Capitolo 2
Materiali sintetici per applicazione nella
deposizione assistita tramite microsiringa (PAM)
2.1 Introduzione
I tessuti e gli organi corporei più complessi, come il fegato, il cuore, e i tessuti neuronali, sono difficili da ingegnerizzare, in quanto presentano una specifica distribuzione cellulare tri-dimensionale, che è legata alle loro funzioni all’interno dell’organismo. Il primo passo verso l’ingegnerizzazione di questi tessuti e organi corporei è una tecnica che consenta di produrre scaffolds con una definita e riproducibile topografia. In letteratura, sono riportate molte tecnologie per l’ottenimento di strutture tridimensionali, e dotate di un’architettura controllabile, ed esse prendono il nome di tecniche di prototipazione rapida [Yeong et al., 2004]. Presso il Centro Piaggio, è stato recentemente sviluppato un sistema di prototipazione rapida, basato sulla deposizione di biopolimeri in strutture ad alta risoluzione, attraverso una siringa attivata da pressione. La tecnica è detta Pressure Assisted
Microsyringe (PAM), e si presenta come un sistema versatile sia per le sue potenziali applicazioni
nell’ingegneria tessutale, sia come valido strumento per studiare la motilità, l’organizzazione cellulare e la risposta delle cellule a topografie diverse. Nel presente capitolo la tecnica è stata applicata dapprima al policaprolattone (PCL), che è un polimero la cui biocompatibilità è ampiamente documentata in letteratura [Serrano et al., 2004; Williamson et al., 2006]. Poi l’attenzione è stata indirizzata ad alcuni polimeri biodegradabili di sintesi: un copolimero a tre blocchi PCL-POE-PCL, già ampiamente ma non completamente caratterizzato [Sbarbati Del Guerra et al., 1995; Cerrai et al., 1994], e una serie di nuovi poliuretani e poliuretani-uree [Rechichi, 2002-2003].
Lo sviluppo di polimeri biodegradabili per applicazioni biomediche, infatti, ha assorbito di recente gli interessi e gli sforzi di molti ricercatori, tuttavia, nella varietà di polimeri biodegradabili sviluppati, i polimeri elastomerici sono stati relativamente pochi [Engleberg et al., 1991]. Con il crescente interesse nel settore del Tissue Engineering, sono sempre più richiesti materiali biodegradabili con un’ampia gamma di proprietà fisiche, tali da integrarsi con i vari tessuti del corpo. In particolare, elastomeri degradabili potrebbero essere utilizzati specificatamente nel settore dell’ingegneria tessutale di tessuti
soft (ad es il sistema vascolare e la pelle artificiale) [Skarja et al., 2000]. Gli elastomeri poliuretanici
segmentati rappresentano un ottimo esempio di polimeri di notevole versatilità chimica e, grazie alla loro relativamente buona biocompatibilità, sono stati adoperati come biomateriali, soprattutto per applicazioni a lungo termine [Lamba et al., 1998]. Al contrario, lo sviluppo di poliuretani biodegradabili ha ricevuto un minore interesse, dato che i diisocianati comunemente utilizzati come precursori forniscono, per degradazione, prodotti tossici o cancerogeni [Yoda et al., 1998]. L’ottenimento di diisocianati basati sulla L-lisina o su altri amminoacidi ha permesso di superare tale ostacolo. Sono state condotte numerose ricerche sulla degradazione di poliuretani non degradabili [Santerre et al., 1993; Pinchuk et al., 1994]. Le ricerche hanno indicato che i gruppi esterei, uretanici, e ureici dei poliuretani sono soggetti ad un limitato grado di idrolisi (sia chimica sia enzimatica) in
mezzo biologico. Questa caratteristica è comune a molti polimeri sintetici i cui segmenti sono incapaci di adattarsi, tramite riarrangiamenti conformazionali, alla geometria del sito attivo dell’enzima. Polimeri volutamente biodegradabili invece sono stati sintetizzati, introducendo unità strutturali disponibili in natura (ad esempio amminoacidi e peptidi) [Lipatava et al., 1983]. Questa strategia presenta due aspetti interessanti. Innanzitutto, i prodotti di degradazione di questi polimeri dovrebbero essere non tossici e facilmente metabolizzati in vivo. Inoltre, potrebbero essere progettati polimeri biodegradabili, in grado di rispondere a cambiamenti nell’ambiente fisiologico, come la variazione di concentrazione enzimatica nel sito di applicazione. Questa premessa racchiude le motivazioni del particolare interesse che è stato rivolto in questo lavoro alla classe innovativa di polimeri biodegradabili costituita da poliuretani e poliuretani-uree. Questi polimeri offrono la possibilità di variare le loro proprietà chimico-fisiche e biologiche e di adattarle alle specifiche dell’applicazione, semplicemente modificando la composizione dei blocchi e/o la loro lunghezza.
L’obiettivo del Capitolo 2 è stato quello di valutare la citotossicità di tutti i polimeri di sintesi (PCL-PEG-PCL, poliuretani, poliuretani-uree) e, successivamente, di realizzare, con i materiali idonei per applicazioni biomediche, microstrutture bi- e tri-dimensionali, adoperando la tecnologia innovativa di deposizione assistita tramite microsiringa (PAM). I materiali di sintesi non adoperabili per la produzione di scaffolds sono stati comunque variamente caratterizzati: essi, infatti, essendo un prodotto di sintesi, sono migliorabili, alla luce dei risultati delle caratterizzazione e dell’uso a cui sono destinati. Precisamente, essi sono stati caratterizzati tramite calorimetria differenziale a scansione, per valutarne le caratteristiche termiche, tramite analisi al microscopio ottico, per analizzarne la morfologia cristallina e, tramite microscopio a forza atomica, per confrontare la topografia superficiale di campioni diversi ottenuti nelle stesse condizioni. I materiali applicabili nella produzione di scaffolds sono stati maggiormente caratterizzati, valutando il grado di bagnabilità superficiale, la densità di carica elettrica superficiale ed eseguendo prove di adesione cellulare sulle microstrutture ottenute e sui film per casting. Sul poliuretano risultato maggiormente biocompatibile, sono state eseguite ulteriori caratterizzazioni. Sono state infatti misurate le proprietà meccaniche di microstrutture tri-dimensionali con diversa geometria, per valutare la compatibilità strutturale degli scaffolds e avere indicazioni sulle possibili applicazioni biomediche del poliuretano analizzato. Con lo stesso poliuretano, infine, sono state realizzate strutture tri-dimensionali che sono state impiantate in vivo con risultati soddisfacenti.
2.2 Parte sperimentale
2.2.1 Materiali
I polimeri usati sono stati un poli(ε-caprolattone) commerciale (PCL) fornito dalla Polysciences, Inc. con peso molecolare medio ponderale (Mw) pari a 45,000 e alcuni polimeri di
sintesi: un copolimero a tre blocchi policaprolattone-polietilenossido-policaprolattone (PCL-POE-PCL) identificato con il codice C27 e alcuni poliuretani e poliuretani-uree, le cui caratteristiche vengono riportate di seguito. Il copolimero PCL-POE-PCL contiene l’85 % in peso di ε-caprolattone e il 15 % in peso di poli(etilene glicol) (Merck, Mn=35,000) ed è stato sintetizzato da Cerrai et al.
attraverso una procedura che non comporta l’uso di iniziatori potenzialmente tossici [Cerrai et al., 1994]. La percentuale molare dei due blocchi del copolimero è la seguente: 66 mol % di PCL e 34 mol % di PEG. Il rapporto in moli CL/PEG vale 1493 e il peso molecolare medio numerale (Mn) del
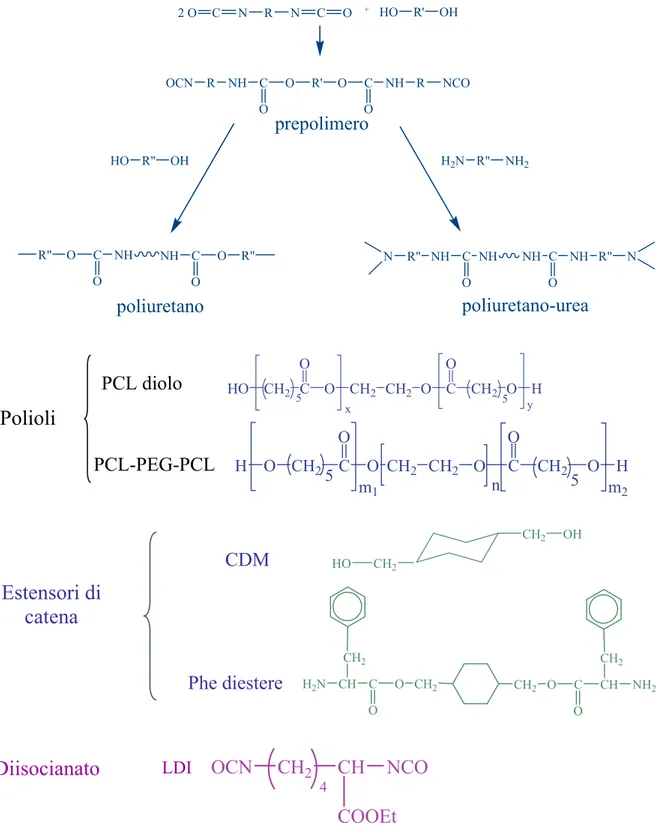
copolimero C27 vale 203,700. I risultati di citotossicità ed emocompatibilità di precedenti studi hanno dimostrato che la biocompatibilità di PCL-POE-PCL è buona [Sbarbati Del Guerra et al., 1995; Cerrai et al., 1994]. I poliuretani e poliuretani-uree utilizzati sono stati sintetizzati presso il Dipartimento di Ingegneria Chimica durante un precedente lavoro di tesi [Rechichi, 2002-2003]. La sintesi, riportata in Figura 2-1, prevede due stadi: nel primo si ha la formazione del prepolimero per reazione del macrodiolo o segmento soft con un diisocianato a base di lisina (LDI). Il segmento soft può essere un PCL commerciale (Mn = 1250, Aldrich) o un copolimero di sintesi a tre blocchi (PCL-PEG-PCL)
ottenuto per reazione tra un polietilenglicole commerciale con Mn pari a 600 (PEG) e il PCL
commerciale sopra descritto. Nel secondo stadio di sintesi dei poliuretani e poliuretani-uree, il prepolimero viene trasformato nel prodotto finale per reazione con l’estensore di catena: usando un diolo come estensore di catena (1,4-cicloesandimetanolo, CDM; Aldrich) si ottiene un poliestere (PU),
adoperando invece una diammina come estensore di catena (1,4-di(L-fenilalanil-ossimetilen)-cicloesano, Phe diestere) si ottiene un poliuretano-urea (PUU). La diammina adoperata, che è un derivato dell’amminoacido L-lisina, è un prodotto di sintesi e, per idrolisi, fornisce etanolo e L-lisina, entrambi biocompatibili. Inoltre, la presenza di un amminoacido in catena principale conferisce ai polimeri una maggiore suscettibilità all’attacco enzimatico [A. Rechichi, 2003].
