamma di meccanismi19.
I. Aumento della capacità di rimozione del farmaco (caso tipico riguardante la P-gp)
II. Disattivazione enzimatica III. Riduzione della permeabilità
IV. Alterazione dei siti di legame del farmaco
V. Vie metaboliche alternative in grado di sopperire alle disfunzioni determinate dai farmaci anti-tumorali
18 Figura 11.Possibili meccanismi di resistenza
7.RUOLO DELLA P-gp NELLE CELLULE NEOPLASTICHE
Il cancro è causato da una disfunzione caratterizzata da una perdita di controllo della replicazione cellulare. Non c’è più equilibrio fra i fenomeni di proliferazione e quelli apoptotici,ovvero,di morte cellulare programmata. Le vie metaboliche che sostengono la crescita tumorale, sono estremamente complesse e regolate da una miriade di oncogeni e geni soppressori tumorali. Lo stesso quadro lo si ritrova per quanto riguarda l’apoptosi. È evidente che,il trattamento del cancro,sia estremamente difficile a causa della grande varietà di mutazioni che possono innescarlo e,contemporaneamente,dei numerosi meccanismi che le cellule neoplastiche possono utilizzare per resistere all’azione dei farmaci.
Questi meccanismi sono la diretta conseguenza di alcuni fenomeni quali : Alterazione delle vie di segnalazione apoptotiche
Amplificazione genica
Attivazione della riparazione del DNA
19 8.LA P-gp NEL PARKINSON
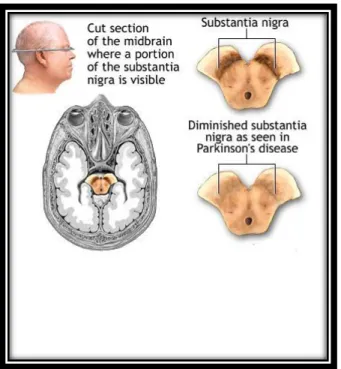
La sindrome di Parkinson (PD) è una patologia neurodegenerativa, causata dalla morte delle cellule dopaminergiche della sostanza nigra nel mesencefalo. Le cause alla base della morte cellulare ancora non sono chiare. I sintomi caratteristici della malattia sono tremori,rigidità,lentezza dei movimenti e, negli stadi più avanzati, demenza e discinesia. Quest’ultimo sintomo, è caratterizzato da contrazioni involontarie della muscolatura volontaria che impediscono, sempre più, al soggetto di controllare i propri movimenti6.
Figura 12.Differenze relative alla sostanza "nigra" in un cervello sano (sopra) e in uno malato (sotto)
La patologia, è caratterizzata da una riduzione della formazione e dell’attività della
dopamina. Contemporaneamente, si verifica la formazione di aggregati di α-Sinucleina a livello dei neuroni definiti Corpi di Lewy. I Corpi di Lewy sono segni
caratteristici della malattia e la loro distribuzione nel cervello affetto da PD varia da individuo ad individuo. Alcuni studi hanno messo in relazione la comparsa di patologie neurodegenerative con alterazioni riguardanti l’espressione e/o l’attività della P-gp a livello cerebrale6. Ad esempio, un decremento funzionale della glicoproteina, dovuto anche a fenomeni di polimorfismo sul C3435T del gene MDR1, potrebbero aumentare notevolmente la percentuale di incidenza del PD, in
20 quanto verrebbe meno l’azione di filtro della P-gp. In questo modo si verificherebbe un eccessivo accumulo di sostanze tossiche o degli stessi xenobiotici a livello cerebrale con conseguente comparsa della patologia6.