Figura 2-1. Sintesi dei poliuretani e dei poliuretani-urea
Polioli
PCL diolo
PCL-PEG-PCL
HO CH2 C O O CH2 CH2 O C CH2 O O H x y 5 5 HO CH2 CH2 OHEstensori di
catena
CDM
Phe diestere
H2N CH CH2 C O O CH2 CH2 O C O CH CH2 NH2Diisocianato
LDI
OCN
CH
2CH
COOEt
NCO
4 C O O CH2 CH2 O C CH2 O H O H O CH25 5 m1 n m2 2 O C N R N C O + HO R' OH OCN R NH C O O R' O C O NH R NCO R" O C O NH NH C O O R" HO R" OH H2N R" NH2 N R" NH C O NH NH C O NH R" Nprepolimero
poliuretano
poliuretano-urea
Le caratteristiche chimico-fisiche dei segmenti soft e dei poliuretani e poliuretani-uree sono riassunte nelle Tabelle 2-1 e 2-2, rispettivamente. In particolare, sono riportati il peso molecolare medio ponderale (Mw), il peso molecolare medio numerale (Mn), la temperatura di transizione vetrosa (Tg),
quella di fusione (Tm) e l’entalpia di fusione (∆Hm). I dati raccolti nelle Tabelle 2-1 e 2-2 sono stati
ricavati da un precedente lavoro di tesi [Rechichi, 2002-2003].
Tabella 2-1 Caratteristiche del PCL e dei copolimeri PCL-PEG-PCL usati come segmenti soft e del PEG adoperato nella sintesi dei copolimeri PCL-PEG-PCL.
Denominazione del copolimero Mn Tm (°C)b ∆Hm (J/g)b
PEG 600 21 127
PCL-PEG-PCL 50/50a 2136 4; 37; 46 109
PCL-PEG-PCL 65/35a 3034 7; 43; 52 83
PCL 1250 39; 46 69
a La denominazione del copolimero è stata fatta sulla base della sua
composizione percentuale molare in caprolattone (CL) e in PEG, rispettivamente.
b La temperatura e l’entalpia di fusione sono state misurate durante una
prima scansione di riscaldamento, eseguita a 10°C/min tra –100°C e 100°C.
Tabella 2-2 Caratteristiche chimico-fisiche dei poliuretani e poliuretani-uree di sintesi Macrodiolo Estensore Polimeri PCL COP 50/50 COP 65/35 HDI CDM Phe dies. Mn Mw (°C) Tg Tm c (°C). ∆H(J/g)m PU4 √ √ 42335 87387 -26.4 42.1 32.4 PU4-Ha √ √ 200000 87387 -24.0 37.5 53.8 PUU4 √ √ 33212 87387 n.d. n.d. n.d. PU5 √ √ 16742 30805 -46.4 47.4 25.9 PUU5 √ √ 23897 48105 -40.7 49.0 26.3 PU6 √ √ 19402 34828 -47.0 52.0. 52.9 PUU6 √ √ 30228 61208 -46 50.0 46.3
a Poliuretano analogo al PU4, ma con un più alto peso molecolare. b La temperatura di fusione e di transizione vetrosa è stata misurata
durante una prima scansione di riscaldamento eseguita a 5°C/min tra –100°C e 100°C.
c E’ stato riportato solo il picco a più alta temperatura.
2.2.1.1. Preparazione di film per casting
Sono stati ottenuti film per casting di PCL, C27, poliuretani e poliuretani-uree da soluzioni in cloroformio con concentrazione pari al 4 % (w/v). Precisamente una quantità pari a 20 ml di soluzione è stata versata dentro piattini di vetro con diametro pari a 5 cm. Il solvente è stato fatto evaporare sotto cappa per 48 h e in stufa ventilata a 37 °C per una settimana.
2.2.1.2. Tecnica di deposizione assistita tramite microsiringa (PAM)
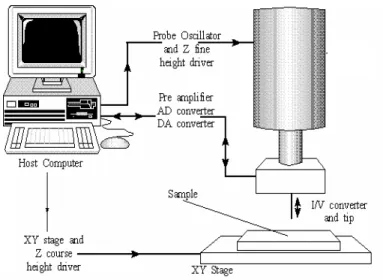
La tecnica di deposizione tramite microsiringa (PAM) è un sistema di prototipazione rapida per la fabbricazione di scaffolds 2-D e 3-D da polimeri biodegradabili [Vozzi et al., 2002-2003]. Il metodo è basato sulla deposizione di strati polimerici tramite una siringa in acciaio inossidabile con ago in vetro da 10-20 µm e con una capacità di circa 10 ml. L’apparecchiatura funziona nel modo descritto di seguito. Per prima cosa, la siringa viene caricata con un certo quantitativo di soluzione polimerica. La siringa è montata in posizione verticale ed è in grado di traslare lungo l’asse z. Al di sotto dell’ago della siringa è posizionato un vetrino (3×3 cm2) per la deposizione delle strutture polimeriche. Esso, a
sua volta, poggia su un basamento mobile nel piano x-y, grazie all’azione di due motori passo-passo. Il sistema di micro-posizionamento tridimensionale presenta una risoluzione di 0.1 µm. Il materiale in forma di soluzione viene estruso dall’ago della siringa grazie all’applicazione di una pressione tramite aria compressa filtrata a 10-300 mmHg. Tutto il sistema è controllato tramite personal computer attraverso una scheda IRIS (Eclypse, Pisa; Italy). Il software che guida il sistema è stato sviluppato in linguaggio C ed è dotato di un’interfaccia grafica che consente di definire la struttura geometrica di ogni strato, prima della deposizione. Generalmente, le piste depositate hanno dimensioni laterali comprese tra 5 e 600 µm, a seconda della pressione applicata, della viscosità della soluzione, della velocità del motore (0.5-5 mm/s) e delle dimensioni dell’ago della siringa [Vozzi et al., 2002].
La Figura 2-2 mostra lo schema di funzionamento dell’apparecchiatura di deposizione tramite microsiringa, mentre la Figura 2-3 mostra il sistema di microposizionamento a tre assi.
Figura 2-2. Schema del sistema di deposizione tramite microsiringa (PAM)
Figura 2-3. Visione del sistema di microposizionamento a tre assi della tecnologia PAM Regolatore di pressione
Piattaforma di posizionamento
Microsiringa Aria compressa
Le soluzioni per la deposizione tramite microsiringa sono state preparate in cloroformio (Aldrich), con le seguenti concentrazioni: 20 % (w/v) per il PCL ed il copolimero C27, e 15 % (w/v) per il PU4-H. Per trovare i parametri ottimali per i diversi materiali (velocità del motore, dimensione dell’ago della siringa e pressione applicata), sono state eseguite prove preliminari, deponendo piste ad una velocità costante del motore (2.5 mm/s per il PU4-H e il copolimero C27) e a pressioni variabili (5-70 mmHg per il PU4-H e 10-100 mmHg per il copolimero C27) o, viceversa, ad una pressione costante (30 mmHg per il PU4-H) e velocità variabili del motore (2.5-5 mm/s per il PU4-H). Nelle prove si è adoperato una ago da 20 µm.
In questo lavoro, sono state realizzate strutture 2-D in forma di griglie con maglia quadrata di lato 1 mm a base di PCL, C27 e PU4-H. Tali semplici strutture sono utili per studiare l’adesione cellulare. I parametri adoperati per la deposizione delle strutture a partire dai tre diversi materiali sono riassunti in Tabella 2-3.
Tabella 2-3. Parametri adoperati per la deposizione tramite PAM dei vari polimeri e la larghezza media delle piste deposte.
Materiale Concentrazione della soluzione (%, w/v) Pressione di deposizione (mmHg) Velocità del motore (mm/s) Dimensioni dell’ago (µm) Larghezza delle piste (µm) PCL 20 10 2.5 20 80 C27 20 50 2.5 20 150 PU4-H 15 30-40 4.5 20 50
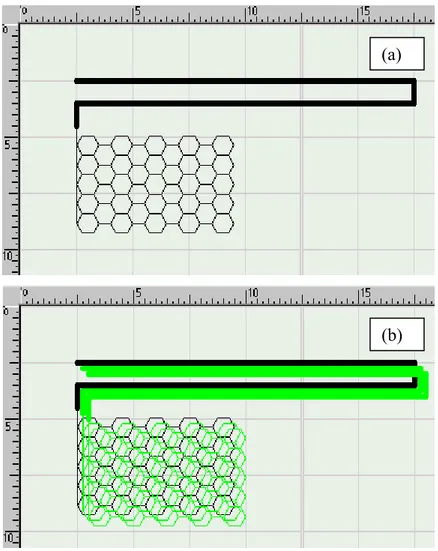
Per il poliuretano PU4-H, inoltre, sono state prodotti anche scaffolds 2-D con geometria della cella elementare esagonale e ottagonale, adoperando i parametri indicati in Tabella 2-3. L’esatta grafica delle geometrie esagonale e ottagonale adoperata per guidare la deposizione degli scaffolds bidimensionali è mostrata in Figura 2-4a e 2-5a.