9.LA P-gp NELL’ALZHEIMER
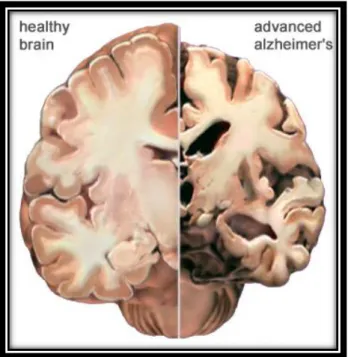
La sindrome di Alzheimer (AD) rappresenta una delle forme più comuni di demenza, la quale è caratterizzata da un costante ed irreversibile peggioramento, soprattutto delle funzioni cognitive, che, in alcuni casi, può condurre alla morte del soggetto affetto. L’AD si evolve e sviluppa con modalità diverse da individuo ad individuo, ma ci sono alcuni sintomi comuni riscontrabili nei diversi casi. I sintomi iniziali possono essere erroneamente attribuiti all’invecchiamento o a fenomeni di stress, in quanto vi è difficoltà nel ricordare eventi occorsi di recente. Col progredire della malattia, il soggetto manifesta fenomeni di confusione,problemi nel linguaggio, aggressività, irritabilità ed,in ultimo, perdita della memoria a lungo termine. Il tutto è anche accompagnato da una graduale perdita delle funzioni corporali6.
Figura 13.Sezione di un cervello sano (destra) e sezione di un cervello affetto da AD (sinistra)
Le cause della sindrome sono, per lo più, sconosciute. Sono stati proposti diversi meccanismi, ma quello che sembra più realistico riguarda la formazione di placche β-amiloidi (accumuli di peptidi Aβ) e taupatie6
21 proteina tau che tende ad accumularsi interferendo con le normali funzioni cognitive). Ciò fa sì che vi sia una alterazione delle normali funzioni cerebrali, soprattutto di quelle cognitive, dovute ad una perdita delle connessioni neuronali. In tutti i casi, si verifica una demielinizzazione delle fibre neuronali. La proteina tau sembra innescare la cascata di eventi che determina, attraverso una sua iperfosforilazione, la formazione di grovigli neurofibrillari che, successivamente, provocano il collasso dell’architettura strutturale del neurone e sua conseguente morte6.
Figura 14.Placche β-amiloidi caratterizzanti il PD
La P-gp sembra essere direttamente coinvolta nella genesi delle placche β-amiloidi. Per la precisione, vi è una correlazione inversamente proporzionale fra l’attività e l’espressione della P-gp e la loro formazione6
. La glicoproteina, infatti, esercita un ruolo protettivo poiché facilita l’eliminazione delle Aβ dai neuroni. Ovviamente, una riduzione della sua espressione o della sua funzione aumentano esponenzialmente la possibilità che tali placche possano formarsi. A sostegno di questa tesi, dati sperimentali indicano che, negli stadi iniziali dell’AD, vi è un up-regulation della P-gp mentre, negli stadi avanzati, la sua espressione è notevolmente ridotta6.
22 10.LA P-gp NELL’EPILESSIA
L’epilessia è caratterizzata da un insieme di cronici e comuni disordini neurologici che, nella maggior parte dei casi,provocano convulsioni. Le convulsioni epilettiche, sono dovute ad una anormale ed eccessiva attività neuronale dei neuroni che fanno parte della sostanza grigia dell’Encefalo. L’aggregato di neuroni dai quali parte la scarica epilettica è definito focolaio epilettogeno. L’ intensità, la frequenza e la gravità delle convulsioni è diversa a seconda del tipo di epilessia e nei casi peggiori vi è una vera e propria crisi.
Figura 15.Stimolazione di un neurone durante un fenomeno epilettico
L’importanza del ruolo della P-gp in questa patologia non riguarda la sua eziologia, come avviene invece nel PD e nell’AD, ma il suo trattamento farmacologico. Infatti, nel 20-30% dei casi, i pazienti affetti da epilessia cronica, manifestano fenomeni di resistenza nei confronti dei farmaci antiepilettici. Sono stati suggeriti diversi meccanismi, fra i quali, uno, riguarderebbe un’iperespressione della glicoproteina a livello della BEE con conseguente drastica riduzione della biodisponibilità cerebrale dei farmaci1,6.
23 Figura 16. Azione della P-gp sul neurone post-sinaptico
Molti farmaci usati come antiepilettici sono substrati del trasportatore. A sostegno di questa tesi vi sono 3 riscontri. Primo, l’iperespressione della P-gp è circoscritta esclusivamente al focus epilettogeno1. Secondo, l’iperespressione si verifica solo in quei soggetti che hanno manifestato farmaco resistenza che può essere dovuta a fenomeni di polimorfismo sul gene ABC B1. Terzo, alcuni farmaci antiepilettici sono substrati della P-gp6.