Tramite la tecnica PAM, si ottengono anche strutture 3-D, che sono quelle necessarie per la rigenerazione dei tessuti. Per produrre strutture tri-dimensionali, si realizza dapprima uno strato bidimensionale. Successivamente, si depone una soluzione di un polimero di supporto -in un non-solvente per il polimero deposto- attorno alle piste e si lascia evaporare il non-solvente del polimero di supporto in modo da ottenere un film per casting. Poi si trasla la siringa verso l’alto di una quantità pari allo spessore dello strato da deporre e si procede con un’altra deposizione. La procedura viene ripetuta per un numero di volte necessario a completare la struttura 3-D dello scaffold. Alla fine, lo
scaffold viene immerso nel solvente del polimero di supporto (che è non solvente per le piste deposte)
per eliminare tale materiale. Il passo successivo consiste nell’essiccamento delle strutture in stufa da vuoto a temperatura ambiente per almeno una settimana.
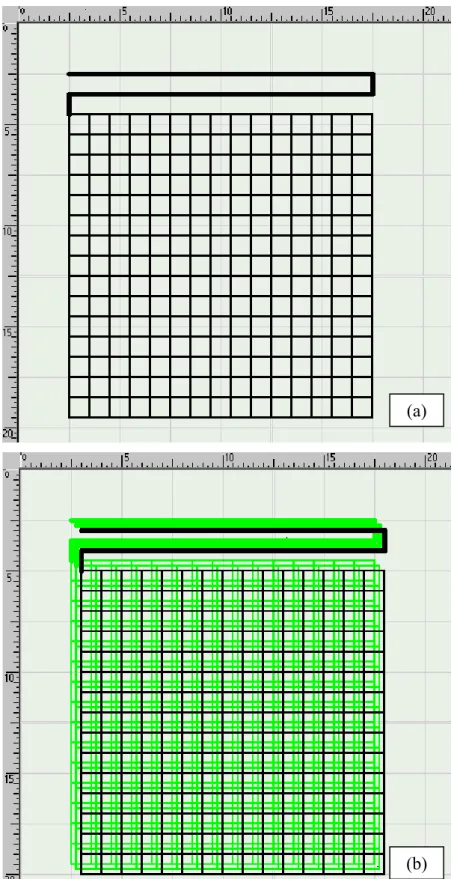
In questo lavoro sono state ottenute strutture 3-D da PU4-H, sovrapponendo, sfalsati, 6 strati in forma di griglia con maglia quadrata di lato 1 mm, ottenuti adoperando i parametri corrispondenti di Tabella 2-3. Inoltre, sono state prodotte anche strutture tridimensionali in PU4-H a tre strati sfalsati, con geometria della cella elementare quadrata, esagonale o ottagonale. Le Figure 2-6a e 2-6b riportano i
files grafici che hanno guidato la deposizione tramite micro-siringa rispettivamente di uno strato con
geometria a griglia quadrata e di uno scaffolds a tre strati, ottenuto sovrapponendo tre strati con geometria a griglia quadrata. Le Figure 2-4b e 2-5b riportano i files grafici che hanno guidato la deposizione di strutture tri-dimensionali a tre strati con geometria della cella elementare rispettivamente esagonale e ottagonale. Il polimero di supporto usato è stata gelatina di tipo A (Sigma), che è stata sciolta in acqua demineralizzata a 37°C, ottenendo una soluzione al 10 % (w/v).
Figura 2-4. Grafica della geometria esagonale adoperata nella tecnica PAM: (a) scaffold 2D; (b)
scaffold 3D. La scala numerica indicata è in mm (10 = 10 mm).
(a)
Figura 2-5. Grafica della geometria esagonale adoperata nella tecnica PAM: (a) scaffold 2D; (b)
scaffold 3D. La scala numerica indicata è in mm (10 = 10 mm).
(a)
Figura 6. Grafica dello scaffold a geometria quadrata realizzato tramite PAM: (a) struttura 2-D; (b) struttura 3-D. La scala numerica indicata è in mm (20 = 20 mm).
(a)
2.2.2 Caratterizzazione fisico-chimica e biologica
2.2.2.1 Calorimetria differenziale a scansione (DSC)
I dati calorimetrici dei poliuretani e poliuretani-uree sono stati ricavati sottoponendo i campioni (del peso di 5-10 mg e sigillati in capsule di Al) a cicli di riscaldamento, raffreddamento e secondo riscaldamento a 5 °C/min tra 5°C e 100°C in un’apparecchiatura Pyris Diamond. In una seconda prova, si è effettuata una prima scansione di riscaldamento a 5 °C/min tra 5°C e 100°C, poi il campione è stato rapidamente estratto dall’apparecchiatura DSC e sottoposto a quench in azoto liquido e, quindi, velocemente inserito nel calorimetro e sottoposto ad un nuovo riscaldamento a 5 °C/min tra 5°C e 100°C.
Analogamente, per il copolimero C27, si è effettuata una scansione di riscaldamento, seguita da una di raffreddamento e da un’altra di riscaldamento a 5 °C/min tra 5 °C e 100 °C. In una seconda prova, il copolimero C27 e stato sottoposto ad un ciclo di riscaldamento/raffreddamento/riscaldamento a 3 °C/min tra 5 °C e 100 °C.
Dalla posizione del massimo dei picchi endotermici registrati e del minimo dei picchi esotermici, si sono ricavate, rispettivamente, le temperature di fusione (Tm) e di cristallizzazione (TC), mentre
dall’area dei vari picchi si sono calcolati i dati di entalpia di fusione (∆Hm)e di cristallizzazione (∆HC).
2.2.2.2 Microscopia ottica
Campioni di film per casting di poliuretani e di copolimero C27 sono stati analizzati tramite microscopio ottico a luce polarizzata (Letz Ortholux II POL-BK) equipaggiato con una piastra riscaldante (Linkam, model THMSE 600).
I campioni sono stati sistemati tra due vetrini, poi riscaldati a 3°C/min fino alla completa fusione (70°C), lasciati 5 minuti a tale temperatura e poi raffreddati a 3°C/min fino a temperatura ambiente. La morfologia dei campioni è stata analizzata nuovamente tramite microscopio ottico, 48 h dopo il raffreddamento ed i campioni sono stati poi nuovamente riscaldati fino a 70°C a 3°C/min, esaminando le variazioni nella loro morfologia. Sono state acquisite immagini dei campioni anche durante le fasi di riscaldamento e di raffreddamento, tramite una telecamera (JVC TK-1085E video camera) e una scheda per l’acquisizione delle immagini (Pinnacle System miro VIDEO DC30), che sono state poi visualizzate tramite programma Adobe Premiere 5.1.
2.2.2.3 Prove di degradazione/erosione
Sono state condotte prove di degradazione/erosione in vitro su un poliuretano-urea contenente un estensore di catena a base di fenilalanina (PUU5) e sul corrispondente poliuretano di riferimento (PU5), adoperando la procedura di seguito riportata. Sono stati ottenuti campioni con dimensioni 1.5 × 1.5 cm2 dai film ottenuti per casting (c.f.r. 2.2.1.1), che sono stati pesati e poi sistemati all’interno di
provette di plastica da 15 ml. Ad ogni provetta sono stati aggiunti:
- 10 ml di soluzione dell’enzima: 500 Uml-1 di chimotripsina in Tris (0.036 M) con CaCl 2
(0.045 M) e NaN3 (0.02 %), pH=8.0;
oppure:
- 10 ml di soluzione tampone: Tris (0.036 M) con CaCl2 (0.045 M) e NaN3 (0.02 %), pH=8.0.
Le provette sono state sistemate in un bagno ad acqua, agitato meccanicamente e mantenuto a 37°C. Le soluzioni enzimatiche e quelle tampone sono state sostituite ogni due giorni, per compensare la perdita di attività enzimatica col tempo di incubazione.
I campioni sono stati rimossi dopo 2, 4, 6, 8, 10 e 12 giorni e ciascuno è stato posto in 15 ml di soluzione Triton-X all’1% (w/v), per rimuovere l’enzima legato irreversibilmente.
I campioni sono stati lavati tre volte con acqua demineralizzata, asciugati con carta assorbente e pesati. Poi sono stati seccati a temperatura ambiente in essiccatore, per almeno tre giorni, e ripesati per determinare la perdita di peso (erosione).
I campioni iniziali e quelli sottoposti a degradazione per 12 giorni sono stati analizzati tramite microscopia elettronica a scansione (SEM; apparecchiatura JEOL JSM 300), per valutare i cambiamenti morfologici avvenuti. Le variazioni di peso molecolare dei polimeri nel corso della prova
(dopo 4, 8 e 12 giorni) sono state misurate tramite cromatografia a permeazione di gel (GPC). Precisamente, i pesi molecolari e le distribuzioni dei pesi molecolari sono stati determinati tramite lo strumento Waters mod. 600 E, equipaggiato di rivelatore a indice di rifrazione (RI) Waters mod. 410 e sistema di acquisizione su personal computer Millennium Chromatography Manager Waters. Come fase stazionaria, sono state utilizzate due colonne PL gel Mixed-C (e solo dove indicato Mixed-D), impiegando come eluente cloroformio. Per la calibrazione sono stati impiegati campioni monodispersi di polistirene.
2.2.2.4 Test di citotossicità
Sono stati eseguiti test di citotossicità sui poliuretani e sul copolimero C27 che, essendo polimeri di sintesi, necessitano di una preliminare verifica della loro idoneità per applicazioni biomediche. Il test consiste nel valutare la presenza di rilasci tossici da parte dei materiali, quando questi sono immersi in terreno di coltura. I test sono stati effettuati su film per casting con dimensioni 1.5×1.5 cm2 e, per
ciascun materiale, si sono adoperati tre campioni per ogni tempo di prova.
I campioni sono stati inizialmente sterilizzati tramite una soluzione al 70 % (v/v) di etanolo (Aldrich) in acqua sterile e poi sottoposti ai raggi UV per 15 min per lato. Successivamente, sono stati immersi in 2 ml di terreno di coltura all’interno dei pozzetti di piastre di coltura da 24 celle. Si è adoperato il mezzo di coltura DMEM (Dulbecco’s modified Eagle’s medium; Cambrex ) con un’alta quantità di glucosio, il 10 % di siero fetale bovino (Cambrex), l’1 % di glutammina (Cambrex), penicillina (200 U/ml; Cambrex) e streptomicina (200 µg/ml; Cambrex).