Di seguito sono riportati alcuni esempi di farmaci anti epilettici che sono substrati della P-gp :
24
FARMACI CHE AGISCONO SULLA P-gp
Finora è stato sottolineato il ruolo della P-gp nel cancro, nelle malattie neurodegenerative e nella genesi dei fenomeni di MDR. È, quindi, logico che la ricerca si sia concentrata su di essa e sullo sviluppo di farmaci in grado di modularne l’attività e/o l’espressione. Le varie indagini hanno evidenziato non solo le caratteristiche chimico-strutturali che la sostanza deve possedere per poter interagire con la P-gp, ma anche i siti di legame della proteina e le caratteristiche dei suoi substrati.
1.POSSIBILI SITI DI LEGAME DEI MODULATORI/INIBITORI
Vi sono ,essenzialmente, 3 possibili siti di legame per un modulatore/inibitore a livello della P-gp:
1) Siti di legame col substrato 2) Il sito NBD
3) I siti allosterici
1) I siti di legame col substrato rappresentano il target classico per gli inibitori che determinano la loro azione bloccando efficientemente il processo di traslocazione del substrato da parte della P-gp. Si deve tener presente, inoltre, il coinvolgimento dei residui responsabili del movimento dei segmenti TM (trasmembrana) che sono il residuo 6 ed il 12. Anche quest’ultimi rappresentano target efficaci per l’azione degli inibitori. Attraverso la modulazione dovuta all’interazione con questi siti, si riesce ad intervenire pesantemente sull’attività della P-gp, impedendo che il suo substrato sia eliminato dalla cellula. Questo rappresenterebbe un grande beneficio per la chemioterapia. Ovviamente, l’inibitore deve presentare un’alta affinità nei confronti di questi siti e, contemporaneamente, una bassa probabilità di dissociazione in presenza del farmaco che competerà con l’inibitore per questi stessi siti. Altra caratteristica deve essere la mancanza di interazioni dell’inibitore col farmaco stesso. Il problema principale per gli inibitori è, senza dubbio, dovuto alla plasticità dei siti di legame della P-gp che possono interagire simultaneamente con diversi tipi di substrato. Ciò, rende estremamente difficile lo sviluppo di un composto strutturalmente in grado di interagire efficacemente con tali siti20.
25 Figura 19.Siti di legame della P-gp con i suoi substrati (indicati con *)
2) Gli NBD sono domini altamente conservati in tutte le proteine ABC. Sono cruciali per il corretto funzionamento della proteina e, di conseguenza, potrebbero rappresentare un ulteriore target per l’azione dei modulatori/inibitori della stessa. Agendo su di essi, si determina un’interruzione del ciclo idrolitico dell’ATP. Gli NBD potrebbero essere suscettibili all’azione di ligandi progettati de novo, specifici nei loro confronti. Inoltre, la presenza dei 2 NBD per ogni trasportatore, aumenta il numero dei potenziali siti di interazione e consente, attraverso l’interazione con uno solo di essi, di bloccare il processo di eliminazione dei farmaci che la P-gp lega. L’ostacolo più grande da superare in questo caso, riguarda la produzione di un composto che sia selettivo esclusivamente per la P-gp e non per gli altri trasportatori ABC, in modo da non correre il rischio che si manifestino effetti collaterali o fenomeni di tossicità20.
26 Figura 20. A)Attività degli NBD B)posizione degli NBD nella P-gp
3) Questi residui determinerebbero, in seguito al legame di uno dei 4 siti della P-gp con un substrato, un riarrangiamento conformazionale a livello degli altri 3. Due esempi, sono il residuo L339 ed F983. Sono importantissimi per lo sviluppo di farmaco-resistenze mediate dalla glicoproteina. Infatti, controllano e comunicano l’eventuale occupazione dei siti di legame per i substrati alla cavità TMD. Se si impedisce che tali residui subiscano modificazioni covalenti, si verifica un’inattivazione della proteina attraverso il blocco dell’attività ATPasica innescata dal substrato20.