I campioni sono stati mantenuti alla temperatura di 4 °C per vari tempi: 1, 2, 3, 7, 10, 15 giorni. Ad ogni tempo, il terreno di coltura è stato sostituito con terreno fresco, mentre sul terreno prelevato sono stati eseguiti test di adesione cellulare. Precisamente, il test di adesione cellulare è stato condotto su ciascun mezzo di coltura a 4, 24 e 48 h, adoperando fibroblasti di topo NIH-3T3. Successivamente, dopo 4, 24 e 48 h, il mezzo è stato aspirato ed è stato valutato il numero di cellule che hanno aderito sulle pareti dei pozzetti di coltura. Precisamente, le cellule sono state fissate con una soluzione al 4 % (v/v) di formaldeide (Sigma; Italy) in tampone fosfato (PBS, Sigma), e colorate con una soluzione di blue di Coomassie (Fluka, Italy). Si sono considerati, come controllo, i fibroblasti aderiti alle pareti di pozzetti nel quale è stato versato terreno di coltura fresco. I vari pozzetti sono stati analizzati al microscopio ottico (Olympus AX70). Il rapporto tra il numero delle cellule nei pozzetti contenenti i mezzi di coltura, nei quali sono stati immersi i film di poliuretani e di poliuretani-uree, e il numero delle cellule nei pozzetti contenenti il terreno di coltura fresco è stato assunto come indice della non-citotossicità dei mezzi di coltura e, quindi, dei corrispondenti campioni con i quali i mezzi di coltura sono stati in contatto. Precisamente, un elevato valore di tale rapporto indica che i campioni sono biocompatibili, viceversa un basso valore del rapporto è indice della citotossicità dei campioni.
2.2.2.5 Microscopia a forza atomica (AFM)
L’analisi tramite microscopia a forza atomica è stata condotta su film sottili depositati su vetrini, tramite la tecnica di spin coating di soluzioni al 4 % (w/v) in cloroformio, a 1000 rpm e a temperatura ambiente, adoperando l’apparecchiatura SPURR Microtech. I film sono stati essiccati sotto cappa per 48 h e in stufa ventilata a 37°C per una settimana. Lo strumento adoperato per la caratterizzazione è un AFM commerciale (Park Scientific Instrument) con una punta di 5 µm. Le misure sperimentali sono state eseguite in modalità “di contatto” della punta dello strumento con la superficie del campione, sia in presenza, sia in assenza di millitensioni (70 e 400 mV) applicate alla punta, e mediante misure dello
shift di risonanza. Maggiori dettagli sui principi di funzionamento della tecnica di microscopia a forza
atomica sono riportati in appendice B.
2.2.2.6 Angoli di contatto
Gli angoli di contatto sono stati misurati su campioni in forma di film ottenuti per spin-coating a 1000 rpm per 30 s, da soluzioni al 4 % (w/v) in cloroformio nell’apparecchiatura SPURR Microtech. Il solvente è stato fatto evaporare in stufa ventilata a 37°C per 48 h. Gli angoli di contatto sono stati misurati a temperatura ambiente attraverso l’apparecchiatura KSV, adoperando una goccia da 5 µl di
acqua doppiamente distillata. Per ogni campione, sono state fatte cinque misurazioni di angolo di contatto in cinque punti diversi della superficie, e in seguito sono state mediate. Maggiori dettagli relativi alla misurazione dell’angolo di contatto sono riportati in appendice C.
2.2.2.7 Tecnica di ellissometria: misura dell’indice di rifrazione, della costante dielettrica
e dello spessore di film polimerici.
L’ellissometria è una tecnica ottica altamente sensibile, utile per le misure di spessore e di densità di films sottili [Mc Crackin et al., 1963]. Precisamente, l’ellissometria è definita come la misura dello stato di polarizzazione di un’onda vettoriale e i principi su cui si basa sono stati raccolti nell’appendice D. La tecnica ha consentito di ottenere la misura degli indici di rifrazione, della costante dielettrica dei materiali e dello spessore dei films utilizzati. Le misure sono state effettuate tramite apparecchiatura ELLIPSOMETER CHECK SAMPLE (Rudolph), su films ottenuti per spin-coating di soluzioni al 4% (w/v) dei vari PU e PUU e del copolimero C27. Le misure non sono state eseguite sul PCL, essendo un polimero commerciale già ampiamente caratterizzato per applicazioni biomediche. Le misure sono state effettuate su tre campioni per ogni materiale e il valore della costante dielettrica, dello spessore e dell’indice di rifrazione del materiale sono stati ricavati come media aritmetica dei risultati delle prove.
2.2.2.8 Misura della carica superficiale (Tecnica Kelvin-Probe)
La tecnica di Kelvin-Probe ha consentito di ottenere misure di carica superficiale dei campioni di poliuretani, poliuretani-uree e del copolimero C27. Il metodo si basa sulla misura della differenza di potenziale tra due piastre di alluminio poste ad una distanza di alcuni millimetri, di cui una è fissa mentre l’altra vibra. La misura è effettuata sia in presenza, sia in assenza di un dielettrico interposto, costituito da un film polimerico deposto su una delle piastre in alluminio. In particolare, si sono deposti film per casting da soluzioni in cloroformio di poliuretani e poliuretani-uree (4 % w/v) e di copolimero C27 (2.5 % w/v) sulle piastre di alluminio adoperate nella prova e aventi un diametro pari a 10 cm. Precisamente, la piastra è stata prima alloggiata in una capsula Petri di vetro con diametro pari a 12 cm e, successivamente, è stato versato sopra la piastra un volume di soluzione pari a 20 ml. Si è lasciato evaporare il solvente in stufa ventilata per 48 h a 37 °C e, in seguito, sono state effettuate le misure di densità di carica superficiale. La Figura 2-7 mostra lo schema di funzionamento dell’apparecchiatura adoperata nella tecnica di Kelvin Probe.
Figura 2-7. Schema dell’apparecchiatura di Kelvin-Probe
Tra le due piastre è imposta una differenza di potenziale e la piastra superiore viene fatta vibrare, dando origine ad una differenza di potenziale oscillante alla stessa frequenza di vibrazione della piastra superiore. L’istante in cui la differenza di potenziale tra le piastre è nulla, la forza elettromotrice imposta al circuito uguaglia il potenziale di contatto tra le piastre. La misura del
potenziale di contatto tra le piastre, permette di ricavare la densità di carica superficiale, tramite un semplice modello elettrico, che fa riferimento ad un sistema costituito da due condensatori circolari posizionati in serie, per i quali il dielettrico di uno è costituito da aria e quello dell’altro da un film di polimero. Lo spessore del film è noto, inoltre si assume che la sua superficie sia perfettamente piana. In base a tale modello, la densità di carica superficiale è data da:
R
d
V
o+
−
⋅
⋅
⋅
⋅
=
)
1
(
2
ε
ε
ε
σ
(2.1)dove σ è la densità di carica superficiale, εo è la costante dielettrica assoluta, ε è la costante dielettrica
del polimero, V è la differenza di potenziale tra le piastre con e senza polimero, d è lo spessore del film di polimero e R è il raggio del disco di alluminio utilizzato come substrato di deposizione per il film di polimero.
Maggiori dettagli sulla tecnica sono riportati nell’appendice D.
2.2.2.9 Caratterizzazione meccanica delle strutture ottenute per deposizione tramite
microsiringa
Le proprietà meccaniche a trazione di campioni di PU4-H sotto forma di strutture bidimensionali depositate tramite microsiringa sono state valutate tramite un trasduttore isotonico di posizione (modello 7006; UGO Basile Biological Research Apparatus, Italy), nel quale la forza applicata presenta una risoluzione di 1 mN.
Le prove di trazione sono state eseguite su strutture a tre strati con diversa geometria della cella elementare depositata: quadrata, esagonale, ottagonale (c.f.r. 2.2.1.2.).
I campioni analizzati hanno una forma rettangolare, con una lunghezza di 2 cm e una larghezza di 0.5 cm. La forza di trazione, infatti, è stata applicata alle estremità più sottili del campione. Lo spessore dei campioni dipende dallo spessore delle piste depositate e, generalmente, è attorno a 100 µm. La geometria dei campioni (in particolare lo spessore) è stata analizzata tramite l’acquisizione di immagini da un microscopio ottico (Olympus AX 70), equipaggiato con una macchina fotografica digitale (ccD Camera HIRES, DTA; Italy). L’analisi al microscopio ottico ha consentito di verificare la direzione di applicazione del carico di trazione, rispetto alla direzione delle piste depositate all’interno dello scaffold bidimensionale e di calcolare lo spessore dei campioni.
Le prove di trazione sono state eseguite su tre campioni per ciascuna tipologia di geometria della cella base depositata. Il modulo di Young di ogni campione è stato calcolato dalla pendenza iniziale della parte lineare delle curve sforzo (σ) – deformazione (ε). Dato il particolare andamento delle curve (σ)– (ε) (che verrà discusso nella relativa parte della sezione Risultati e Discussione), si è proceduto anche al calcolo di una seconda pendenza delle curve sforzo-deformazioe, per alti valori della deformazione.
2.2.2.10 Test di adesione cellulare su film per casting e microfabbricazioni 2D
Gli scaffold bidimensionali (griglie ottenute tramite PAM e films per casting) sono stati preparati alla coltura cellulare secondo la procedura seguente. I campioni asciutti sono stati sterilizzati tramite una soluzione al 70 % (v/v) di etanolo in acqua sterile, sottoposti ai raggi UV per 15 min per lato. Successivamente sono stati rivestiti esternamente con uno strato di gelatina di tipo A derivata da pelle di maiale (Sigma), lasciando le strutture a contatto con una soluzione all’1% (w/v) di gelatina in acqua demineralizzata, a 37°C per una notte.