27 Figura 21. Posizione dei residui di regolazione allosterica a livello della cavità TMD. le bande in rosso
rappresentano lo spessore della membrana cellulare
28 2.SUBSTRATI DELLA P-gp
È stato verificato che i substrati della P-gp, inizialmente, riescono a penetrare liberamente nella cellula e solo dopo sono riconosciuti ed eliminati. È noto che i siti di legame per i substrati si trovano all’interno della membrana plasmatica. Quindi, una sostanza per poter interagire con la P-gp deve presentare delle caratteristiche chimico-fisiche tali che le permettano di diffondere attraverso il doppio strato fosfolipidico, riconoscere i siti di legame della glicoproteina e interagire con essi. Alcuni studi SAR2, mettendo in relazione la struttura di un composto con la sua attività, hanno dimostrato che la P-gp interagisce in maniera diversa differenti tipologie di substrati. Questi studi sono stati effettuati anche con inibitori e substrati noti della P-gp (Clorochina, Vinblastina, Reserpina, Verapamile) ed hanno evidenziato alcune caratteristiche chimico-strutturali che un substrato tipico della glicoproteina deve necessariamente possedere : estrema liposolubilità a pH fisiologico con un logP>2,92, peso molecolare basso (compreso tra 250 Da e 900 Da), struttura planare aromatica ed un ammino-gruppo terziario, gruppi elettron-donatori o accettori di legami ad idrogeno2,6.
29 Tabella 1: Esempi di substrati della P-gp
ANTITUMORALI Ca2+-BLOCCANTI MORFINE Actinomicina-D Daunorubicina Etapaside Mitomicina-C Paclitaxel Tamoxifen Topotectan Vinblastina Vincristina Diltiazem Nicardipina Verapamil Morfina-6-glucuronide Morfina Loperamide
ANTIALLERGICI CARDIACI PEPTIDI Terfenadina Propafenone Amiodarone Quinidina Digossina Gramicina-D Valinomicina
ANTIBIOTICI ATTIVI SUL SNC STEROIDI Cefazolin Cefoperazone Domperidone Flufenazina Ondansetron Perfenazina Fenoxazina Fenitoina Aldosterone Dexametasone Idrocortisone
30
2.1-INIBITORI DI PRIMA GENERAZIONE
Gli inibitori di prima generazione annoverano composti come il verapamile, un Ca2+ bloccante, in grado di determinare la regressione dei fenomeni di MDR2. Tale farmaco sensibilizzava le cellule leucemiche all’azione della vincristina e vinblastina, annullando, così, i fenomeni di MDR manifestati. L’uso del verapamile fu giustificato grazie alla sua capacità di modulazione non specifica di diverse vie secretive; di alterazione delle proteine di trasporto Ca2+ dipendenti e grazie alla capacità di intercalarsi nel doppio strato fosfolipidico. Successivamente, fu dimostrato che la ristabilita citotossicità degli alcaloidi della vinca, era effettivamente dovuta ad un’inibizione della P-gp da parte di questo farmaco. I medesimi effetti, furono riprodotti utilizzando composti funzionalmente legati ad esso. Si utilizzarono diversi farmaci ad attività vasodilatatoria in grado di modulare l’attività dei canali al Ca2+: diltiazem, nicardipina, ecc2,6. Fu stabilito anche che un composto, per essere considerato un inibitore, deve produrre 1 o più dei seguenti effetti :
Aumentare la potenza del farmaco contro cellule resistenti Aumentare la sua concentrazione all’interno di tali cellule
Dimostrare interferenza con la P-gp durante le analisi di “photoaffinity labeling” (tecnica utilizzata per evidenziare i siti di legame delle proteine)
Attraverso tali tecniche,furono scoperte numerose sostanze, anche strutturalmente molto diverse fra loro, in grado di modulare l’attività della P-gp. Alcuni esempi di inibitori : antagonisti della calmodulina, ciclosporina A, analoghi della chinina, tamossifene (antiestrogeno). Alcuni esempi di modulatori furono gli ormoni steroidei, che rappresentarono la prima generazione di modulatori poiché erano in grado di invertire i fenomeni di MDR6.