Fibroblasti di topo NIH-3T3 sono stati coltivati in piastre di coltura da 25 cm2 di area contenenti il
mezzo di coltura DMEM (Dulbecco’s modified Eagle’s medium; Cambrex ) con un’alta quantità di glucosio, il 10 % di siero fetale bovino (Cambrex), l’1 % di glutammina (Cambrex), penicillina (200 U/ml; Cambrex), e streptomicina (200 µg/ml; Cambrex). La coltura è stata mantenuta in un incubatore equilibrato con il 5% di CO2 a 37°C. Una volta che le cellule sono giunte a confluenza, il mezzo di
coltura è stato rimosso sotto cappa sterile, e le fiasche di coltura sono state lavate due volte con tampone fosfato (PBS, Sigma). Per staccare le cellule aderite alle fiasche, si sono aggiunti 3 ml di una soluzione allo 0.01 % di tripsina (Cambrex)/0.01 % di EDTA (Cambrex) e il sistema è stato mantenuto
in incubazione per 3 min a 37°C. La tripsina è stata inattivata aggiungendo una quantità di mezzo di coltura tre volte maggiore di quello di soluzione di tripsina/EDTA.
I pellets cellulari sono stati recuperati dalla dispersione cellulare mediante centrifugazione (centrifuga Haereous, Switzerland) a 1000 rpm per 10 min, e successiva dispersione in un nuovo mezzo di coltura alla concentrazione di 100,000 cellule/ml. Le strutture polimeriche sono state seminate con la sospensione cellulare in pozzetti di polistirene da 24 celle. Le misure di adesione cellulare sono state effettuate su tre campioni ad ogni tempo (2, 4, 24 h per il PCL e il copolimero C27, e 4, 8, 48 h per il poliuretano PU4-H). Ad ogni tempo il mezzo di coltura è stato aspirato e rimosso, e i substrati con le cellule attaccate sono stati lavati in tampone fosfato. Le cellule aderite sugli scaffold sono state fissate con una soluzione al 4 % (v/v) di formaldeide (Sigma) in PBS per 10 min, e poi colorate con una soluzione di blu di Coomassie (Fluka) per 10 min. I campioni sono stati analizzati al microscopio ottico (Olympus AX 70). L’indice della densità cellulare è stato calcolato come rapporto tra l’area occupata dalle cellule aderite sul substrato e l’intera area del substrato. Per calcolare l’area occupata dalle cellule, è stato adoperato un software sviluppato in ambiente Matlab e basato su un processo di segmentazione. La densità cellulare sulle strutture è stata paragonata a quella di un campione di riferimento (un film di gelatina di tipo A). La morfologia cellulare è stata studiata su ogni campione, tramite foto al microscopio ottico con ingrandimento pari a 40 X (Olympus AX 70).
2.2.2.11 Test in vivo con strutture 3D ottenute per deposizione tramite microsiringa
I test in vivo sono stati effettuati adoperando strutture tri-dimensionali in PU4-H (descritte al paragrafo 2.2.1.2), campioni (1×1 cm2) di films per casting di PU4-H e microstrutture in copolimero poli(acidolattico-co-acido glicolico) (PLGA) ottenute come descritto in letteratura [Vozzi et al., 2002]. Il PLGA è stato adoperato per fare un confronto diretto del tempo di biodegradazione del poliuretano di sintesi. Per le prove (effettuate presso i laboratori biologici del CNR, Pisa), sono stati utilizzati quattro topi, dopo avere verificato il loro perfetto stato di salute. L’intervento chirurgico è stato condotto in anestesia totale. I topi sono stati fissati su un piano, in posizione supina, e sono state individuate tre posizioni nella zona lombare del topo, dove effettuare l’impianto sottocutaneo (tra derma ed epidermide) delle tre strutture selezionate. Per ogni topo, la posizione relativa degli scaffolds impiantati è stata cambiata, per verificare la dipendenza dei risultati dal sito d’impianto. Il quarto topo è stato operato per avere un margine di sicurezza, nel caso si potesse verificare qualche inconveniente per i tre topi necessari per la prova. Le tre incisioni sulla zona lombare del topo sono state poi richiuse con tre punti di sutura, di cui solo uno non bioassorbibile, per permettere l’individuazione precisa della zona operata. L’esame istologico delle varie strutture è stato effettuato su ciascun topo dopo 1, 3 e 6 mesi. Precisamente, in corrispondenza dei siti d’impianto, la struttura è stata prelevata effettuando un’incisione tramite un apposito bisturi circolare, detto punch.
Poi i campioni sono stati fissati con una soluzione di formaldeide al 4 % w/v in tampone fosfato (PBS), lavati con una soluzione di PBS e fissati con una soluzione di tetrossido di osmio (Aldrich) al 4 % w/v per 2 h. Dopo i campioni sono stati disidratati con soluzioni di acetone (Aldrich) a concentrazioni crescenti e, poi, inseriti all’interno della resina epossidica Spurr. A questo punto si sono ricavate tramite microtomo sezioni sottili di 1 µm di spessore, che sono state colorate tramite blu di toluidina (Fluka) nel caso dello scaffold in PU4-H e con ematossilina-eosina (Fluka) nel caso del
film di PU4-H e della struttura in PLGA. Infine, i campioni sono stati esaminati al microscopio ottico
2.3. Risultati e Discussione
In questa sezione si riportano i risultati relativi alla caratterizzazione chimico-fisica dei polimeri adoperati: poliuretani, poliuretani-uree e copolimero a tre blocchi PCL-POE-PCL. La valutazione delle caratteristiche termiche e morfologiche ha un ruolo importante, in quanto è strettamente legata alla risposta dei materiali alle prove meccaniche e a quelle di degradazione enzimatica eseguita in
vitro. Un’ulteriore caratterizzazione ha riguardato la valutazione di caratteristiche specifiche per la
risposta cellulare, attraverso prove di citotossicità, misura dell’angolo di contatto, misura della densità di carica superficiale, valutazione della rugosità superficiale, prove di adesione cellulare e, infine, prove in vivo. Parallelamente a tali caratterizzazioni, nella sezione vengono riportati esempi di microfabbricazioni eseguite tramite microdeposizione da soluzione polimerica. Lo scopo delle caratterizzazioni eseguite è stato quello di identificare, tra tutti i materiali esaminati, quello più adatto per applicazioni biomediche, e anche quello di fornire utili indicazioni sulle proprietà generali dei polimeri studiati, nella prospettiva di un loro utilizzo come materiali a basso impatto ambientale.
2.3.1 Analisi calorimetria (DSC)
I risultati dell’analisi DSC dei poliuretani e dei poliuretani-uree, relativi al primo riscaldamento a 5 °C/min tra 5°C e 100°C, sono riportati in Tabella 2-4.
Tabella 2-4. Dati DSC relativi al primo riscaldamento dei poliuretani e poliuretani-uree tra 5 °C e 100°C a 5°C/min Polimero Tm (°C) ∆Hm (J/g) PU4-H 36.9 11.7 PU4 34.0 - 38.7 30.9 PUU4 (34.0) - 40.0 21.2 PU5 34.0 - 42.7 39.7 PUU5 36.5 - 47.1 32.0 PU6 (39.0) - 48.9 53.7 PUU6 44 - 49.1 45.1
I poliuretani e i poliuretani-uree hanno mostrato una bassa velocità di cristallizzazione: il raffreddamento eseguito a 5°C/min tra 100°C e 5°C non ha permesso ai materiali di cristallizzare. Le scansioni DSC di raffreddamento e di secondo riscaldamento, infatti, non hanno registrato eventi di cristallizzazione e di fusione. La prima scansione di riscaldamento, invece, ha rivelato, per alcuni campioni, la presenza di un picco singolo di fusione, generalmente allargato e caratterizzato da una “spalla” a temperatura minore, e per i rimanenti campioni la presenza di un doppio picco di fusione. Per indurre i polimeri a cristallizzare in fase di raffreddamento, dopo il primo iniziale riscaldamento che ha la funzione di annullare la storia termica del polimero, i campioni sono stati sottoposti ad un
quench in azoto liquido e, poi, sono stati riscaldati a 5°C/min, all’interno dell’apparecchiatura DSC,
per rilevare eventuali eventi termici di fusione. La Tabella 2-5 raccoglie i dati relativi al riscaldamento a 5°C/min dei campioni di PU e PUU precedentemente sottoposti a quench.
Tabella 2-5. Dati DSC relativi al riscaldamento tra 5 °C e 100°C a 5°C/min dei poliuretani e poliuretani-uree sottoposti a quench.
Polimero Tm (°C) ∆Hm (J/g) PU4-H 35.5 6.0 PU4 30.0 – 37.5 19.8 PUU4 n.d. n.d. PU5 34.9 – 41.4 32.0 PUU5 31.4 – 40.4 25.2 PU6 27.0 – 46.9 48.1 PUU6 44.0 – (46.5) 36.2
Come evidenziato dai dati riportati in Tabella 2-5, i fenomeni endotermici di fusione dei campioni che hanno subito un quench hanno mostrato valori di temperatura e di entalpia vicini a quelli dei campioni sottoposti al primo riscaldamento (Tabella 2-4). I campioni sottoposti a quench, tuttavia, hanno registrato un lieve abbassamento delle temperature di fusione ed uno più marcato delle entalpie di fusione: queste variazioni sono state attribuite al fatto che i materiali sono cristallizzati per effetto di un abbassamento termico rilevante, in un tempo relativamente breve, quindi, la frazione cristallizzata e il grado di perfezione dei cristalli sono risultati entrambi inferiori a quanto registrato nella prima scansione di riscaldamento.
Sulla base dei dati calorimetrici delle Tabelle 2-4 e 2-5 e del loro confronto con i dati calorimetrici della Tabella 2-1, è emerso che i PU e i PUU sono polimeri semicristallini, con grado di cristallinità variabile, legato alla cristallizzazione del segmento soft. A parità di composizione chimica del segmento soft (confronto tra la serie PU e PUU), il grado di cristallinità è risultato minore, adoperando come estensore di catena il Phe diestere (serie PUU). Tale risultato è stato attribuito al maggiore ingombro sterico del Phe diestere rispetto al CDM, caratteristica che ha ostacolato l’impacchettamento delle catene in domini cristallini. Il picco di fusione relativo al blocco PEG dei segmenti soft PCL-PEG-PCL non è stato registrato dai relativi spettri DSC dei PU e dei PUU. In nessun caso si sono rilevati fenomeni endotermici dovuti alla fusione di domini cristallini formatisi a partire dai segmenti
hard. Il diisocianato adoperato (LDI), infatti, è asimmetrico e contiene un gruppo estereo in catena
laterale, che impedisce un efficace impacchettamento delle catene. Il confronto tra i dati DSC dei campioni ottenuti per precipitazione (Tabella 2-2) e quelli dei campioni ottenuti per casting (Tabella 2-4) ha mostrato differenze sostanziali solo per i campioni PU5 e PUU5: per tali polimeri la procedura di evaporazione lenta del solvente (48 h) probabilmente ha favorito un impacchettamento più efficace delle catene polimeriche in strutture cristalline. Tale comportamento può essere attribuito alla maggiore flessibilità conformazionale delle catene cristallizzabili, legata alla presenza di un maggiore quantitativo di PEG nei segmenti soft.
In Tabella 2-6 sono raccolti i dati DSC relativi al copolimero C27.
Tabella 2-6. Dati DSC relativi al copolimero C27, ricavati attraverso scansioni di riscaldamento e di raffreddamento nell’intervallo 5-100°C. Scansione Velocità di scansione (°C/min) Tm (°C) ∆Hm (J/g) I riscaldamento 5 45.6 – 61.5 94.3 I riscaldamento 3 56.5 – 63.7 120.8 I raffreddamento 3 38.9 87.4 II riscaldamento 3 55.1 – 57.7 90.7
Il copolimero C27 ha mostrato un doppio picco di fusione dovuto alla fusione separata dei blocchi di PCL e dei blocchi di POE, mentre la scansione di raffreddamento ha evidenziato un unico picco di cristallizzazione allargato, che potrebbe essere il risultato della sovrapposizione di due picchi di cristallizzazione molto ravvicinati. Nel primo riscaldamento a 3 °C/min, si è registrato un valore elevato dell’entalpia di fusione, molto maggiore di quello misurato durante il secondo riscaldamento. Questo risultato ha mostrato che il copolimero C27 ha una velocità di cristallizzazione
sufficientemente elevata da cristallizzare durante la scansione di raffreddamento a 3°C/min eseguita tra 100°C e 5°C, tuttavia, la sua frazione cristallina (e il grado di perfezione dei cristalli) crescono con il tempo di permanenza a temperatura ambiente.
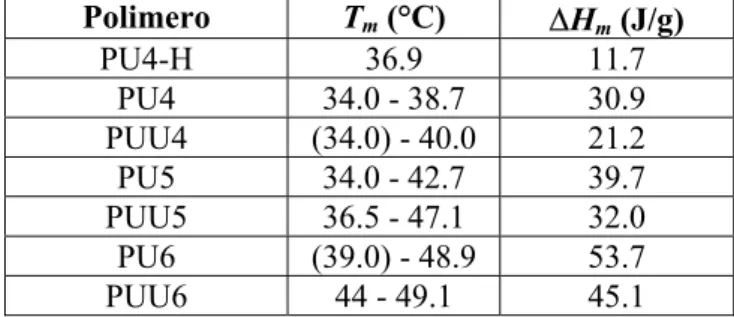
La Figura 2-8 riporta le scansioni calorimetriche di primo riscaldamento, raffreddamento e secondo riscaldamento a 3 °C/min relative al copolimero C27. Le scansioni di Figura 2-8 mostrano con chiarezza la riduzione del grado di cristallinià del copolimero in seguito al primo riscaldamento e al successivo raffreddamento. Inoltre, l’analisi delle scansioni DSC riportate in Figura 2-8 suggerisce che la tipologia dei cristalli si è modificata dopo il primo riscaldamento ed il successivo raffreddamento, dal momento che il rapporto tra le intensità dei due picchi di fusione, parzialmente sovrapposti, è risultato differente nella seconda scansione di riscaldamento a confronto con la prima scansione di riscaldamento. Nel Capitolo 6 (paragrafo 6.1.3.3.) sono riportati i dati calorimetrici relativi al polietilenossido (PEO) (Tm = 68°C; ∆Hm = 137 J/g) e ad un PCL ad alto peso molecolare (Tm = 57°C;
∆Hm = 58 J/g), ricavati durante un secondo riscaldamento a 10°C/min. Il confronto tra i dati
calorimetrici del copolimero C27 e quelli del PEO e del PCL puri ha permesso di attribuire il picco di fusione del C27 a più alta temperatura alla fusione dei segmenti poliossietilenici, e quello a bassa temperatura alla fusione dei segmenti di PCL. La variazione nell’intensità dei due picchi parzialmente sovrapposti nella prima e nella seconda scansione di riscaldamento a 3°C/min è stata attribuita alla variazione nel rapporto tra la porzione cristallina a base di PCL e quella a base di PEO. In seguito al raffreddamento a 3°C/min, i segmenti a base di PCL, probabilmente, hanno cristallizzato più velocemente dei segmenti a base di POE del copolimero. Probabilmente, la permanenza a temperatura ambiente del copolimero comporterebbe la crescita del grado di cristallinità e la progressiva cristallizzazione dei segmenti POE, come è stato suggerito dai dati relativi alla prima scansione di riscaldamento. Il valore diminuito dell’entalpia di fusione durante il secondo riscaldamento, quindi, è attribuibile alla minore cristallizzazione dei segmenti di POE, che presentano un’entalpia di fusione molto maggiore rispetto a quella dei segmenti a base di PCL.
Infine, le caratteristiche calorimetriche del copolimero C27 sottoposto al ciclo termico a 3°C/min sono state di aiuto nella comprensione dei dati ricavati tramite osservazione al microscopio ottico, commentati nel paragrafo successivo.
Figura 2-8. Scansioni calorimetriche di primo riscaldamento (a), raffreddamento (b) e secondo riscaldamento (c) a 3°C/min tra 5°C e 100°C, relative al copolimero C27.
2.3.2 Analisi tramite microscopio ottico
La morfologia cristallina dei campioni di poliuretani e poliuretani-uree, in generale, è risultata sensibile al tempo di permanenza a temperatura ambiente: i materiali presentano infatti un punto di
(a)
(b)
fusione (Tm) basso, quindi la forza motrice del processo di cristallizzazione a temperatura ambiente
(∆T = Tm-Tamb) è ridotta.
Le Figure 2-9 e 2-10 riportano le immagini ottenute al microscopio ottico, relative ai campioni di poliuretani e poliuretani-uree in forma di films, prima del riscaldamento a 3 °C/min e, dopo il ciclo di riscaldamento (fino a fusione)/raffreddamento/mantenimento a temperatura ambiente per 48 h.
Precisamente, i film di PUU4 e PU4 (Figura 2-9 a-b, Figura 2-10 a-b), che differiscono tra loro per il tipo di estensore i catena e per il peso molecolare, hanno presentato una diversa morfologia cristallina. I campioni di PUU4 hanno mostrato cristalli di dimensioni inferiori e dalle forme più irregolari e disomogenee, rispetto al caso del PU4. Sottoposti a riscaldamento a 3°C/min , i film di PU4 e di PUU4 hanno mostrato una fusione completa a circa 38 °C. In fase di riscaldamento, la morfologia dei cristalli è risultata più facilmente distinguibile: in particolare, nel caso del PU4, si sono evidenziati cristalli di forma poligonale, con struttura allungata e dimensioni variabili nell’intervallo 10-35 µm. I campioni di PU4 e di PUU4 non sono cristallizzati nella fase di raffreddamento, mentre hanno cristallizzato durante le 48 h di permanenza a temperatura ambiente, sviluppando una morfologia simile, con cristalli più fini di quelli originali. Per riscaldamento a 3°C/min, i campioni hanno mostrato un punto di fusione di circa 37°C:
I campioni di PU5 e PUU5 (Figura 2-9 c-d e Figura 2-10 c-d), che, analogamente ai precedenti, differiscono per il tipo di estensore di catena e per il peso molecolare, hanno presentato una morfologia cristallina molto diversa. Il PU5 ha mostrato zone cristalline omogenee, di dimensioni pari a 60-80 µm, mentre il PUU5 ha evidenziato la presenza di una struttura cristallina più fine e complessivamente disomogenea, con dimensioni dei cristalli variabili nell’intervallo 20-40 µm. I film di PU5 e di PUU5, riscaldati a 3°C/min, hanno registrato la fusione rispettivamente a 44°C e a 47°C. I films di PU5 e PUU5 non hanno cristallizzato per raffreddamento, ma durante il tempo di permanenza a temperatura ambiente, acquisendo una morfologia più omogenea e fine rispetto a quella dei films iniziali. La temperatura di fusione dei film di PU5 e PUU5, riscaldati nuovamente a 3 °C/min, è stata misurata rispettivamente a 44 °C e a 42°C.
I film di PU6 e PUU6 (Figura 2-9 e-f, Figura 2-10 e-f), che, analogamente alle coppie di campioni precedentemente studiati, differiscono per il tipo di estensore di catena e per il peso molecolare, hanno mostrato una morfologia differente. Il PU6 è risultato caratterizzato da cristalli di dimensioni maggiori (75-85 µm) e dalle forme più omogenee, mentre il PUU6 ha mostrato una struttura cristallina disomogenea con cristalli dalle dimensioni e forme variabili (35-40 µm). Per riscaldamento a 3 °C/min, entrambi i films sono fusi a 49 °C. Come per i precedenti campioni, i films di PUU6 e PU6 non hanno cristallizzato per raffreddamento, mentre, lasciati a temperatura ambiente, hanno sviluppato una morfologia cristallina più fine e omogenea rispetto a quella dei campioni iniziali. Nel caso del PU6, i cristalli ottenuti hanno mostrato dimensioni di circa 40 µm, mentre i cristalli del PUU6 hanno evidenziato forme più irregolari con dimensioni di circa 10-20 µm. La fusione dei campioni di PU6 e il PUU6, riscaldati nuovamente a 3 °C/min, è avvenuta a 46 °C e 49 °C, rispettivamente.