Ma la co-somministrazione di questi inibitori con, ad esempio, farmaci antitumorali non determinò alcun miglioramento nel trattamento delle patologie. Ciò a causa di :
a. Mancato raggiungimento di una concentrazione plasmatica sufficiente per inibire la P-gp
31 Figura 23. Esempi di inibitori di prima generazione : verapamile
32
2.2-INIBITORI DI SECONDA GENERAZIONE
Successivamente, gli inibitori di prima generazione furono utilizzati come base di partenza per lo sviluppo di nuove molecole attraverso gli studi QSAR, grazie ai quali si evidenziarono le relazioni quantitative strattura-attività. Tali studi furono condotti marcando alcuni substrati noti della P-gp con isotopi radioattivi (ad esempio, il trizio [3H] ). Rilevando la radioattività fu possibile una migliore caratterizzazione della struttura della glicoproteina e delle proprietà chimico-fisiche necessarie ad un composto per sviluppare un’interazione migliore con essa2
. Quindi, i composti sintetizzati, pur essendo analoghi degli inibitori di prima generazione, presentavano alcune caratteristiche chimico-fisiche particolari in grado di migliorare la loro azione inibitoria e la selettività: liposolubilità a pH fisiologico con un logP>2,92, peso molecolare basso (compreso tra 250 Da e 900 Da), struttura planare aromatica ed un ammino-gruppo terziario, gruppi elettron-donatori o accettori di legami ad idrogeno. Erano derivati che presentavano anche minore cardiotossicità. Alcuni esempi sono il dexverapamil, il valspodar ed il biricodar. Però anche questa classe di composti non determinò gli effetti sperati a causa di numerose interazioni farmacocinetiche causate da un’eccessiva inibizione del CYP4506. Questo determinava un eccessivo accumulo del farmaco somministrato in associazione con questi inibitori, ad esempio un antitumorale, provocando la genesi di effetti collaterali. Un esempio relativo a questa problematica è l’associazione biricodar/doxorubicina la quale determinava un aumento esponenziale dell’incidenza di neutropenie2,6
33 Figura 25.Biricodar
34
2.3-INIBITORI DI TERZA GENERAZIONE
Ancora una volta il Verapamile rappresentò una buona base di partenza per la messa a punto di nuovi modulatori/inibitori della P-gp perché consentì di evidenziare l’importanza della presenza di gruppo accettore di legami ad idrogeno (es. un alcossi-sostituente) e di uno “spacer” alchilico. Inoltre, la sostituzione nel verapamile di gruppo dimetossifenil-4-ciano-4-isopropilbutilico con una porzione azapentaciclica ha rivelato il ruolo fondamentale giocato dagli orbitali π dei residui aromatici nella formazione dei legami con la P-gp. L’introduzione di anelli aromatici ha condotto,di conseguenza, alla sintesi di inibitori più potenti e con minori effetti cardiotossici. Si rilevò anche che l’inserzione di un atomo di azoto in grado di essere protonato nel “linker” alchilico, aumentava la selettività nei confronti dei siti di legame propri della P-gp. Allo stesso modo, una parziale riduzione della flessibilità della molecola determinava un aumento dell’assorbimento e della selettività.
Grazie alla Trifluoroperazina fu chiaro che la presenza di un’ammina terziaria legata ad un nucleo triciclico attraverso una catena metilenica, determinava un aumento della capacità di inibizione della P-gp.
L’Arcidonecacarbossiammide manifestando attività inibitoria sia sulla P-gp che sul BCRP, consentì di valutare quali fossero le caratteristiche strutturali in grado di determinare questo simultaneo riconoscimento. Un altro aspetto importante era legato alla riduzione dei fenomeni di MDR qualora nella molecola si riscontrava la presenza di un nucleo piperazinico o piperidinico legato ad un N-azaxantone. Infatti, il nucleo basico, protonato a pH fisiologico, facilitava l’accumulo del farmaco. Anche alcuni analoghi dell’Antranilammide che presentavano un gruppo tetraidroisochinolinetilfenilamminico agivano sia sulla P-gp che sul BCRP. Tale attività si rilevava anche su derivati maggiormente idrosolubili presentanti metossilazioni sulla porzione antranilammidica.