In conclusione, i poliuretani hanno mostrato una morfologia cristallina regolare e omogenea, con cristalli di dimensioni pari a circa 60-80 µm, il cui punto di fusione è risultato dipendente dalla composizione del segmento soft, precisamente esso è risultato crescente al decrescere del contenuto di PCL all’interno del segmento soft.
I poliesteriuretani, invece, hanno mostrato cristalli più disomogenei, con forme irregolari e dimensioni complessivamente più piccole rispetto al caso dei poliuretani (10-40 µm). Anche per i poliesteriuretani il punto di fusione è risultato leggermente crescente al decrescere del contenuto di PCL all’interno dei segmenti soft.
Per i poliuretani e per i poliesteriuretani la cinetica di cristallizzazione è risultata lenta a temperatura ambiente e, dopo una permanenza di 48 h a condizioni ambiente, successiva al raffreddamento dallo stato fuso, le dimensioni dei cristalli formatisi si sono rivelate inferiori a quelle dei cristalli iniziali.
Figura 2-9. Immagini registrate al microscopio ottico a luce polarizzata, relative ai campioni di poliuretani in forma di films, prima del ciclo di riscaldamento-raffreddamento (a, c, e), e dopo il ciclo di riscaldamento-raffreddamento - permanenza a temperatura ambiente per 48 h (b, d, f): PU4 (a, b); PU6 (c, d) e PU5 (e, f). La barra indica 100 µm.
(a)
(b)
(c)
(d)
Figura 2-10. Immagini registrate al microscopio ottico a luce polarizzata, relative ai campioni di poliuretani-uree in forma di films, prima del ciclo di riscaldamento-raffreddamento (a, c, e), e dopo il ciclo di riscaldamento-raffreddamento - permanenza a temperatura ambiente per 48 h (b, d, f): PUU4 (a, b); PUU6 (c, d) e PUU5 (e, f). La barra indica 100 µm.
(a)
(b)
(c)
(d)
L’analisi al microscopio ottico a luce polarizzata è stata effettuata anche sul poliuretano PU4-H. La Figura 2-11 a-b riporta le immagini, ottenute tramite microscopio ottico a luce polarizzata, relative ad un campione in forma di film di PU4-H, prima e dopo il ciclo di riscaldamento - raffreddamento - permanenza a temperatura ambiente.
Figura 2-11. Immagini registrate al microscopio ottico a luce polarizzata, relative a un campione di poliuretano PU4-H in forma di film: (a) prima, (b) dopo il ciclo di riscaldamento – raffreddamento - permanenza a temperatura ambiente per 48 h. La barra indica 100 µm.
Il poliuretano PU4-H, dopo il ciclo termico a cui è stato sottoposto, ha mostrato una morfologia cristallina più fine, in analogia a quanto osservato per gli altri campioni di PU e PUU precedentemente analizzati. La scarsa risoluzione delle immagini non ha permesso di effettuare misure sulle dimensioni dei cristalli di PU4-H.
Il copolimero C27 ha presentato, a temperatura ambiente, la morfologia cristallina riportata in Figura 2-12a. Riscaldato a 3 °C/min, la fusione completa del campione è avvenuta a circa 60°C. Il polimero è stato poi raffreddato a temperatura ambiente a 3°C/min. L’immagine riportata in Figura 2-12b evidenzia la struttura cristallina del C27 a temperatura ambiente, dopo il raffreddamento. La morfologia del copolimero è risultata completamente modificata in seguito al ciclo termico: come mostrato dall’analisi calorimetrica, in seguito al riscaldamento e al raffreddamento a 3°C/min, i segmenti di PCL sono cristallizzati più velocemente di quelli poliossietilenici. La permanenza a temperatura ambiente comporta la progressiva cristallizzazione dei segmenti poliossietilenici con la formazione della morfologia cristallina mostrata nella Figura 2-12a.
Figura 2-12. Morfologia cristallina del copolimero C27 a temperatura ambiente (a) e dopo il ciclo di riscaldamento – raffreddamento – permanenza a temperatura ambiente per 48 h (b). Ingrandimento 32 X.
(a)
(b)
(a) (b)
100 µm
100 µm
2.3.3 Prove di degradazione/erosione
Esistono diverse strategie di sintesi di poliuretani segmentati biodegradabili, contenenti in catena legami idrolizzabili. La strategia più comune consiste nell’adoperare segmenti soft idrolizzabili [Bruin et al., 1990; Storey et al., 1993; Kylma et al., 1997; Hatakeyama et al., 1998], ottenendo così poliuretani lineari o reticolati, con proprietà fisiche e di degradazione variabili. Un metodo alternativo consiste nell’introdurre gruppi idrolizzabili nel segmento hard: tuttavia, la scelta dei diisocianati commerciali per la sintesi dei poliuretani è piuttosto ridotta. L’alternativa più perseguita per la sintesi di poliuretani degradabili consiste nell’adoperare estensori di catena idrolizzabili.
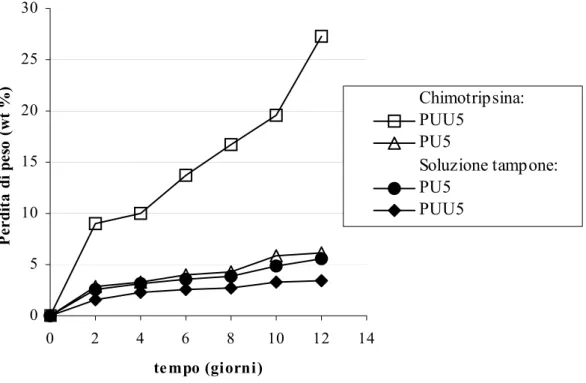
I poliuretani, generalmente, posseggono un’alta percentuale di segmenti soft, che sono quindi fondamentali per le proprietà generali del materiale. La preparazione di poliuretani degradabili, con segmenti hard idrolizzabili, permette l’utilizzo di segmenti soft di varia natura, in relazione alle proprietà richieste al materiale in una specifica applicazione, mantenendo invariata la sua capacità di degradarsi (che dipende dai segmenti hard). In particolare, l’estensore di catena Phe diestere adoperato per la sintesi dei poliuretani-uree (PUU), conferisce ai materiali caratteristiche di biodegradabilità, in quanto è nota la sua suscettibilità all’attacco dell’enzima α-chimotripsina [Skarja et al., 2000]. In Figura 2-13 sono riportate le curve di perdita di peso dei poliuretani PU5 e PUU5 nel corso delle prove di degradazione/erosione in soluzione enzimatica e in soluzione tampone, rispettivamente. Entrambi i polimeri hanno mostrato una maggiore perdita di peso percentuale, quando sono stati esposti alla soluzione di chimotripsina, rispetto al caso della soluzione tampone. L’aumentata erosione del poliuretano PU5 in soluzione enzimatica suggerisce che l’enzima è capace di catalizzare l’idrolisi dei legami esterei del PCL e/o uretanici, essendo questi gli unici legami facilmente idrolizzabili, presenti nel poliuretano. 0 5 10 15 20 25 30 0 2 4 6 8 10 12 14 tempo (giorni) P er dit a di pe so ( w t % ) Chimotripsina: PUU5 PU5 Soluzione tampone: PU5 PUU5
Figura 2-13. Curve di perdita di peso dei poliuretani PU5 e PUU5 nel corso delle prove di degradazione/erosione in soluzione enzimatica e in soluzione tampone.
Il grado di erosione del PUU5 è più marcato di quello del PU5 in soluzione enzimatica, data la presenza di un estensore di catena (Phe diestere) maggiormente suscettibile all’attacco enzimatico. L’inclusione di residui di fenilalanina adiacenti ai legami esterei dovrebbe migliorare l’interazione dell’enzima con il sito di scissione idrolitica. In natura, infatti, la chimotripsina catalizza la scissione dei legami peptidici adiacenti a residui contenenti gruppi aromatici in catena laterale, come Phe o Tyr. E’ stato ipotizzato, sulla base di vari studi, che la degradazione mediata da chimotripsina di polimeri
contenenti fenilalanina deriva da una specifica interazione enzima-substrato, analoga all’idrolisi di proteine e peptidi in vivo [Skarja et al., 2001]. Inoltre, l’inserimento in catena del Phe diestere dovrebbe influenzare la degradazione enzimatica come risultato dell’aumento del contenuto di gruppi esterei. Il PUU5, infatti, contiene due legami esterei idrolizzabili in più per unità ripetitive, rispetto al PU5. Del resto, se la più alta concentrazione di legami esterei fosse la causa della maggiore erosione, si dovrebbe osservare un effetto simile anche per l’idrolisi in tampone.
La Figura 2-13 mostra che, in soluzione tampone, i gradi di erosione del PU5 e del PUU5 hanno mostrato differenze meno marcate che in soluzione enzimatica, tuttavia la perdita di peso percentuale del PU5 è risultata maggiore. Questo comportamento è stato attribuito al carattere maggiormente idrofilo del poliuretano PU5, che è stato precedentemente valutato da prove di assorbimento d’acqua [Rechichi, 2002-2003].
Le immagini SEM delle superfici dei campioni degradati per 12 giorni hanno consentito di valutare il grado di erosione superficiale, mentre quelle relative alle sezioni fratturate in azoto liquido, hanno permesso di estendere l’indagine alle zone interne.
La Figura 2-14 c-d, che si riferisce alla biodegradazione di un campione di PUU5, mostra una marcata alterazione superficiale, tipica dell’attacco enzimatico, con crepe profonde visibili anche nella sezione (Figura 2-14 d). Il materiale di partenza, al contrario, è un materiale compatto (Figura 2-14 a-b). Le Figure 2-14 e-f si riferiscono alla degradazione del PUU5 in soluzione tampone: tali immagini non mostrano segni rilevanti di alterazione della morfologia, confermando che la chimotripsina è responsabile dell’erosione del PUU5.
Per il PU5, la valutazione del grado di erosione è risultata in accordo con i risultati dell’analisi gravimetrica.
La Figura 2-15 c-d, relativa ad un provino di PU5 sottoposto a degradazione in soluzione enzimatica, mostra un livello di erosione superficiale maggiore rispetto alla degradazione in tampone (Figura 2-15 e-f).
Figura 2-14. Immagini SEM, relative alla superficie (a, c, e) e alla sezione (b, d, f) di campioni in forma di films per casting di PUU5: (a, b) prima della prova di degradazione; (c, d) dopo prova di degradazione in soluzione enzimatica; (e, f) dopo la prova di degradazione in soluzione tampone.
(a)
(b)
(c)
(e)
(d)
(f)
Figura 2-15. Immagini SEM, relative alla superficie (a, c, e) e alla sezione (b, d, f) di campioni in forma di films per casting di PU5: (a, b) prima della prova di degradazione; (c, d) dopo prova di degradazione in soluzione enzimatica; (e, f) dopo la prova di degradazione in soluzione tampone.
L’entità della degradazione polimerica è stata determinata tramite analisi GPC su campioni esposti al trattamento per 4, 8, 12 giorni. Non è stato registrato nessun cambiamento significativo del peso molecolare medio ponderale dei campioni di PU5 e di PUU5 in soluzione tampone e del campione di PU5 in soluzione enzimatica. In questi tre casi, infatti, si sono registrati solo fenomeni di erosione superficiale, mentre l’analisi GPC rileva fenomeni di degradazione di massa. Al contrario, il PUU5 in soluzione enzimatica ha mostrato una diminuzione nel peso molecolare medio ponderale, crescente al crescere del tempo di esposizione in soluzione enzimatica (Figura 2-16). La degradazione enzimatica, infatti, avviene tramite erosione superficiale; successivamente, si verificano fenomeni di degradazione di massa, a seguito della formazione di profonde crepe sui campioni, come confermato dall’analisi SEM.
(a)
(b)
(e)
(c)
(d)
Figura 2-16. Andamento del peso molecolare del PUU5 in funzione del tempo di esposizione in soluzione enzimatica di chimotripsina.
2.3.4 Test di citotossicità
I test di citotossicità sono test di adesione cellulare sui mezzi di coltura nei quali sono stati immersi i campioni dei polimeri di sintesi per un certo tempo: valutare l’affinità delle cellule verso tali mezzi di coltura significa verificare l’assenza di rilasci tossici da parte dei materiali. Su ogni mezzo di coltura (nel quale sono stati immersi per un certo tempo i campioni) sono stati eseguiti test di adesione cellulare a 4, 8, 24 h.
La Figura 2-17 riporta i risultati del test di citotossicità eseguito sul copolimero C27. Nel caso del copolimero, è evidente l’assenza di rilasci nocivi alle cellule da parte del polimero: l’adesione cellulare è elevata su tutti i mezzi di coltura prelevati e arriva al 100% dopo 8 ore di coltura cellulare. Questi risultati confermano la biocompatibilità del copolimero C27 e, quindi, la sua idoneità ad impieghi nel settore della Tissue Engineering.
La Figura 2-18 mostra i risultati dei test di citotossicità per il poliuretano ad alto peso molecolare (PU4-H). I test di citotossicità per gli altri poliuretani e poliuretani-uree hanno fornito risultati analoghi a quelli del PU4-H, pertanto non sono stati riportati i relativi grafici.
La Figura 2-18 mostra che, per i mezzi di coltura prelevati a tempi brevi (4 giorni), eventuali rilasci da parte del polimero PU4-H hanno limitato l’adesione cellulare. Per i mezzi prelevati a tempi successivi (7 giorni), il comportamento cellulare è risultato molto buono, ma ha subito un peggioramento per i mezzi di coltura prelevati dopo 10 e 15 giorni. Questi risultati hanno suggerito la necessità di allontanare gli eventuali monomeri e oligomeri residui del processo di sintesi, attraverso una serie di dissoluzioni e precipitazioni successive del polimero.
Prima del loro impiego in forma di scaffolds, quindi, i PU e PUU sono stati purificati. 0 20 40 60 80 100 0 4 8 12 tempo (giorni) [M w (t ) / M w (t0 )] x 100
C27 80 85 90 95 100 105 0 5 10 15 20 giorni
(di prelievo dei mezzi di coltura)
% ar ea oc cup at a d alle c ellu le 4 ore 8 ore 24 ore
Figura 2-17. Risultati dei test di citotossicità per il copolimero C27: andamento della percentuale di cellule vitali sui terreni di coltura che sono stati in contatto con campioni di C27 per il tempo indicato in ascissa, dopo una coltura cellulare di 4, 8, 24 h.
PU4-H 0 20 40 60 80 100 120 0 5 10 15 20 Giorni
(di prelievo dei mezzi di coltura)
% a rea o ccu p at a d alle c ellu le 4 ore 8 ore 24 ore
Figura 2-18. Risultati dei test di citotossicità per il poliuretano PU4-H: andamento della percentuale di cellule vitali sui terreni di coltura che sono stati in contatto con campioni di PU4-H per il tempo indicato in ascissa, dopo una coltura cellulare di 4, 8, 24 h.
2.3.5 Analisi tramite microscopio a forza atomica (AFM)
La tecnica di microscopia a forza atomica è stata applicata allo scopo di studiare la morfologia di film ottenuti per spin-coating di poliuretani, poliuretani-uree e del copolimero C27. La rugosità della superficie dei campioni, infatti, influenza l’adesione cellulare [Vance et al., 2004].
Tutti i campioni ottenuti per casting hanno mostrato una superficie rugosa, il cui parametro caratteristico è stata la rugosità quadratica media.
La Tabella 2-7 riporta i valori di rugosità quadratica media (r.m.s.) per tutti i campioni analizzati. Si osserva che, per i poliuretani, il parametro r.m.s. è risultato maggiore rispetto al caso dei poliuretani-uree, a parità di segmento soft. Per i poliuretani il cui segmento soft è costituito da PCL (PU4-H e
Tempo dei test cellulari:
Tempo dei test cellulari:
PU4), la rugosità quadratica media è risultata crescente con il peso molecolare. Il valore più basso di rugosità è stato misurato per il campione di copolimero C27.
La Figura 2-19 mostra la morfologia dei campioni esaminati, la cui area di scansione vale 5×5 µm2.
Tabella 2-7 Valori della rugosità quadratica media di film ottenuti per spin-coating Campione Rugosità quadratica media, r.m.s.
(nm) PU4-H 20.9 PU4 16.3 PU5 15.4 PU6 24.4 PUU4 4.8 PUU5 6.1 PUU6 6.2 C27 2.8
PU4-H PU4 PU5
PU6 PUU4 PUU5
PUU6 C27
Figura 2-19. Immagini morfologiche ottenute tramite AFM di film per casting di poliuretani, poliuretani-uree e copolimero C27, area di scansione 5µm×5µm.
Le acquisizioni sono state fatte, applicando alla punta AFM potenziali diversi (70 mV e 400 mV): in entrambi i casi, tuttavia, non si sono riscontrate differenze nella topografia superficiale, perché, in modalità di contatto, il contributo della forza elettrostatica è molto inferiore alla forza di contatto. Il
valore della forza elettrostatica è stato ricavato dalla misura degli shift di risonanza: per shift misurati (∆ω) dell’ordine di circa 104 Hz e per distanze dell’ordine dei 10 nm, si è ottenuta una forza
elettrostatica dell’ordine di 0.7 nN. In generale, i valori ricavati di forza elettrostatica, pure essendo viziati da grosse fluttuazioni, sono risultati dell’ordine di 0.1-0.9 nN per tutti i campioni. Per tali valori di forza elettrostatica e per un potenziale applicato alla punta di 400 mV, si ottiene una capacità (C) di circa 5×10-17 Farad.
2.3.6 Misurazione dell’angolo di contatto
L’adesione cellulare dipende da vari fattori tra cui il grado di idrofilia superficiale: le superfici con bagnabilità intermedia favoriscono l’adesione cellulare [Saltzman et al., 1997; Vogler et al., 1998; Van Wachem et al., 1985].
I film ottenuti per spin-coating di PCL e dei polimeri di sintesi hanno mostrato superfici idrofile, i cui angoli di contatto sono riportati in Tabella 2-8. Le caratteristiche idrofile dei campioni sono risultate favorevoli per l’adesione cellulare.
Tabella 2-8. Valori dell’angolo di contatto per i campioni indicati
Campione Angolo di contatto
PCL 65° C27 40° PU4-H 84°
2.3.7 Risultati derivanti dalla tecnica di ellissometria
La tecnica di ellissometria è stata utilizzata per misurare l’indice di rifrazione e lo spessore dei films del copolimero di sintesi C27 e del poliuretano PU4-H. La misura dell’indice di rifrazione è di fondamentale importanza perché permette di calcolare il valore della costante dielettrica del materiale in questione (che si può approssimare come il quadrato dell’indice di rifrazione). Questo è un dato difficilmente reperibile senza il quale risulta impossibile la valutazione della carica superficiale. La misura della carica superficiale di un biopolimero, a sua volta, è un importante e utile indicatore dell’interazione superficiale del materiale con le cellule: le cellule tendono ad avere un contatto continuo con substrati caricati positivamente, dal momento che la membrana cellulare è caratterizzata da una densità di carica superficiale negativa.
La Tabella 2-9 riporta i valori dell’indice di rifrazione e della costante dielettrica dei materiali esaminati.
Tabella 2-9. Valori dell’indice di rifrazione e della costante dielettrica per i polimeri esaminati. Indice di rifrazione, n Costante dielettrica, ε Materiali Valore Deviazione standarda Valore Deviazione standarda
PU4-H 1.46 0.00231 2.12 5.94E-6
C27 1.46 0.00390 2.14 1.52E-5
aI valori sono stati mediati su 27 misure.
La tecnica ellissometrica permette anche di calcolare lo spessore dei films polimerici. Lo spessore dei
films polimerici analizzati è risultato pari a 9.6 (± 1.15) µm per il PU4-H e 0.1 (± 0.01) µm per il copolimero C27. Per il C27, films più spessi del valore utilizzato sono risultati opachi e, pertanto, non adatti all’analisi ellissometrica.
Mediante un software sviluppato in Matlab, è stato possibile ottenere mappe che riportassero gli andamenti dell’indice di rifrazione e dello spessore dei films polimerici (Figure 2-20, 2-21).