Si valutò, inoltre, l’inserzione di anelli pirrolidinici, cicloesilici e arilpiperazinici a livello dello “spacer” alchilico. I risultati più incoraggianti sono stati ottenuti in seguito all’inserzione di una porzione arilpiperazinica e di sostituenti metossilici sul nucleo antranilammidico. Alcuni esempi di farmaci sintetizzati seguendo queste linee giuda sono tariquidar ed elacridar. La caratteristica innovativa di questa classe di farmaci rispetto alle precedenti è la possibilità di determinare un’efficace
35 modulazione della P-gp a concentrazioni nano-molari. Presentano anche una maggiore durata d’azione e una selettività per la P-gp.
Figura 27. Tariquidar
36
2.4-DIMERI DERIVANTI DA FLAVONOIDI
I flavonoidi sono composti polienolici abbondanti in frutta, fiori, vegetali. Sono associati a numerose proprietà benefiche per la salute come quella antiossidante, antivirale o antiinfiammatoria. Studi recenti, hanno proposto che possano anche svolgere un’attività di modulazione sulla P-gp. Infatti, alcuni flavonoidi sono buoni inibitori di proteine ATP-dipendenti. Essi legano direttamente il dominio citosolico NBD2 della P-gp. Però per avere un’inibizione rilevante della P-gp e riduzione dei fenomeni di MDR, è necessaria la somministrazione di dosi che possono determinare effetti secondari21. Qualora si dovesse aumentare il loro carattere lipofilo, si verifica un potenziamento della inibizione poiché si rende possibile un’interazione sia con il sito di legame per gli xenobiotici sia col sito di legame per gli steroidi. È proprio puntando su questa bivalenza funzionale che si potrebbero evitare gli eventuali effetti secondari. Il meccanismo d’azione di questi composti si basa sulla competizione con l’ATP per i siti ATPasici sugli NBD, impedendo il legame dell’ATP si blocca anche l’attività della pompa. Questo potrebbe costituire un ulteriore passo avanti nello sviluppo di modulatori/inibitori della P-gp in grado di non presentare marcati effetti collaterali, determinando un più ampio margine d’azione terapeutico nel trattamento delle patologie21.
37
2.5-TIOXANTONI
I tioxantoni sono S-eterocicli con uno “scaffold” di dibenzo-γ-tiopirone.
Figura 30.Struttura dibenzo-γ-tiopironica.
Questi composti hanno mostrato diverse proprietà antitumorali. Per la precisione, sono in grado di espletare due attività molto interessanti22 :
i. Inibizione della P-gp
ii. Inibizione della proliferazione cellulare
Però i dati sperimentali sulla capacità di inibizione della P-gp sono discordanti. Alcuni composti mostrano un’inibizione competitiva della proteina. Presentano strutture simili al verapamile e alla chinidina e aumentano i fenomeni idrolitici dell’ATP. Altri determinano un’inibizione non competitiva, riducono l’utilizzo di ATP da parte della glicoproteina legando o i suoi siti di legame o bloccando i siti allosterici necessari al suo funzionamento22.
Per quanto riguarda l’inibizione della proliferazione cellulare, test effettuati su diverse linee cellulari in presenza di doxorubicina hanno evidenziato la possibilità che tale effetto, in alcuni casi, non sia dovuto all’inibizione della P-gp ed a fenomeni di sensibilizzazione alla doxorubicina, ma sia riconducibile esclusivamente all’azione del tioxantone. I meccanismi alla base di questa dualità di effetti non sono del tutto chiari, comunque, questi composti potrebbero rappresentare in futuro una buona via per ottenere farmaci in grado di essere impiegati oltre che nella chemioterapia, anche nel controllo dei fenomeni di MDR22.
38 Figura 31. Hycantone